
TRIM21 Promotes Rabies Virus Production by Degrading IRF7 through Ubiquitination
 , and
, and
Abstract
:1. Introduction
2. Results
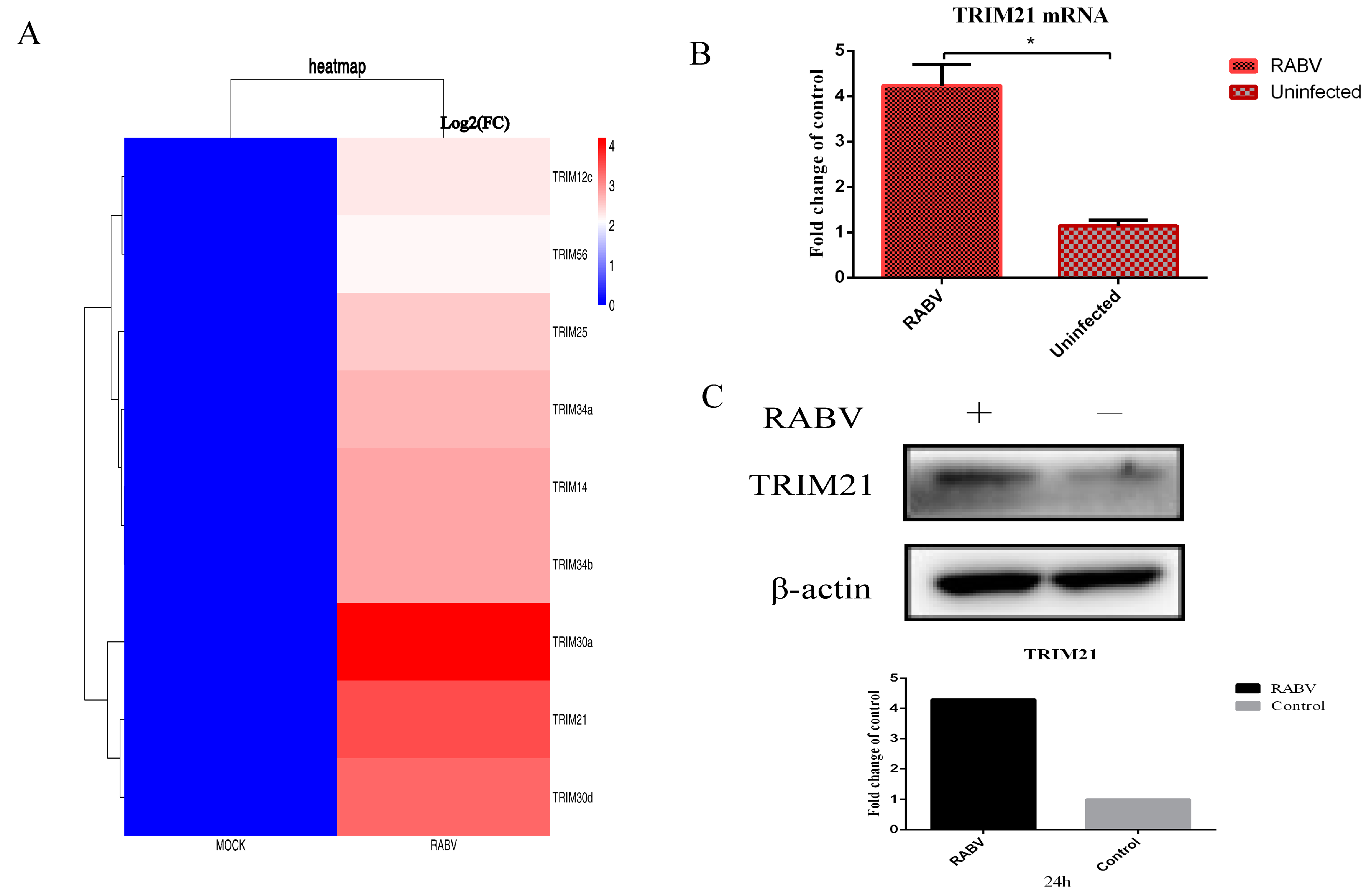
2.1. The Infection of RABV Results in Increased Expression of TRIM21 in NA Cells
2.2. TRIM21 Positively Regulates RABV Replication
2.3. TRIM21 Promotes RABV by Increasing the Expression of Type-I Interferon
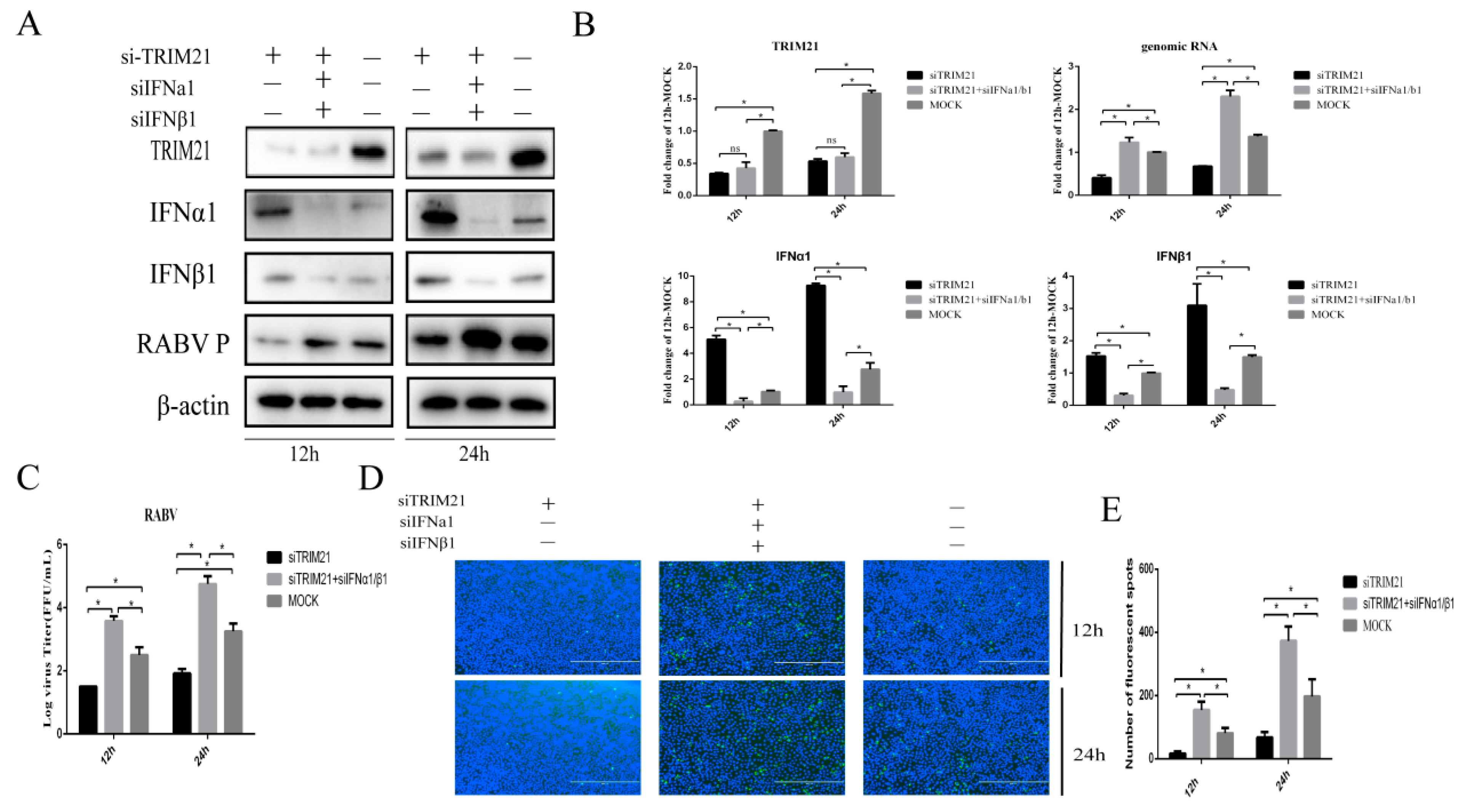
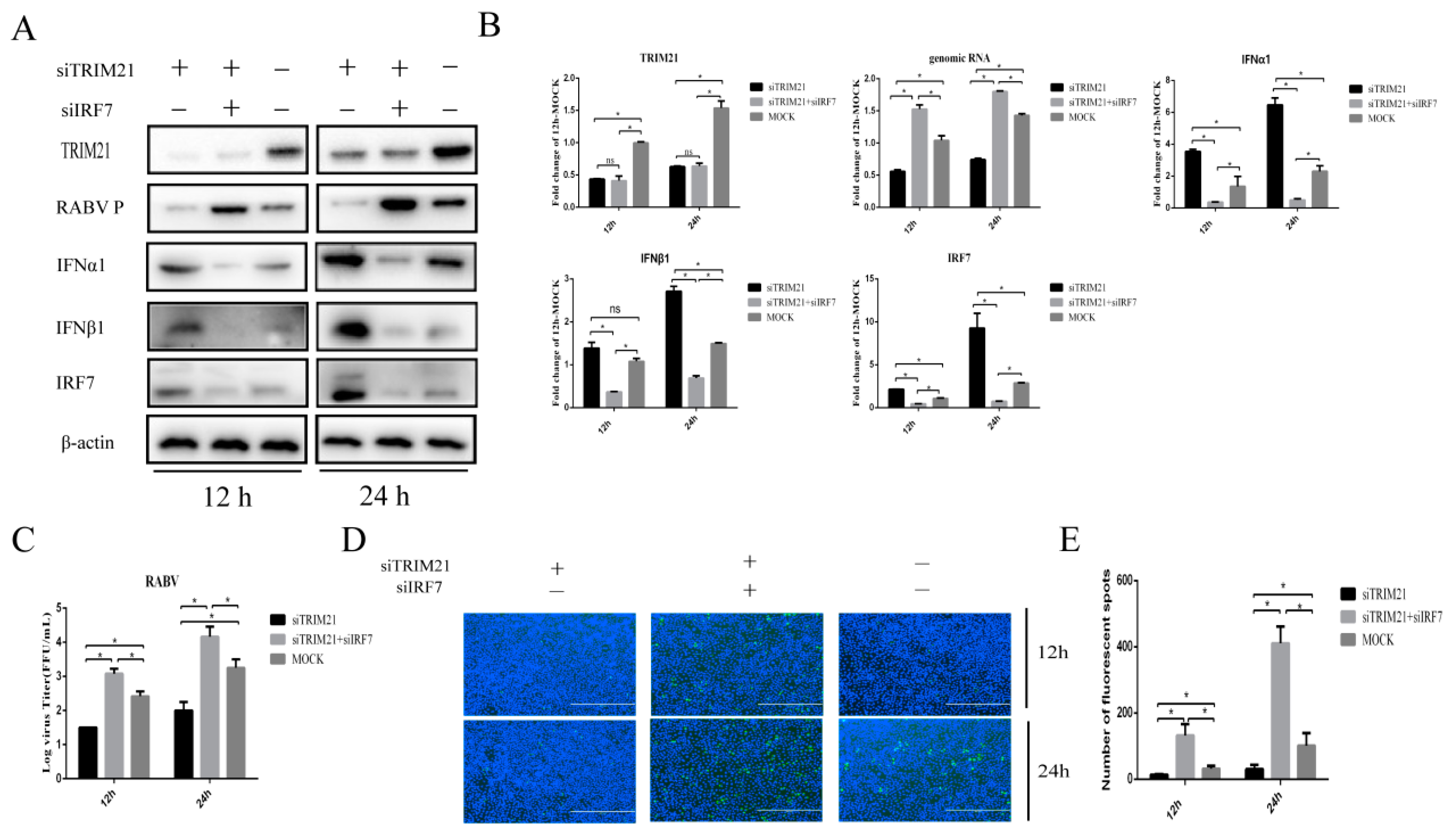
2.4. TRIM21 Regulates IFN Expression through IRF7 during RABV Infection
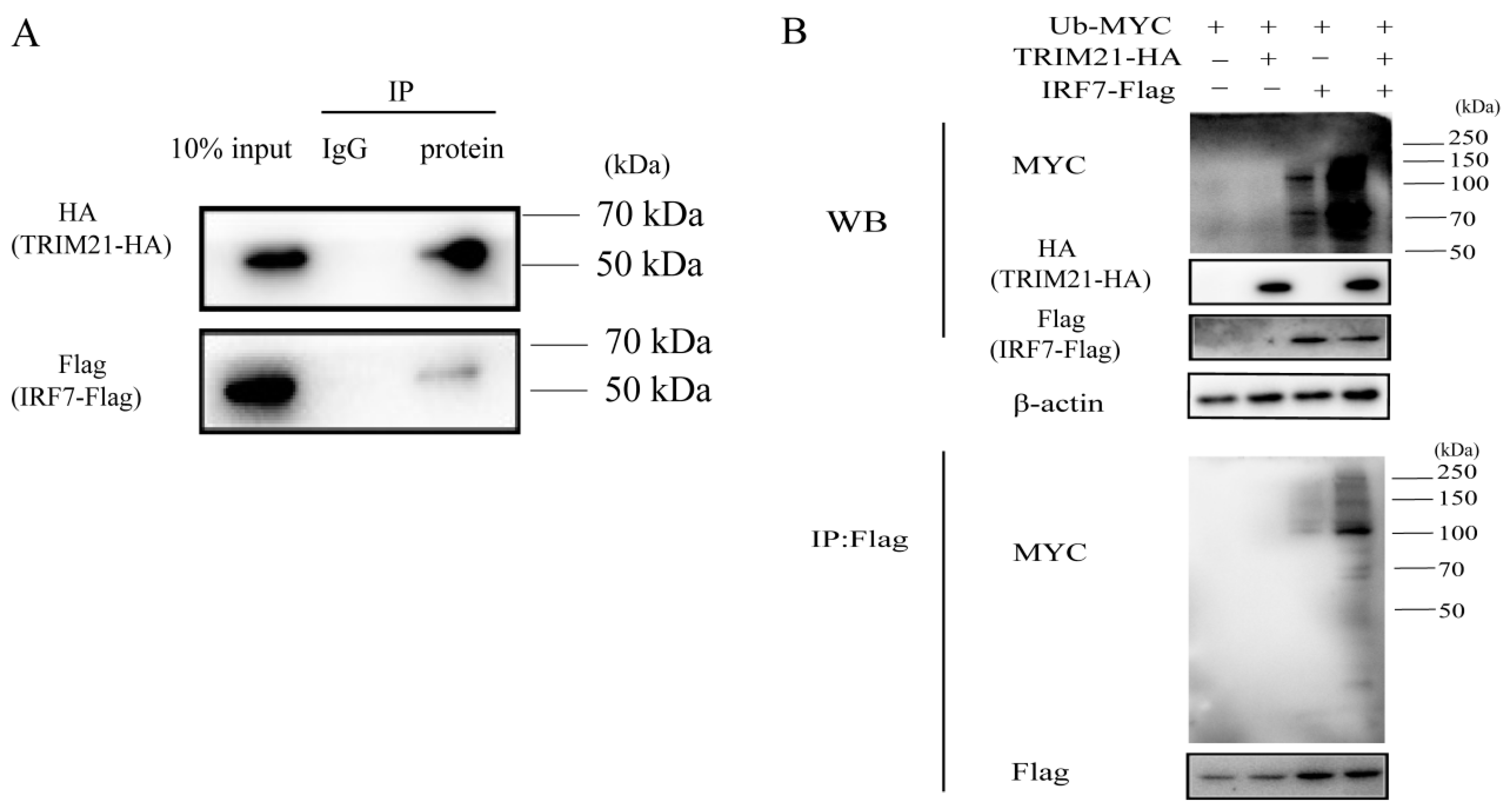
2.5. TRIM21 Ubiquitination Degrades IRF7
3. Discussion
4. Materials and Methods
4.1. Cells and Viruses
4.2. Plasmids, siRNAs, and Antibodies
4.3. RABV Titration
4.4. Immunofluorescence Assay
4.5. Plasmid DNA and siRNA Transfection
4.6. Co-Immunoprecipitation
4.7. Western Blotting
4.8. Quantitative Real-Time PCR Analysis
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hampson, K.; Coudeville, L.; Lembo, T.; Sambo, M.; Kieffer, A.; Attlan, M.; Barrat, J.; Blanton, J.D.; Briggs, D.J.; Cleaveland, S.; et al. Estimating the global burden of endemic canine rabies. PLoS Neglect. Trop. Dis. 2015, 9, e3709. [Google Scholar] [CrossRef] [Green Version]
- Finke, S.; Conzelmann, K.K. Replication strategies of rabies virus. Virus Res. 2005, 111, 120–131. [Google Scholar] [CrossRef]
- Albertini, A.A.; Ruigrok, R.W.; Blondel, D. Rabies virus transcription and replication. Adv. Virus. Res. 2011, 79, 1–22. [Google Scholar] [CrossRef]
- Brunker, K.; Mollentze, N. Rabies Virus. Trends Microbiol. 2018, 26, 886–887. [Google Scholar] [CrossRef]
- Leroy, M.; Pire, G.; Baise, E.; Desmecht, D. Expression of the interferon-alpha/beta-inducible bovine Mx1 dynamin interferes with replication of rabies virus. Neurobiol. Dis. 2006, 21, 515–521. [Google Scholar] [CrossRef]
