
The Role of Activation of PI3K/AKT/mTOR and RAF/MEK/ERK Pathways in Aggressive Pituitary Adenomas—New Potential Therapeutic Approach—A Systematic Review

 , , and
, , and
Abstract
:1. Introduction
1.1. Current Knowledge of Pituitary Adenoma Pathogenesis
1.2. The Challenge
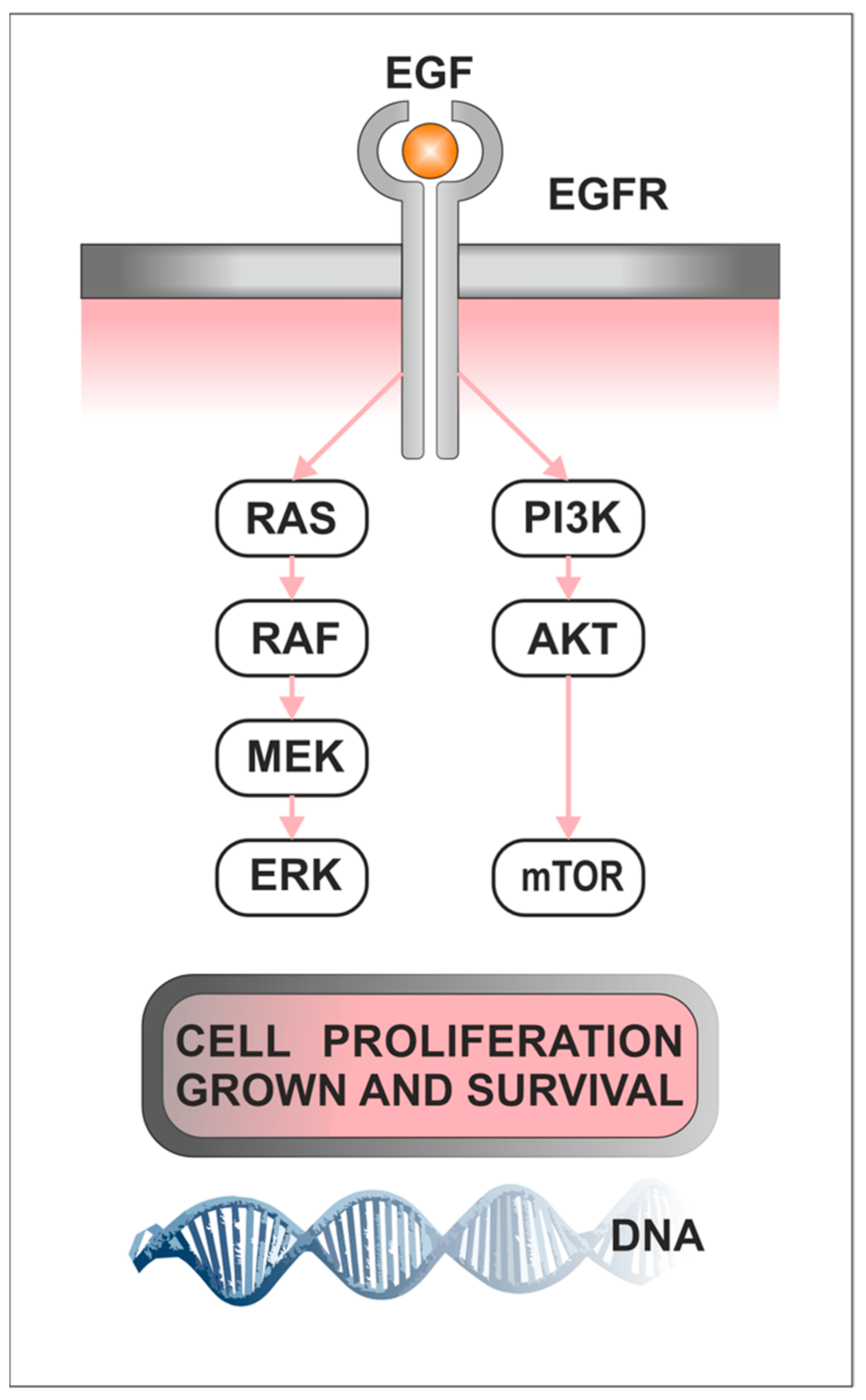
1.3. Role of PI3K/AKT/mTOR Pathway in Cancerogenesis
1.4. Role of Raf/MEK/ERK in Cancerogenesis
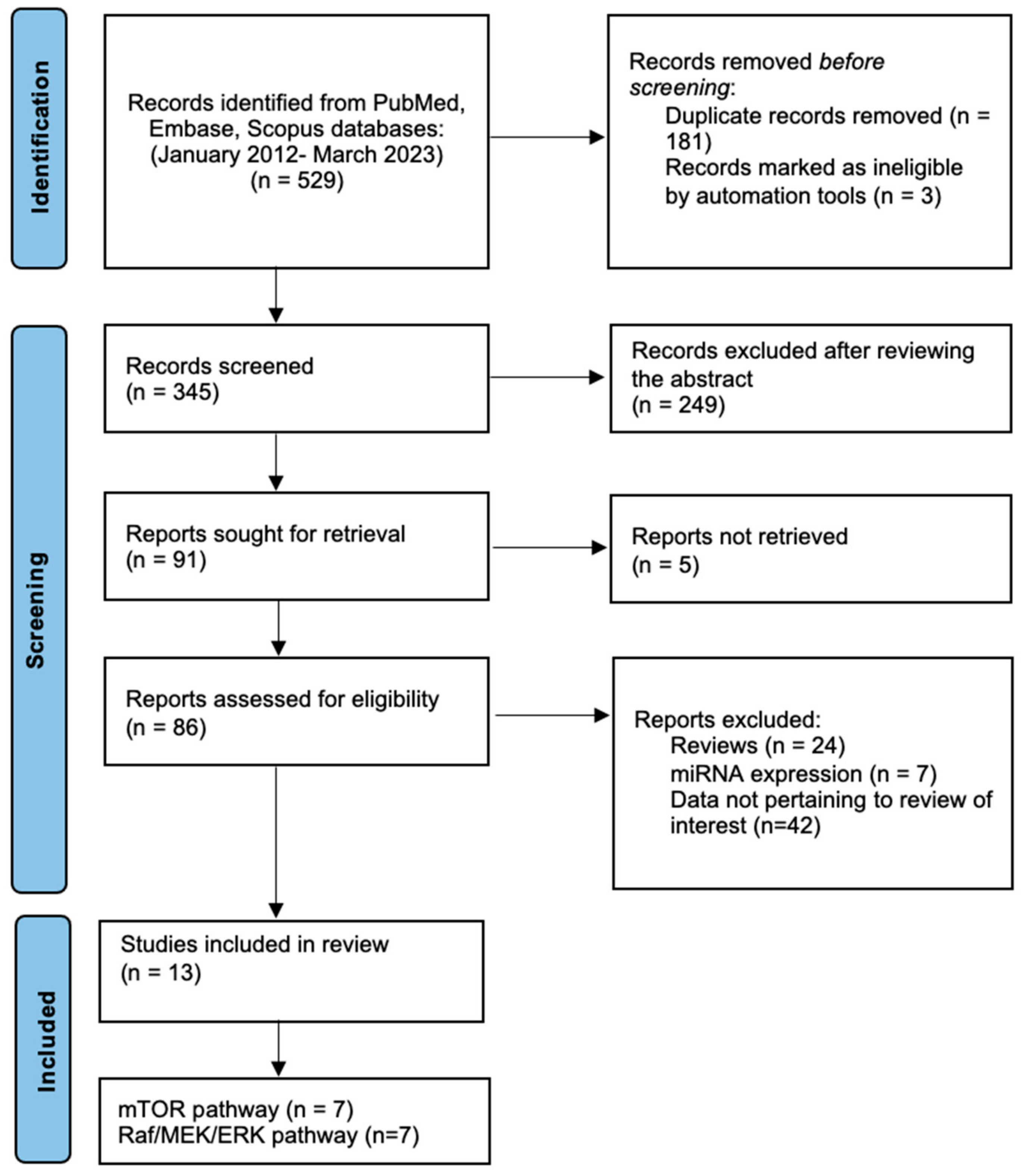
2. Materials and Methods
3. Results
3.1. PI3K/AKT/mTOR Pathway in Pituitary Adenomas (Table 1)

{kind=link}
{kind=link}
{kind=link}
| Study | Country | Clinical Data | Method and Sample | Results |
|---|---|---|---|---|
| Sajjad et al., 2013 [116] | Poland | 53 Pituitary adenomas: 14 GHomas, 33 NFPAs, 6 ACTHomas Male/Female GHomas: 4/10 NFPAs: 21/12 ACTHomas: 2/4 Age: 54.6 ± 15.7 | Tissue samples Western blot Primary cell culture | The level of mTOR kinase activity was calculated as pS6rp/elF4E ratio in all tissue samples. GHomas had the highest level of mTOR activity in comparison to NFPA (p = 0.04). The level of mTOR activity did not show any significant correlation with any of the parameters (tumor volume, tumor largest dimension, Knosp’s grading, Ki-67%, and pErk activity). All primary cell culture lines showed mTOR inhibition in response to rapamycin. |
| Di Pasquale et al., 2018 [121] | Italy | Not applicable | Cell culture (GH3 and GH4C1 rat pituitary adenoma cell lines) GH secretion RNA extraction and qRT-PCR Western blot | GH3 cell viability was significantly induced by IGF1 (+30%; p < 0.01 vs. untreated control cells) and reduced by everolimus and NVP-BEZ235 up to 30% (p < 0.01 vs. untreated control cells). GH4C1 cell viability was not influenced by IGF1 but was significantly reduced by everolimus (−60%; p < 0.01 vs. control treated cells) and by NVP-BEZ235 (−46%; p < 0.01 vs. untreated control cells). IGF1 significantly inhibited GH secretion (−40%; p < 0.01 vs. untreated control cells). IGF1 reduced GH mRNA expression (−37%; p < 0.01 vs. untreated control cells). |
