
Transcriptomic and Metabolomic Analyses Reveal Differences in Flavonoid Pathway Gene Expression Profiles between Two Dendrobium Varieties during Vernalization

 , ,
, ,
Abstract
:1. Introduction
2. Results
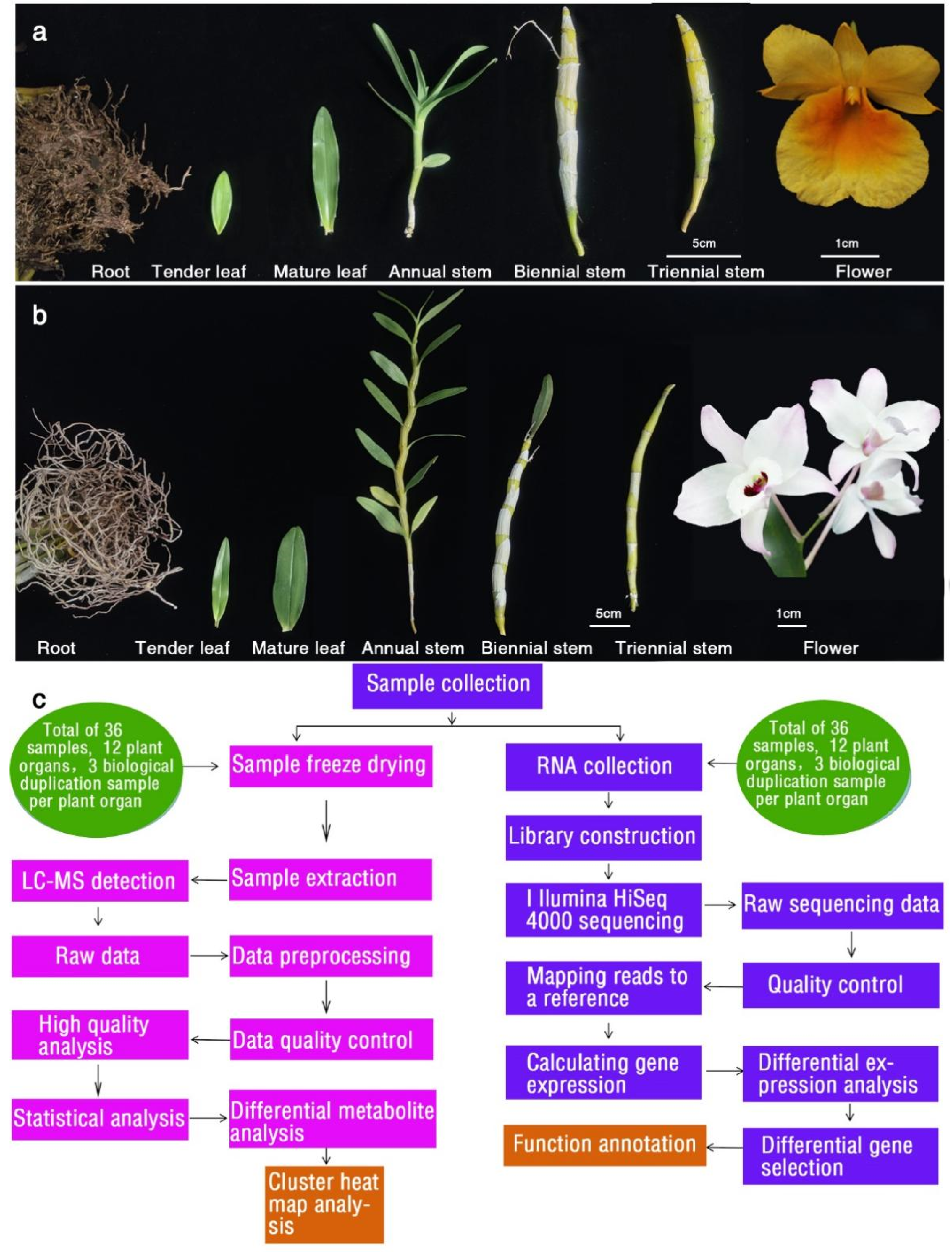
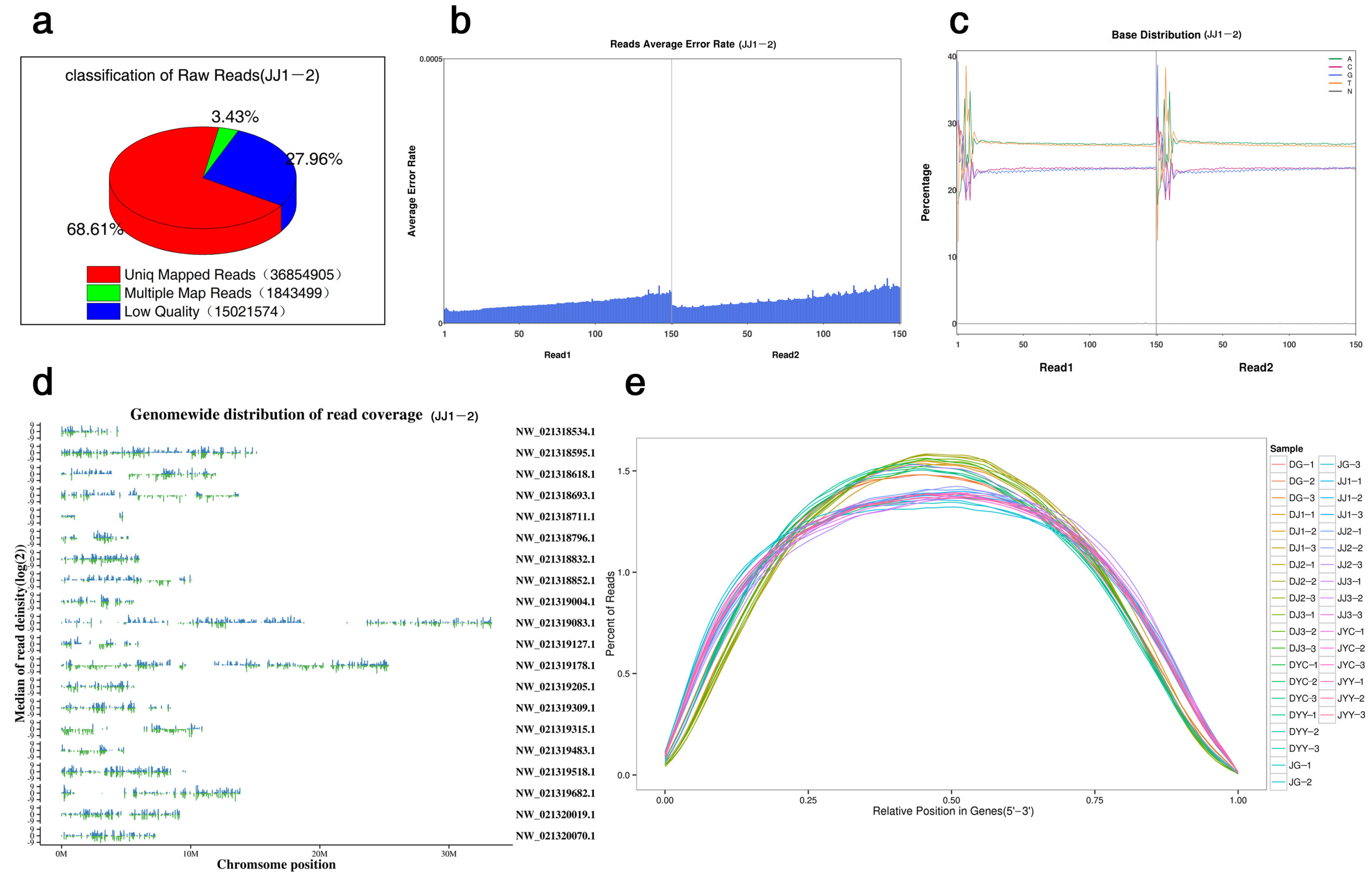
2.1. General Description of the RNA Sequencing Data
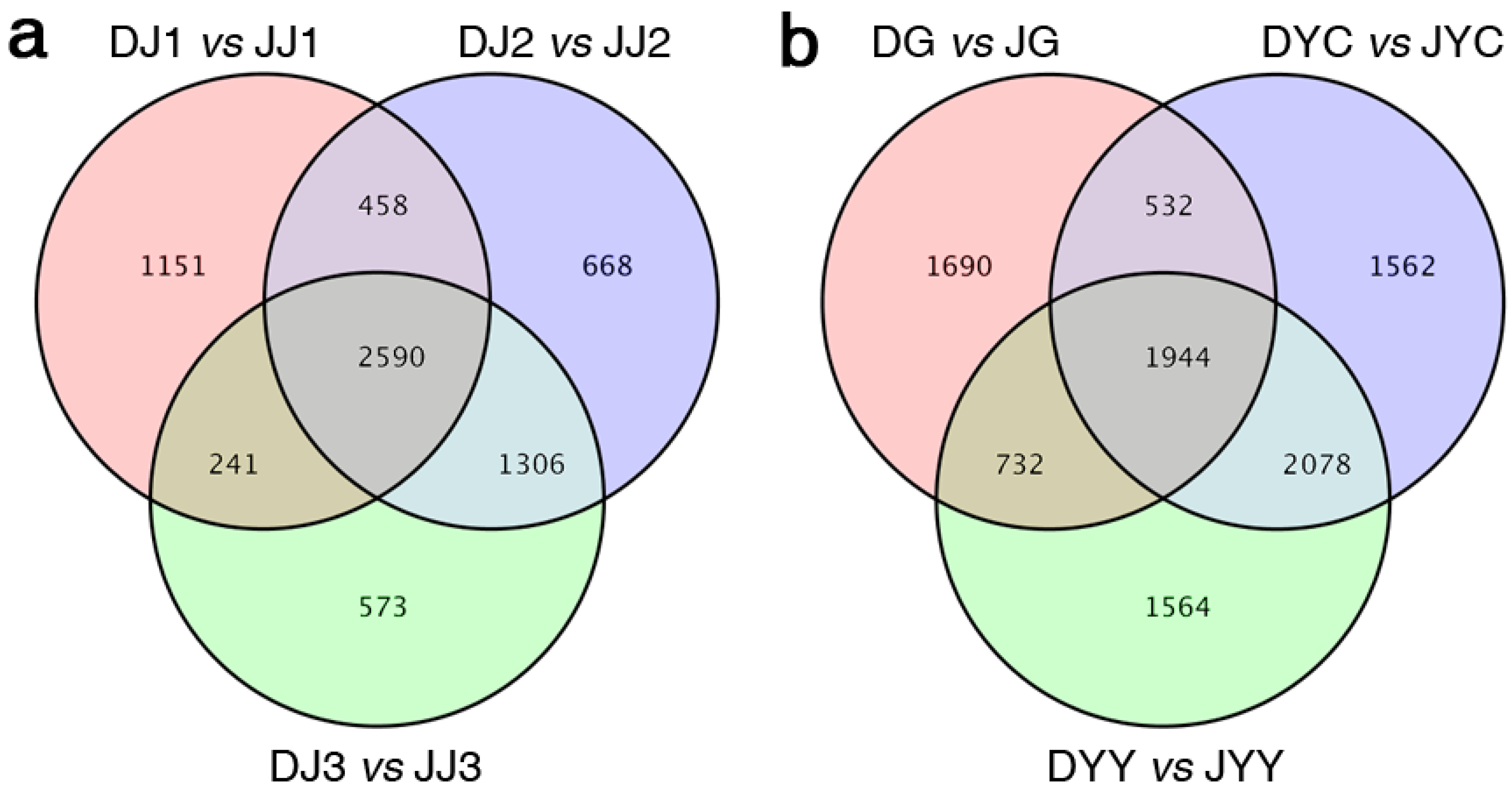
2.2. Functional Enrichment Analysis of DEGs
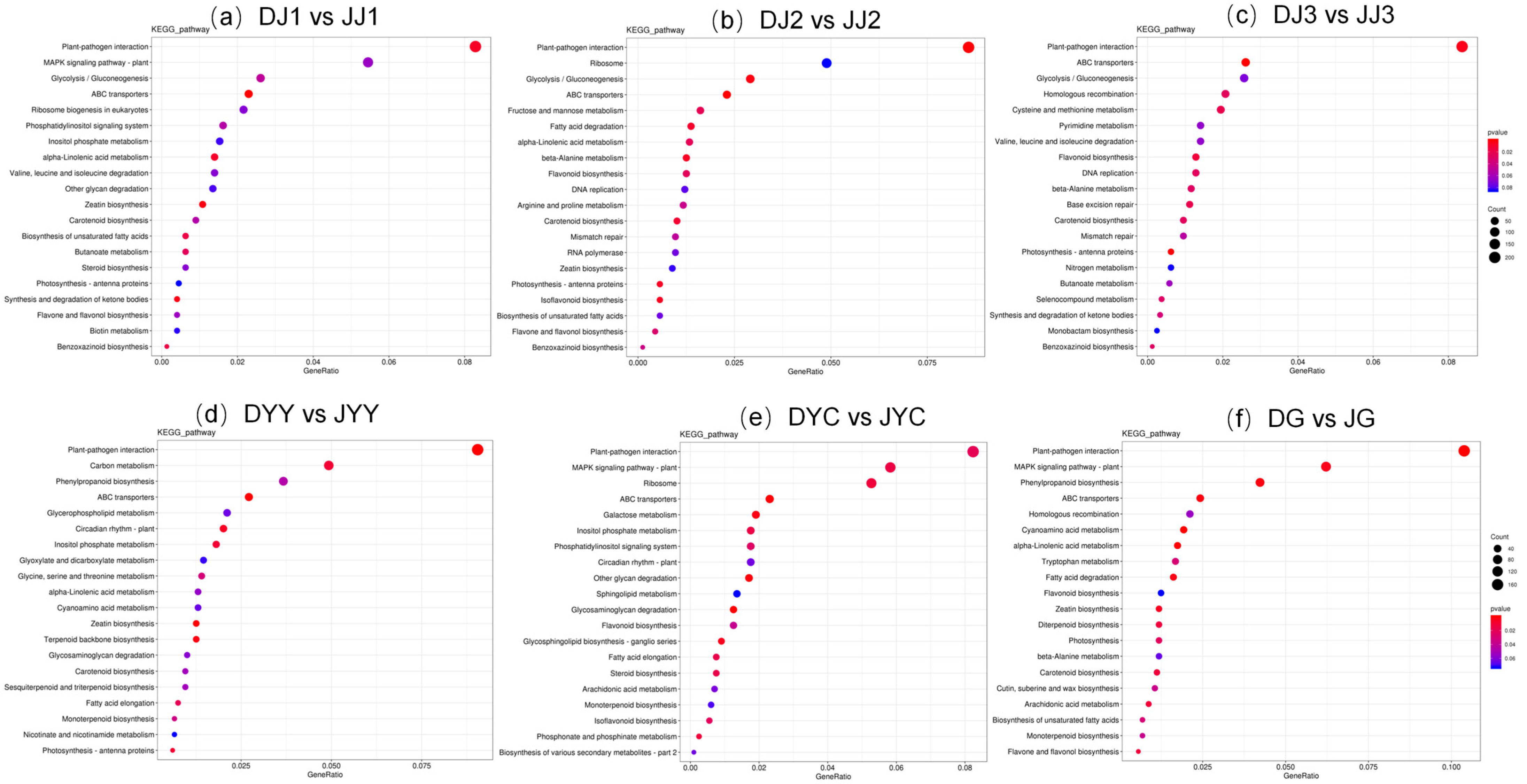
2.3. KEGG Pathway Enrichment Analyses of DEGs
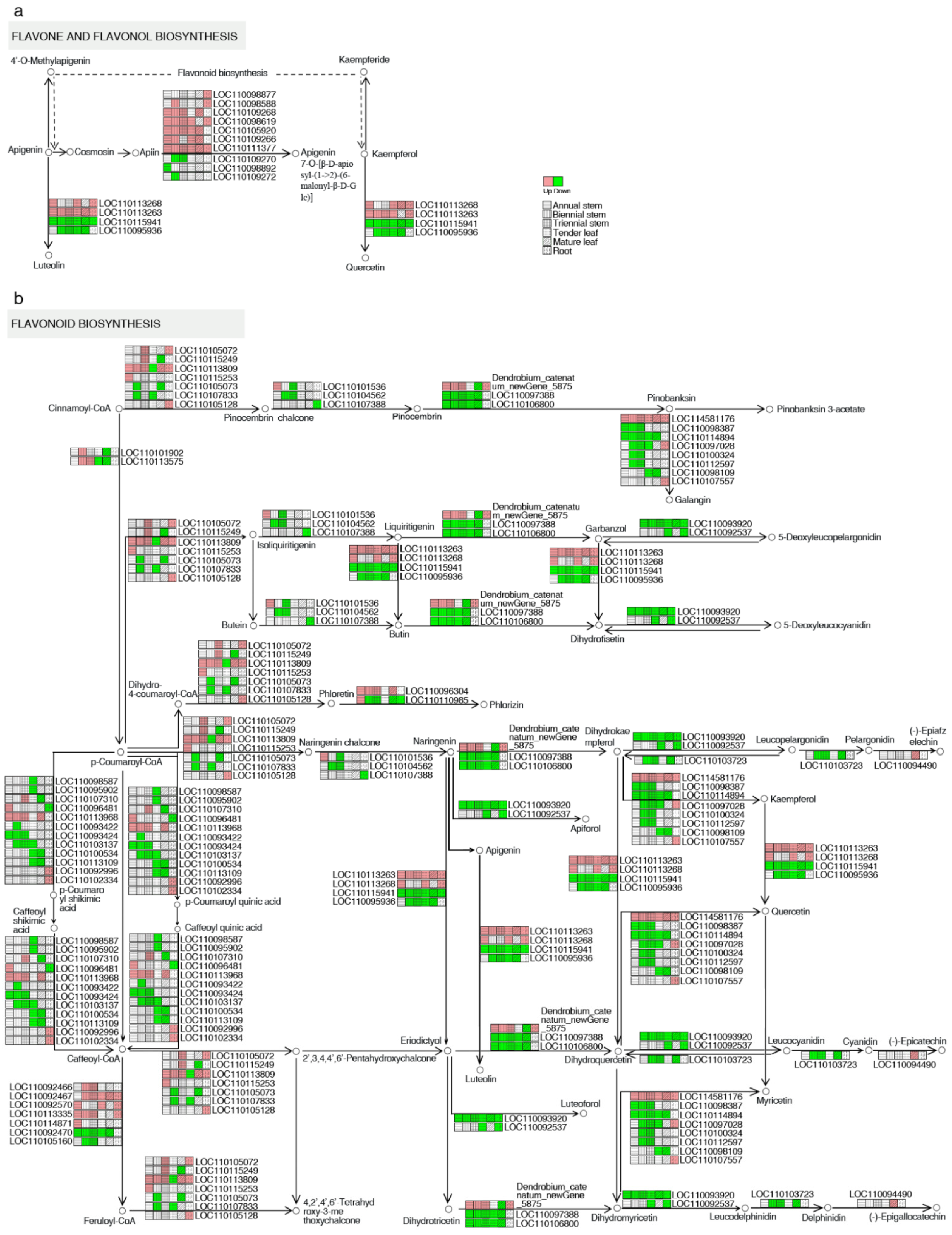
2.4. Analysis of DEGs Related to the Flavonoid Biosynthesis Pathway
2.5. Validation of DEGs by qRT–PCR
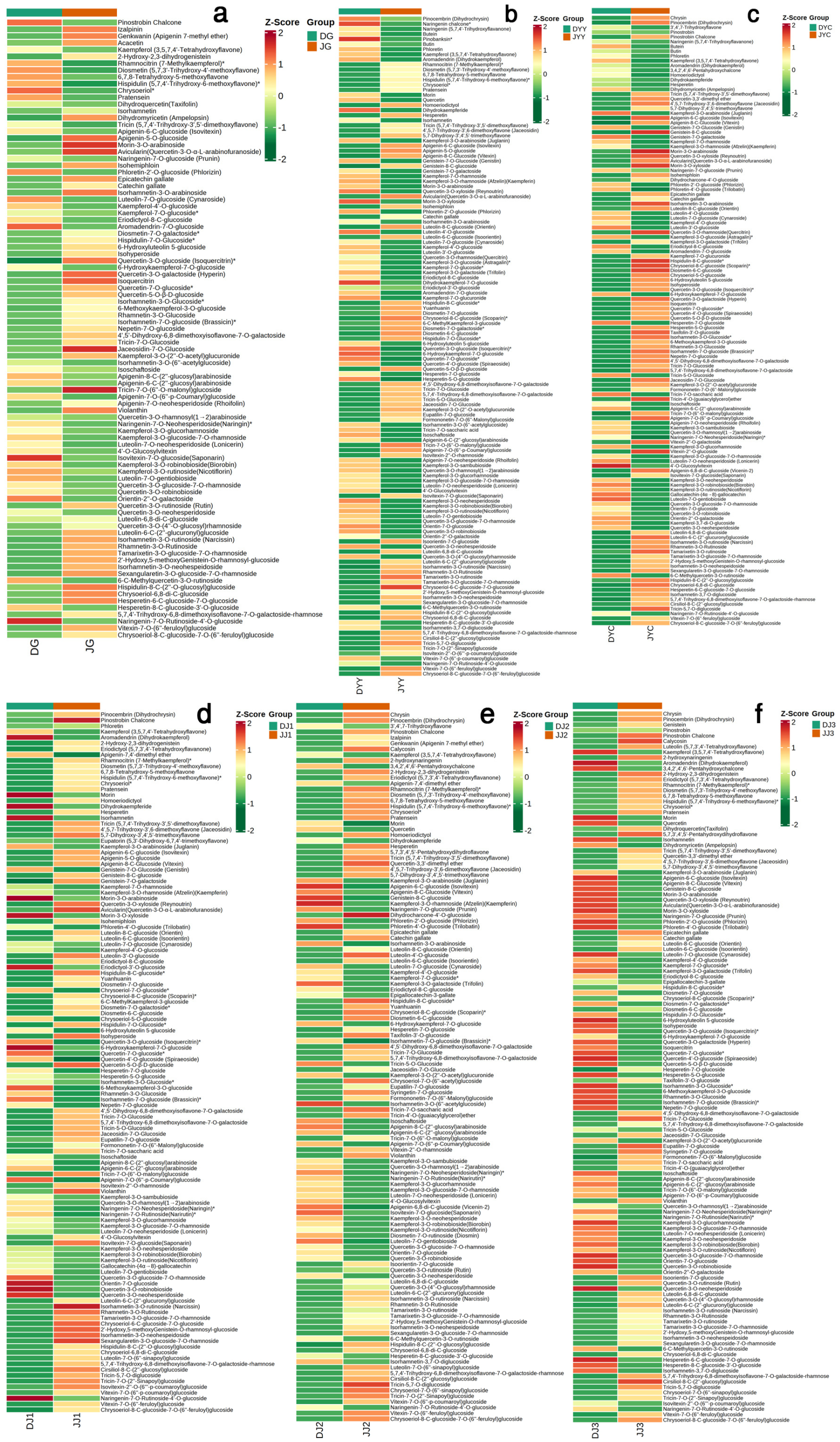
2.6. Differential Metabolite (DAM) Analysis in Different Tissues during Vernalization
2.7. Integrated Analysis of Transcriptomics and Metabolomics
3. Discussion
4. Materials and Methods
4.1. Overview of Experimental Design and Sample Preparation
4.2. Sample Extraction and Analysis of Flavonoid Metabolites
4.3. RNA Extraction, Library Construction, and RNA Sequencing (RNA-Seq)
4.4. Quantification of Gene Transcription Level and Analysis of DEGs
4.5. Quantitative Real-Time PCR (qRT–PCR) Validation
4.6. Statistical Analysis
4.7. Data Records
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wen, Z.; Guo, W.; Li, J.; Lin, H.; He, C.; Liu, Y.; Zhang, Q.; Liu, W. Comparative Transcriptomic Analysis of Vernalization- and Cytokinin-Induced Floral Transition in Dendrobium nobile. Sci. Rep. 2017, 7, 