- Mendonca, R.Z.; Pereira, C.A. Relationship of interferon synthesis and the resistance of mice infected with street rabies virus. Braz. J. Med. Biol. Res. 1994, 27, 691–695. [Google Scholar] [PubMed]
- Chopy, D.; Detje, C.N.; Lafage, M.; Kalinke, U.; Lafon, M. The type I interferon response bridles rabies virus infection and reduces pathogenicity. J. Neurovirol. 2011, 17, 353–367. [Google Scholar] [CrossRef]
- Vitour, D.; Doceul, V.; Ruscanu, S.; Chauveau, E.; Schwartz-Cornil, I.; Zientara, S. Induction and control of the type I interferon pathway by Bluetongue virus. Virus Res. 2014, 182, 59–70. [Google Scholar] [CrossRef]
- Wang, Z.W.; Sarmento, L.; Wang, Y.; Li, X.Q.; Dhingra, V.; Tseggai, T.; Jiang, B.; Fu, Z.F. Attenuated rabies virus activates, while pathogenic rabies virus evades, the host innate immune responses in the central nervous system. J. Virol. 2005, 79, 12554–12565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marosi, A.; Forgach, P.; Gyuranecz, M.; Sulyok, K.M.; Bakonyi, T. Evaluation of in vitro inhibitory potential of type-I interferons and different antiviral compounds on rabies virus replication. Vaccine 2019, 37, 4663–4672. [Google Scholar] [CrossRef]
- Rieder, M.; Conzelmann, K.K. Interferon in rabies virus infection. Adv. Virus. Res. 2011, 79, 91–114. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Zhou, M.; Yang, Y.; Yu, L.; Luo, Z.; Tian, D.; Wang, K.; Cui, M.; Chen, H.; Fu, Z.F.; et al. Lab-Attenuated Rabies Virus Causes Abortive Infection and Induces Cytokine Expression in Astrocytes by Activating Mitochondrial Antiviral-Signaling Protein Signaling Pathway. Front. Immunol. 2017, 8, 2011. [Google Scholar] [CrossRef] [PubMed]
- Prehaud, C.; Megret, F.; Lafage, M.; Lafon, M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J. Virol. 2005, 79, 12893–12904. [Google Scholar] [CrossRef] [Green Version]