| Zhu et al., 2021 [119] | China | Not applicable | Rat prolactinoma cell lines (GH3 cells) Western blot Cell counting kit (CCK)-8 assay | ACT001 induced autophagic cell death in cabergoline CAB-resistant GH3 cells by AMPK-mTOR pathway. The cell viability of the CAB + ACT001 group was lower than that of the CAB group (35.30 ± 3.33% vs. 59.63 ± 1.76%, p < 0.001) and ACT001 group (35.30 ± 3.33% vs. 84.10 ± 3.90%, p < 0.001). The number of autophagosomes in the CAB + ACT001 group was significantly higher than that in the CAB group (p < 0.001) and ACT001 group (p < 0.001). |
| Tang et al., 2019 [118] | China | Not applicable | Cell culture (MMQ cells and GH3 cells) Western blot | Levels of p-AKT (p = 0.0034) and p-mTOR (p = 0.0005) were significantly lower in the group treated by cabergoline than in the control group. |
| Mangili et al., 2022 [123] | Italy | NF-PitNETs:14 Male/Female: 6/8 Age: 60.7 ± 13.8 | Tissue samples Primary cell culture RT-PCR analysis | Everolimus treatment was effective in reducing cell proliferation in 5 out of 14 NF-PitNET primary cultured cells (−39.2 ± 25.8% at 1 nM, p < 0.01 vs. basal). In NF-PitNETs resistant to Everolimus, the coadministration of cabergoline was effective in inhibiting cell proliferation in 7 out of 9 tumors (−31.4 ± 9.9%, p < 0.001 vs. basal). |
| Jia et al., 2013 [113] | China | 95 Pituitary adenomas: NFPAs 59 PRLomas 5, GHomas 8, LHomas 2, FSHomas 2, TSHomas 4, ACTHomas 6, Mixed 9 Male/Female: 51/44 Age: <45 45 >45 50 | Tissue samples Quantitative gene transcript analyses | Correlation between RICTOR expression and tumor size, namely p = 0.0012 and p = 0.0055 for tumors 1–2 cm and tumors >3 cm compared with tumors < 1 cm. Higher levels of mTOR were seen in tumors with cystic lesions (p = 0.044). Levels of mTOR were found to be significantly correlated with levels of both RAPTOR (p = 0.000234) and RICTOR (p = 0.0000002). RAPTOR and RICTOR levels were also significantly correlated (p < 0.0000002). |