45748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, X.; Qi, J.; Zhou, B.; Mao, B. Metabolomic and transcriptomic analyses reveal the regulation of pigmentation in the purple variety of Dendrobium officinale. Sci. Rep. 2020, 10, 17700. [Google Scholar] [CrossRef]
- Cheng, J.; Dang, P.P.; Zhao, Z.; Yuan, L.C.; Zhou, Z.H.; Wolf, D.; Luo, Y.B. An assessment of the Chinese medicinal Dendrobium industry: Supply, demand and sustainability. J. Ethnopharmacol. 2019, 229, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhang, B.; Tang, X.; Zhang, J.; Lin, J. Comparative Transcriptome Analysis of Different Dendrobium Species Reveals Active Ingredients-Related Genes and Pathways. Int. J. Mol. Sci. 2020, 21, 861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.G.; Hu, Y.D.; Zhao, R.X.; Yan, S.; Zhang, X.Q.; Zhao, T.M.; Chun, Z. Genome-wide researches and applications on Dendrobium. Planta 2018, 248, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.-E.; Schwarzacher, T.; Othman, R.Y.; Harikrishna, J.A. dsRNA silencing of an R2R3-MYB transcription factor affects flower cell shape in a Dendrobium hybrid. BMC Plant Biol. 2015, 15, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, F.; Zhao, L.; Wu, X.; Song, Z.; Li, Y. Metabolome and transcriptome analyses of the molecular mechanisms of flower color mutation in tobacco. BMC Genom. 2020, 21, 611. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Yuan, Y. A Comparison of the Flavonoid Biosynthesis Mechanisms of Dendrobium Species by Analyzing the Transcriptome and Metabolome. Int. J. Mol. Sci. 2022, 23, 11980. [Google Scholar] [CrossRef]