- Menager, P.; Roux, P.; Megret, F.; Bourgeois, J.P.; Le Sourd, A.M.; Danckaert, A.; Lafage, M.; Prehaud, C.; Lafon, M. Toll-like receptor 3 (TLR3) plays a major role in the formation of rabies virus Negri Bodies. PLoS Pathog. 2009, 5, e1000315. [Google Scholar] [CrossRef]
- Faul, E.J.; Wanjalla, C.N.; Suthar, M.S.; Gale, M.; Wirblich, C.; Schnell, M.J. Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling. PLoS Pathog. 2010, 6, e1001016. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Marie, I.; Smith, E.; Prakash, A. Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J. Interferon Cytokine Res. 2002, 22, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001, 19, 623–655. [Google Scholar] [CrossRef]
- Barnes, B.; Lubyova, B.; Pitha, P.M. On the role of IRF in host defense. J. Interferon Cytokine Res. 2002, 22, 59–71. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef]
- Wang, C.; Lv, L.; Wu, Q.; Wang, Z.; Luo, Z.; Sui, B.; Zhou, M.; Fu, Z.F.; Zhao, L. The role of interferon regulatory factor 7 in the pathogenicity and immunogenicity of rabies virus in a mouse model. J. Gen. Virol. 2021, 102. [Google Scholar] [CrossRef]
- Zhou, Y.; He, C.; Wang, L.; Ge, B. Post-translational regulation of antiviral innate signaling. Eur. J. Immunol. 2017, 47, 1414–1426. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Mehta, D.; Mishra, N.; Nayak, D.; Sunil, S. Role of Host-Mediated Post-Translational Modifications (PTMs) in RNA Virus Pathogenesis. Int. J. Mol. Sci. 2020, 22, 323. [Google Scholar] [CrossRef]
- de Brevern, A.G.; Rebehmed, J. Current status of PTMs structural databases: Applications, limitations and prospects. Amino Acids 2022, 54, 575–590. [Google Scholar] [CrossRef]
- Kessler, B.M.; Edelmann, M.J. PTMs in conversation: Activity and function of deubiquitinating enzymes regulated via post-translational modifications. Cell Biochem. Biophys. 2011, 60, 21–38. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.P.; Zhang, M.; Li, Y.; Diao, F.C.; Chen, D.; Zhai, Z.; Shu, H.B. Differential regulation of IKK alpha-mediated activation of IRF3/7 by NIK. Mol. Immunol. 2008, 45, 1926–1934. [Google Scholar] [CrossRef]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef]