| Li et al., 2017 [122] | China | 11 Gonadotrophin adenomas Male/Female: 7/4 Age: 43.9 ± 16 | Tissue samples RNA-seq analysis qRT-PCR | Genome-wide analysis of lncRNAs and mRNAs obtained from gonadotrophin adenomas. Co-expression involving 126 lncRNAs interacting with 14 mRNAs of the mTOR pathway (PCC > 0.80, p < 0.001), which might promote the pathogenesis of the gonadotrophin tumor. |
3.2. Raf/MEK/ERK Pathway in Pituitary Adenomas (Table 2)
| Study | Country | Clinical Data | Method and Sample | Results |
|---|---|---|---|---|
| Sajjad et al., 2013 [116] | Poland | 53 patients Male/Female: 27/26 Age: 54.6 ± 15.7 14 GHPA, 33 NFPAs, 5 ACTHomas | Tissue/cell culture IHC, WB | Erk was activated in most pituitary samples, including control samples Erk activity was the highest in control pituitary samples (p = 0.003) In NFPAs, the activity of pErk showed a medium level of inverse correlation with Knosp’s grading (R Spearman= −0.31, p = 0.018) In GHomas, pErk showed a strong level of correlation with somatostatin receptor subtype 2 A (SSTR2A) expression (R Spearman = 0.57, p = 0.04) |
| Liu et al., 2019 [125] | China | 52 patients with CD (22 with tumor recurrence, 30 without) Age: 35.2 ± 12.4 6 HC | Tissue IHC, WB | EGFR immunoreactivity in 29 of 52 (55.8%) pituitary corticotroph adenomas (14 EGFR-positive adenomas in 20 (70%) recurrent adenomas and 15 EGFR-positive adenomas in 32 (46.9%) non-recurrent adenomas) and in 1 of 6 (16.6%) normal pituitary glands EGFR levels in the recurrent corticotroph adenomas were significantly increased compared to those in the non-recurrent ones p-EGFR and p-Erk were upregulated in recurrent adenomas but were not upregulated in non-recurrent adenomas or in normal pituitary glands, while the total Erk, total AKT, and p-AKT levels were unchanged EGFR protein was found to be significantly associated with the recurrence status (p = 0.005), cortisol level (p = 0.009), and ACTH level (p = 0.008) but was not related to the sex, age, or symptom duration of the patient (p = 0.280, p = 0.351 and p =0.142, respectively). |