- Lou, H.; Hu, L.; Lu, H.; Wei, T.; Chen, Q. Metabolic Engineering of Microbial Cell Factories for Biosynthesis of Flavonoids: A Review. Molecules 2021, 26, 4522. [Google Scholar] [CrossRef]
- Zheng, B.Q.; Deng, X.M.; Li, K.; Miao, K.; Wang, Y. Effects of Temperature Treatment on Flower Bud Differentiation and Development of Dendrobium. For. Res. 2017, 30, 460–464. [Google Scholar]
- Yen, C.Y.T.; Starman, T.; Wang, Y.T.; Niu, G. Effects of cooling temperature and duration on flowering of the nobile Dendrobium orchid. Hortsci. A Publ. Am. Soc. Hortic. Sci. 2008, 43, 1765–1769. [Google Scholar]
- Liang, S.; Ye, Q.S.; Li, R.H.; Leng, J.Y.; Li, M.R.; Wang, X.J.; Li, H.Q. Transcriptional Regulations on the Low-Temperature-Induced Floral Transition in an Orchidaceae Species, Dendrobium nobile: An Expressed Sequence Tags Analysis. Comp. Funct. Genom. 2012, 2012, 757801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.; Starman, T.W.; Wang, Y.T.; Niu, G.H.; Cothren, J.T. Deferring flowering of nobile dendrobium hybrids by holding plants under low temperature after vernalization. Sci. Hortic. 2011, 130, 869–873. [Google Scholar] [CrossRef]
- Yeh, Y.-T.; Shen, R.-S.; Huang, K.-L.; Miyajima, I. Effect of Two-stage Vernalization and Temperature Treatment at the Stage of Floral Bud Development on Flowering in Nobile Dendrobium. J. Fac. Agric. Kyushu Univ. 2021, 66, 163–171. [Google Scholar]