- Maarifi, G.; Smith, N.; Maillet, S.; Moncorge, O.; Chamontin, C.; Edouard, J.; Sohm, F.; Blanchet, F.P.; Herbeuval, J.P.; Lutfalla, G.; et al. TRIM8 is required for virus-induced IFN response in human plasmacytoid dendritic cells. Sci. Adv. 2019, 5, eaax3511. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Hayward, G.S. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 2010, 33, 863–877. [Google Scholar] [CrossRef] [Green Version]
- Siednienko, J.; Jackson, R.; Mellett, M.; Delagic, N.; Yang, S.; Wang, B.; Tang, L.S.; Callanan, J.J.; Mahon, B.P.; Moynagh, P.N. Pellino3 targets the IRF7 pathway and facilitates autoregulation of TLR3- and viral-induced expression of type I interferons. Nat. Immunol. 2012, 13, 1055–1062. [Google Scholar] [CrossRef]
- Correia, S.; Ventura, S.; Parkhouse, R.M. Identification and utility of innate immune system evasion mechanisms of ASFV. Virus Res. 2013, 173, 87–100. [Google Scholar] [CrossRef]
- Liang, Q.; Deng, H.; Li, X.; Wu, X.; Tang, Q.; Chang, T.H.; Peng, H.; Rauscher, F.R.; Ozato, K.; Zhu, F. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol. 2011, 187, 4754–4763. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, W.; Cen, M.; Yang, L.; Zhang, W.; Xia, J.; Xu, F. NMI Facilitates Influenza A Virus Infection by Promoting Degradation of IRF7 through TRIM21. Am. J. Respir. Cell. Mol. Biol. 2021, 65, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Cai, X.; Niu, S.; Wei, J.; Jiang, N.; Deng, H.; Wang, W.; Zhang, J.; Shen, S.; Ma, Y.; et al. TRIM21 promotes ubiquitination of SARS-CoV-2 nucleocapsid protein to regulate innate immunity. J. Med. Virol. 2023, 95, e28719. [Google Scholar] [CrossRef]