| Liu et al., 2018 [124] | China | 48 GHPA | Tissue/ Cell culture- Rat GHPA cell GH3 and mouse GT1.1 pituitary adenoma cells, WB, IHC, TMA | EGFL7 positive staining in invasive GHPAs was significantly higher (2-fold higher) than that in non-invasive GHPAs positive staining of total EGFR was higher in invasive GHPAs than that in non-invasive GHPA tissues average expression level of p-EGFR in invasive GHPAs was 3.5-fold higher than that in non-invasive GHPAs positive staining of p-EGFR was closely related with high-level EGFL7 in invasive GHPAs knockdown of EGFL7 expression significantly suppressed p-EGFR expression in GH3 cells p-AKT and p-ERK expression was decreased in EGFL7 knockdown cells after 48 h treatment with 50 ng mL−1 rhEGFL7 the level of EGFR, AKT, and ERK phosphorylation in GH3 cells was significantly increased, as compared with PBS control knockdown of EGFL7 effectively suppressed activation of EGFR signaling cascades in GH3 cells, including p-EGFR, p-AKT, and p-ERK |
| De Dios et al., 2019 [127] | Argentina | Not applicable | Cell culture—GH3 somatolactotrope cells, RT-PCR, WB | ERK1/2 inhibition mediates the apoptotic effect induced by PRLR activation in GH3 cells Inhibition of PRL signaling increased ERK1/2 phosphorylation in a time-dependent manner |
| Roof et al., 2018 [126] | USA | 4 prolactinoma samples and 4 HC | Tissue/Cell culture—HEK 293T and BOSC cells GH4C1 rat somatolactotrope cells, WB, RT-PCR | p-ERK1/2 was undetectable in normal pituitary sample, in prolactinoma samples, p-ERK1/2 was expressed in all samples All prolactinoma samples and HC expressed t-ERK1/2 p-ERK/t-ERK expression ratio was increased in prolactinoma samples compared with normal pituitary tissue inhibition of the ERK1/2 signaling pathway promotes a decrease in the PRL/GH ratio inhibition of ERK1/2 signaling resulted in a small but significant increase in GH4T2 cell proliferation and colony formation ERK and PI3K signaling is dysregulated in human prolactinoma inhibition of Raf/MEK/ERK signaling increases GH4T2 cell proliferation |
| Booth et al., 2014 [128] | USA | Not applicable | Cell culture- GH4T2 cells | the duration of MAPK activation is critical in dictating the biological response activation of pMAPK leads to GH4 pituitary somatolactotrope differentiation to a lactotrope phenotype increase in the PRL to GH ratio observed both in vitro and in vivo suggests the differentiation of GH4 somatolactotrope cells into a lactotrope phenotype |