- Monteiro, M.S.; Carvalho, M.; Bastos, M.L.; de Pinho, P.G. Metabolomics analysis for biomarker discovery: Advances and challenges. Curr. Med. Chem. 2013, 20, 257–271. [Google Scholar] [CrossRef]
- Yuan, Y.; Zuo, J.; Zhang, H.; Zu, M.; Yu, M.; Liu, S. Transcriptome and metabolome profiling unveil the accumulation of flavonoids in Dendrobium officinale. Genomics 2022, 114, 110324. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Z.; Wu, Y.; Zheng, L.; Zhang, G. Regulatory Mechanisms of Anthocyanin Biosynthesis in Apple and Pear. Int. J. Mol. Sci. 2021, 22, 8441. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, S.; Tong, M.; Lu, J.; Wang, T.; Xu, Y.; Li, W.; Wang, L. Physiological and genetic analysis of leaves from the Resprouters of an old Ginkgo biloba tree. Forests 2021, 12, 1255. [Google Scholar] [CrossRef]
- Belkheir, A.K.; Gaid, M.; Liu, B.; Hänsch, R.; Beerhues, L. Benzophenone Synthase and Chalcone Synthase Accumulate in the Mesophyll of Hypericum perforatum Leaves at Different Developmental Stages. Front. Plant Sci. 2016, 7, 921. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Zhang, J.; Liu, X.; Meng, M.; Wang, J.; Lin, J. Tissue-specific transcriptome for Dendrobium officinale reveals genes involved in flavonoid biosynthesis. Genomics 2020, 112, 1781–1794. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, N.; Wang, P.; Liu, J.; Sun, J.; Ye, W.; Fan, B. Flavonoid biosynthesis in four Dendrobium species based on transcriptome sequencing and metabolite analysis. Mol. Biol. Rep. 2022, 49, 2047–2057. [Google Scholar] [CrossRef] [PubMed]