- Kong, X.; Lu, X.; Wang, S.; Hao, J.; Guo, D.; Wu, H.; Jiang, Y.; Sun, Y.; Wang, J.; Zhang, G.; et al. Type I interferon/STAT1 signaling regulates UBE2M-mediated antiviral innate immunity in a negative feedback manner. Cell Rep. 2023, 42, 112002. [Google Scholar] [CrossRef]
- Guo, M.; Cao, W.; Chen, S.; Tian, R.; Xue, B.; Wang, L.; Liu, Q.; Deng, R.; Wang, X.; Wang, Z.; et al. TRIM21 Regulates Virus-Induced Cell Pyroptosis through Polyubiquitination of ISG12a. J. Immunol. 2022, 209, 1987–1998. [Google Scholar] [CrossRef]
- Mishra, R.; Kumawat, K.L.; Basu, A.; Banerjea, A.C. Japanese Encephalitis Virus infection increases USP42 to stabilize TRIM21 and OAS1 for neuroinflammatory and anti-viral response in human microglia. Virology 2022, 573, 131–140. [Google Scholar] [CrossRef] [PubMed]
- McEwan, W.A.; James, L.C. TRIM21-dependent intracellular antibody neutralization of virus infection. Prog. Molec. Biol. Transl. Sci. 2015, 129, 167–187. [Google Scholar] [CrossRef]
- Luo, Z.; Li, Y.; Zhou, M.; Lv, L.; Wu, Q.; Chen, C.; Zhang, Y.; Sui, B.; Tu, C.; Cui, M.; et al. Toll-Like Receptor 7 Enhances Rabies Virus-Induced Humoral Immunity by Facilitating the Formation of Germinal Centers. Front. Immunol. 2019, 10, 429. [Google Scholar] [CrossRef]
- Tateda, K.; Okazaki, S.; Nagoya, S.; Katada, R.; Mizuo, K.; Watanabe, S.; Yamashita, T.; Matsumoto, H. The suppression of TRIM21 and the accumulation of IFN-alpha play crucial roles in the pathogenesis of osteonecrosis of the femoral head. Lab. Invest. 2012, 92, 1318–1329. [Google Scholar] [CrossRef] [Green Version]
- Young, J.A.; Sermwittayawong, D.; Kim, H.J.; Nandu, S.; An, N.; Erdjument-Bromage, H.; Tempst, P.; Coscoy, L.; Winoto, A. Fas-associated death domain (FADD) and the E3 ubiquitin-protein ligase TRIM21 interact to negatively regulate virus-induced interferon production. J. Biol. Chem. 2011, 286, 6521–6531. [Google Scholar] [CrossRef] [Green Version]