| Treppiedi et al., 2021 [129] | Italy | Not applicable | Cell culture—Murine pituitary corticotroph tumor cells, AtT-20 cells (ATCC CRL-1795™) WB | EGF is able to stimulate ERK phosphorylation in WT USP8 transfected cells was transient with a peak of phosphorylation reached at 24 h and strongly reduced at 48 h incubation in cells expressing S718del and G664R USP8, a persistent activation of ERK was observed at 48 h incubation with EGF (p < 0.05) Overexpression of USP8 G664R resulted in high levels of active phosphorylated ERK1/2 after 48 h of EGF stimulation, thus confirming that the novel USP8 variant sustains EGFR-MAPK signaling to promote corticotrophs ACTH production and cell growth |
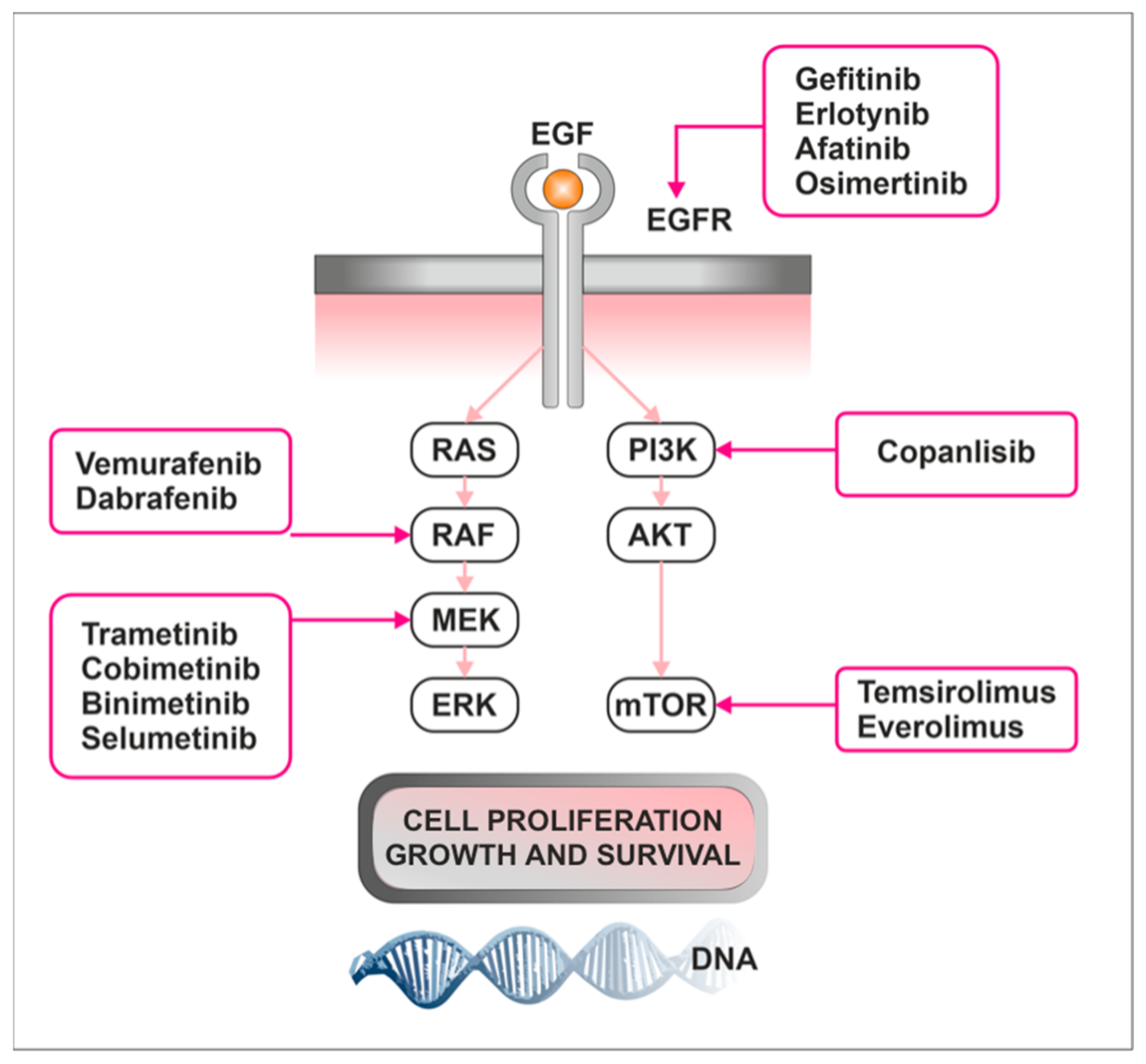
3.3. Treatment
3.3.1. mTOR Inhibitors
3.3.2. Tyrosine Kinase Inhibitors
3.3.3. MAPK Inhibitors
| Drug Name | Publication | Drug Mechanism | Material | Results |
|---|---|---|---|---|
| celastrol | Cai et al. (2022) [139] | second-generation mTOR inhibitor | IN VITRO ACTH-secreting adenoma cell lines AtT20 IN VIVO -on mouse AtT20 tumor xenografts | IN VITRO - blockade of cells in GO/G1 phase - induction of apoptosis and autophagy through downregulation of AKT/mTOR IN VIVO -decrease in tumor volume and weight in mice |
| buparlisib NVP-BEZ235 | Chanal et al. (2016) [140] | dual PI3K/mTOR inhibitor | IN VITRO -GH3 cell lines -Human prolactinomas in primary cell culture IN VIVO On rat SMtTW3 tumor xenograft | IN VITRO NVPBEZ235: -GH3 cell lines: induction of apoptosis, and cytostatic effect by accumulation of cells in G1 -reduction in cell viability and hormone secretion in primary cell culture Buparlisib: GH3 cell lines: limited effect primary cell culture: limited effect IN VIVO NVPBEZ235: -no effect on tumor growth Buparlisib: -decrease in tumor weight |