- Xing, A.; Wang, X.; Nazir, M.F.; Zhang, X.; Wang, X.; Yang, R.; Chen, B.; Fu, G.; Wang, J.; Ge, H.; et al. Transcriptomic and metabolomic profiling of flavonoid biosynthesis provides novel insights into petals coloration in Asian cotton (Gossypium arboreum L.). BMC Plant Biol. 2022, 22, 416. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jiang, X.; Chen, L.; Li, W.-W.; Lai, S.; Fu, Z.; Liu, Y.; Qian, Y.; Gao, L.; Xia, T. Functional analyses of Flavonol synthase genes from Camellia sinensis reveal their roles in anther development. Front. Plant Sci. 2021, 12, 753131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, Y.; Adejobi, O.I.; Wang, Y.; Liu, A. Research Advances in Multi-Omics on the Traditional Chinese Herb Dendrobium officinale. Front. Plant Sci. 2022, 12, 808228. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, C.; Di Ferdinando, M.; Fini, A.; Pollastri, S.; Tattini, M. Flavonoids as antioxidants and developmental regulators: Relative significance in plants and humans. Int. J. Mol. Sci. 2013, 14, 3540–3555. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, X.; Wang, X.; Chang, T.; Peng, Z.; Guan, C.; Guan, M. Flavonoid Synthesis-Related Genes Determine the Color of Flower Petals in Brassica napus L. Int. J. Mol. Sci. 2023, 24, 6472. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, L.-J.; Wang, Y.; Geng, Z.; Liu, S.; Chen, C.; Chen, S.; Jiang, J.; Chen, F. CmMYB9a activates floral coloration by positively regulating anthocyanin biosynthesis in chrysanthemum. Plant Mol. Biol. 2022, 108, 51–63. [Google Scholar] [CrossRef]