- Higgs, R.; Lazzari, E.; Wynne, C.; Ni, G.J.; Espinosa, A.; Wahren-Herlenius, M.; Jefferies, C.A. Self protection from anti-viral responses—Ro52 promotes degradation of the transcription factor IRF7 downstream of the viral Toll-Like receptors. PLoS ONE 2010, 5, e11776. [Google Scholar] [CrossRef] [Green Version]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef] [Green Version]
- Pagani, I.; Poli, G.; Vicenzi, E. TRIM22. A Multitasking Antiviral Factor. Cells 2021, 10, 1864. [Google Scholar] [CrossRef]
- Peng, C.; Zhao, C.; Wang, P.F.; Yan, L.L.; Fan, S.G.; Qiu, L.H. Identification of a TRIM32 from Penaeus monodon is involved in autophagy and innate immunity during white spot syndrome virus infection. Dev. Comp. Immunol. 2021, 123, 104169. [Google Scholar] [CrossRef]
- Wang, S.; Yu, M.; Liu, A.; Bao, Y.; Qi, X.; Gao, L.; Chen, Y.; Liu, P.; Wang, Y.; Xing, L.; et al. TRIM25 inhibits infectious bursal disease virus replication by targeting VP3 for ubiquitination and degradation. PLoS Pathog. 2021, 17, e1009900. [Google Scholar] [CrossRef]
- Diaz-Beneitez, E.; Cubas-Gaona, L.L.; Candelas-Rivera, O.; Benito-Zafra, A.; Sanchez-Aparicio, M.T.; Miorin, L.; Rodriguez, J.F.; Garcia-Sastre, A.; Rodriguez, D. Interaction between chicken TRIM25 and MDA5 and their role in mediated antiviral activity against IBDV infection. Front. Microbiol. 2022, 13, 1068328. [Google Scholar] [CrossRef]
- Wei, Y.; Zeng, S.; Zou, C.; Zhang, H.; Peng, O.; Xue, C.; Cao, Y. Porcine TRIM21 RING-finger E3 ubiquitin ligase is essential for anti-PRRSV activity. Vet. Microbiol. 2021, 256, 109043. [Google Scholar] [CrossRef]
- Watkinson, R.E.; McEwan, W.A.; Tam, J.C.; Vaysburd, M.; James, L.C. TRIM21 Promotes cGAS and RIG-I Sensing of Viral Genomes during Infection by Antibody-Opsonized Virus. PLoS Pathog. 2015, 11, e1005253. [Google Scholar] [CrossRef] [Green Version]