| metformin | Jin et al. (2018) [141] | antihyperglycemic agent anti-tumor mechanism mainly includes the activation AMPK, thus inhibiting the mTOR pathway | IN VITRO -AtT20 cell lines | IN VITRO Inhibition of proliferation and induction of apoptosis by ac- activating AMPK/mTOR and inhibiting IGF-1R/AKT/mTOR |
| NVP-BEZ235 everolimus | Lee et al. (2011) [142] | dual PI3K/mTOR inhibitor mTOR inhibitor | IN VITRO - GH3 cell lines - embryonic primary fibroblast cells -rat pituitary adenoma cells in primary culture | IN VITRO NVP-BEZ235 -Inhibition of PI3K pathway upstream and downstream of AKT - triggering of apoptosis due to decreasing AKT and S6 phosphorylation - reduction in cell viability more effective than everolimus |
| ACT001 | Zhu et al. (2021) [119]. | attenuation of the function of MnSOD increase ROS concentration in tumor cells | IN VITRO -GH3 and MMQ cell lines | IN VITRO - Possible reversal of CAB resistance in GH3 cells by inhibition of mTOR signaling pathway and induction of cell death attributed to autophagy - possible reversal of BRC resistance in MMQ cells by activation of EGR1 signaling pathway and induction of cell death due to apoptosis. |
| XL765 with temozolomide | Dai et al. (2013) [143] | dual-PI3K/mTOR inhibitor | IN VITRO -GH3, T3-1, and MMQ cell lines IN VIVO On rat GH3 tumor xenograft | IN VITRO - synergistic inhibition of growth of cell lines and induction of apoptosis IN VIVO -synergistic inhibition of tumor growth |
| Nelfinavir and radiation | Zeng et al. (2011) [144] | Radiosensitizer HIV protease inhibitor | IN VITRO -GH3, MMQ and AtT20 cell lines IN VIVO On rat GH3 tumor xenograft | IN VITRO -sensitization of PA cells to radiation, resulting in increased apoptosis -inhibition of the PI3K-AKT-mTOR pathway. IN VIVO -synergistic negative effect of radiotherapy and nelfinavir on tumor growth |
| BIM-23A760 | Peverelli et al. (2010) [145] | dopamine-somatostatin chimeric compound | IN VITRO human non-functioning pituitary tumors cells in primary culture | IN VITRO -Activation of ERK1/2 and p38 pathways - antiproliferative and the pro-apoptotic effects on the cells |