- Zou, H.; Zhou, L.; Han, L.; Lv, J.; Jia, Y.; Wang, Y. Transcriptome profiling reveals the roles of pigment formation mechanisms in yellow Paeonia delavayi flowers. Mol. Genet. Genom. MGG 2023, 298, 375–387. [Google Scholar] [CrossRef]
- Zhu, H.-H.; Yang, J.-X.; Xiao, C.-H.; Mao, T.-Y.; Zhang, J.; Zhang, H.-Y. Differences in flavonoid pathway metabolites and transcripts affect yellow petal colouration in the aquatic plant Nelumbo nucifera. BMC Plant Biol. 2019, 19, 277. [Google Scholar] [CrossRef]
- Tapas, D.A.; Sakarkar, D.M.; Kakde, R.B. Flavonoids as nutraceuticals: A review. Trop. J. Pharm. Res. 2008, 7, 1089–1099. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Gao, Y.; Chen, W.; Wang, W.; Gong, L.; Liu, X.; Luo, J. Spatiotemporal distribution of phenolamides and the genetics of natural variation of hydroxycinnamoyl spermidine in rice. Mol. Plant 2015, 8, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Meyer, R.S.; Yang, T.; Whitaker, B.D.; Trouth, F.; Shangguan, L.; Huang, J.; Litt, A.; Little, D.P.; Ke, H.; et al. A novel hydroxycinnamoyl transferase for synthesis of hydroxycinnamoyl spermine conjugates in plants. BMC Plant Biol. 2019, 19, 261. [Google Scholar] [CrossRef] [PubMed]
- Elejalde-Palmett, C.; de Bernonville, T.D.; Glevarec, G.; Pichon, O.; Papon, N.; Courdavault, V.; St-Pierre, B.; Giglioli-Guivarc’h, N.; Lanoue, A.; Besseau, S. Characterization of a spermidine hydroxycinnamoyltransferase in Malus domestica highlights the evolutionary conservation of