- Watkinson, R.E.; Tam, J.C.; Vaysburd, M.J.; James, L.C. Simultaneous neutralization and innate immune detection of a replicating virus by TRIM21. J. Virol. 2013, 87, 7309–7313. [Google Scholar] [CrossRef] [Green Version]
- Bottermann, M.; James, L.C. Intracellular Antiviral Immunity. Adv. Virus. Res. 2018, 100, 309–354. [Google Scholar] [CrossRef]
- Manocha, G.D.; Mishra, R.; Sharma, N.; Kumawat, K.L.; Basu, A.; Singh, S.K. Regulatory role of TRIM21 in the type-I interferon pathway in Japanese encephalitis virus-infected human microglial cells. J. Neuroinflammation 2014, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Dinh, P.X.; Pattnaik, A.K. Trim21 regulates Nmi-IFI35 complex-mediated inhibition of innate antiviral response. Virology 2015, 485, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wan, M.; Cai, L.; Hou, A.; Sun, B.; Zhou, Y.; Gao, F.; Su, W.; Jiang, C. Interferon Inhibition Enhances the Pilot-Scale Production of Rabies Virus in Human Diploid MRC-5 Cells. Viruses 2021, 14, 49. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Servant, M.J.; Tenoever, B.; Lin, R. Overlapping and distinct mechanisms regulating IRF-3 and IRF-7 function. J. Interferon Cytokine Res. 2002, 22, 49–58. [Google Scholar] [CrossRef]
- Tamura, T.; Yanai, H.; Savitsky, D.; Taniguchi, T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 2008, 26, 535–584. [Google Scholar] [CrossRef]
- Luo, J.; Zhang, Y.; Zhang, Q.; Wu, Y.; Zhang, B.; Mo, M.; Tian, Q.; Zhao, J.; Mei, M.; Guo, X. The Deoptimization of Rabies Virus Matrix Protein Impacts Viral Transcription and Replication. Viruses 2019, 12, 4. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Zhang, Y.; Wang, Y.; Liu, Q.; Chen, L.; Zhang, B.; Luo, Y.; Huang, S.; Guo, X. Rhabdovirus Infection Is Dependent on Serine/Threonine Kinase AP2-Associated Kinase 1. Life-Basel 2020, 10, 170. [Google Scholar] [CrossRef]