| fulvestrant | Gao eta al. (2017) [146] | antiestrogen | Tissue samples 289 PAs cases IN VITRO GH3 and JT1-1 cell lines IN VIVO rat model of prolactinoma (injection of 17b-estradiol) | Tissue samples -estrogen receptor alpha present in more than 50% of cases IN VITRO -Reduction in cell viability IN VIVO - inhibition of tumor growth by modulation of PTEN/MAPK signaling, including ERK pathway |
4. Discussion
4.1. Raf/MEK/ERK and PI3K/AKT/mTOR Pathways Are Involved in Pituitary Tumorigenesis and Aggressiveness
4.2. The Activation of EGFR-Signaling Cascades Plays an Important Role in Cell Proliferation, Migration, and Invasion in PitNETs
4.3. Targeting Raf/MEK/ERK and mTOR Pathways as a Novel Therapeutic Approach
4.4. Limitations and Further Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Araujo-Castro, M.; Berrocal, V.R.; Pascual-Corrales, E. Pituitary Tumors: Epidemiology and Clinical Presentation Spectrum. Hormones 2020, 19, 145–155. [Google Scholar] [CrossRef]
- Ezzat, S.; Asa, S.L.; Couldwell, W.T.; Barr, C.E.; Dodge, W.E.; Vance, M.L.; McCutcheon, I.E. The Prevalence of Pituitary Adenomas: A Systematic Review. Cancer 2004, 101, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Asa, S.L.; Mete, O.; Cusimano, M.D.; McCutcheon, I.E.; Perry, A.; Yamada, S.; Nishioka, H.; Casar-Borota, O.; Uccella, S.; Rosa, S.L.; et al. Pituitary Neuroendocrine Tumors: A Model for Neuroendocrine Tumor Classification. Mod. Pathol. 2021, 34, 1634–1650. [Google Scholar] [CrossRef]
- Vallecillos, F.J.T.; Fernández, S.O. Histopathological Features of Post-Mortem Pituitaries: A Retrospective Analysis. Rev. Assoc. Med. Bras. 2016, 62, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrow, G.N.; Wortzman, G.; Rewcastle, N.B.; Holgate, R.C.; Kovacs, K. Microadenomas of the Pituitary and Abnormal Sellar Tomograms in an Unselected Autopsy Series. N. Engl. J. Med. 1981, 304, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Coopmans, E.C.; Korbonits, M. Molecular Genetic Testing in the Management of Pituitary Disease. Clin. Endocrinol. 2022, 97, 424–435. [Google Scholar] [CrossRef]