trihydroxycinnamoyl spermidines in pollen coat of core Eudicotyledons. J. Exp. Bot. 2015, 66, 7271–7285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Z.; Zhu, F.; Fan, Y.; Li, C.; Zhang, B.; Zhu, S.; Hou, Z.; Wang, M.; Yang, J.; Xue, Q.; et al. The chromosome-level reference genome assembly for Dendrobium officinale and its utility of functional genomics research and molecular breeding study. Acta Pharm. Sin. B 2021, 11, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Lu, H.; Tian, C.; Xu, T.; Song, C.; Wang, W.; Wei, P.; Gu, F.; Liu, D.; Cai, Y.; et al. Selection of Suitable Reference Genes for Gene Expression Normalization Studies in Dendrobium huoshanense. Genes 2022, 13, 1486. [Google Scholar] [CrossRef]
- Feng, G.; Wu, J.; Yi, H. Global tissue-specific transcriptome analysis of Citrus sinensis fruit across six developmental stages. Sci. Data 2019, 6, 153. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Gene ID | Upregulated | Downregulated |
|---|---|---|---|
| Flavonoid 3′-monooxygenase | LOC110113263 | all five groups | |
| Isoflavone 7-O-glucoside-6″-O-malonyltransferase | LOC110111377 | all five groups | |
| Flavonol synthase 6 | LOC114581176 | all five groups | |
| Flavonoid 3′-monooxygenase | LOC110115941 | all five groups | |
| Dihydroflavonol 4-reductase/flavanone 4-reductase | LOC110093920 | all five groups | |
| Shikimate O-hydroxycinnamoyltransferase | LOC110093422 | 2-year-old stems for all five groups | |
| Isoflavone 7-O-glucoside-6″-O-malonyltransferase | LOC110098588 | 2-year-old stems for three stem tissues | |