- Luo, J.; Zhang, B.; Wu, Y.; Tian, Q.; Zhao, J.; Lyu, Z.; Zhang, Q.; Mei, M.; Luo, Y.; Guo, X. Expression of interleukin-6 by a recombinant rabies virus enhances its immunogenicity as a potential vaccine. Vaccine 2017, 35, 938–944. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| siRNA | Sense (5′–3′) | Antisense (5′–3′) |
|---|---|---|
| TRIM21-1 | GGAGCCUAUGAGUAUCGAATT | UUCGAUACUCAUAGGCUCCTT |
| TRIM21-2 | CCAAUAGACAUAUAGCCAATT | UUGUUAGGUGAGAAGUGGGTT |
| TRIM21-3 | CCUGGACACGUUAGAUAUUTT | AAUAUCUAACGUGUCCAGGTT |
| IRF7-1 | CCGCAUAAGGUGUACGAACUUTT | AAGUUCGUACACCUUAUGCGGTT |
| IRF7-2 | CCUGGAAGCAUUUCGGUCGUATT | UACGACCGAAAUGCUUCCAGGTT |
| IRF7-3 IFNα-1 IFNα-2 IFNα-3 | CUUCGACUUCAGCACUUUCUUTT GAGCCAGAUUAUCUCUUUCUATT CGUCAUUGAAUCACACCUGAUTT CAGUCAUUGAAAGCCUAGAAATT | AAGAAAGUGCUGAAGUCGAAGTT UAGAAAGAGAUAAUCUGGCUCTT AUCAGGUGUGAUUCAAUGACGTT UUUCUAGGCUUUCAAUGACUGTT |
| IFNβ-1 | GCAGAAGAGUUACACUGCCUUTT | AAGGCAGUGUAACUCUUCUGCTT |
| IFNβ-2 | AGCCCUCUCCAUCAACUAUAATT | UUAUAGUUGAUGGAGAGGGCUTT |
| IFNβ-3 | GCUCUCCACUUGAAGAGCUAUTT | AUAGCUCUUCAAGUGGAGAGCTT |
| NC | UUCUCCGAACGUGUCACGUTT | ACGUGACACGUUCGGAGAATT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Cai, T.; He, H.; Huang, X.; Chen, G.; Lai, Y.; Luo, Y.; Huang, S.; Luo, J.; Guo, X. TRIM21 Promotes Rabies Virus Production by Degrading IRF7 through Ubiquitination. Int. J. Mol. Sci. 2023, 24, 10892. https://doi.org/10.3390/ijms241310892
Zhang B, Cai T, He H, Huang X, Chen G, Lai Y, Luo Y, Huang S, Luo J, Guo X. TRIM21 Promotes Rabies Virus Production by Degrading IRF7 through Ubiquitination. International Journal of Molecular Sciences. 2023; 24(13):10892. https://doi.org/10.3390/ijms241310892
Chicago/Turabian StyleZhang, Boyue, Ting Cai, Hongling He, Xuezhe Huang, Guie Chen, Yanqin Lai, Yongwen Luo, Shile Huang, Jun Luo, and Xiaofeng Guo. 2023. "TRIM21 Promotes Rabies Virus Production by Degrading IRF7 through Ubiquitination" International Journal of Molecular Sciences 24, no. 13: 10892. https://doi.org/10.3390/ijms241310892