| Trans-cinnamate 4-monooxygenase | LOC110101902 | 2-year-old stems for three stem tissues | |
| Isoflavone 7-O-glucoside-6″-O-malonyltransferase | LOC110109272 | 2-year-old stems for three stem tissues | |
| Chalcone synthase | LOC110105073 | 2-year-old stems for three stem tissues | |
| Chalcone synthase 8 | LOC 110107833 | 2-year-old stems for three stem tissues |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, W.; Shi, M.; Zhang, Q.; Xie, W.; Chu, L.; Qiu, M.; Li, L.; Zeng, Z.; Han, L.; Sun, Z. Transcriptomic and Metabolomic Analyses Reveal Differences in Flavonoid Pathway Gene Expression Profiles between Two Dendrobium Varieties during Vernalization. Int. J. Mol. Sci. 2023, 24, 11039. https://doi.org/10.3390/ijms241311039
Shu W, Shi M, Zhang Q, Xie W, Chu L, Qiu M, Li L, Zeng Z, Han L, Sun Z. Transcriptomic and Metabolomic Analyses Reveal Differences in Flavonoid Pathway Gene Expression Profiles between Two Dendrobium Varieties during Vernalization. International Journal of Molecular Sciences. 2023; 24(13):11039. https://doi.org/10.3390/ijms241311039
Chicago/Turabian StyleShu, Wenbo, Meirong Shi, Qiqi Zhang, Wenyu Xie, Liwei Chu, Mingxuan Qiu, Linyan Li, Zhixin Zeng, Lei Han, and Zhenyuan Sun. 2023. "Transcriptomic and Metabolomic Analyses Reveal Differences in Flavonoid Pathway Gene Expression Profiles between Two Dendrobium Varieties during Vernalization" International Journal of Molecular Sciences 24, no. 13: 11039. https://doi.org/10.3390/ijms241311039