- Kasuki, L.; Raverot, G. Definition and Diagnosis of Aggressive Pituitary Tumors. Rev. Endocr. Metab. Disord. 2020, 21, 203–208. [Google Scholar] [CrossRef]
- Melmed, S.; Kaiser, U.B.; Lopes, M.B.; Bertherat, J.; Syro, L.V.; Raverot, G.; Reincke, M.; Johannsson, G.; Beckers, A.; Fleseriu, M.; et al. Clinical Biology of the Pituitary Adenoma. Endoc. Rev. 2022, 43, 1003–1037. [Google Scholar] [CrossRef]
- Molitch, M.E. Diagnosis and Treatment of Pituitary Adenomas: A Review. JAMA 2017, 317, 516–524. [Google Scholar] [CrossRef]
- Lin, A.L.; Donoghue, M.T.A.; Wardlaw, S.L.; Yang, T.J.; Bodei, L.; Tabar, V.; Geer, E.B. Approach to the Treatment of a Patient with an Aggressive Pituitary Tumor. J. Clin. Endocrinol. Metab. 2020, 105, 3807–3820. [Google Scholar] [CrossRef]
- McCormack, A.; Dekkers, O.M.; Petersenn, S.; Popovic, V.; Trouillas, J.; Raverot, G.; Burman, P.; ESE Survey Collaborators. Treatment of Aggressive Pituitary Tumours and Carcinomas: Results of a European Society of Endocrinology (ESE) Survey 2016. Eur. J. Endocrinol. 2018, 178, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, M.B.S. The 2017 World Health Organization Classification of Tumors of the Pituitary Gland: A Summary. Acta Neuropathol. 2017, 134, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Lopes, M.B. Overview of the 2017 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2017, 28, 228–243. [Google Scholar] [CrossRef]
- Burcea, I.F.; Năstase, V.-N.; Poiană, C. Pituitary Transcription Factors in the Immunohistochemical and Molecular Diagnosis of Pituitary Tumours—A Systematic Review. Endokrynol. Polska 2021, 72, 53–63. [Google Scholar] [CrossRef]
- Tebani, A.; Jotanovic, J.; Hekmati, N.; Sivertsson, Å.; Gudjonsson, O.; Edén Engström, B.; Wikström, J.; Uhlèn, M.; Casar-Borota, O.; Pontén, F. Annotation of Pituitary Neuroendocrine Tumors with Genome-Wide Expression Analysis. Acta. Neuropathol. Commun. 2021, 9, 181. [Google Scholar] [CrossRef] [PubMed]
- Pulichino, A.-M.; Vallette-Kasic, S.; Tsai, J.P.-Y.; Couture, C.; Gauthier, Y.; Drouin, J. Tpit Determines Alternate Fates during Pituitary Cell Differentiation. Genes Dev. 2003, 17, 738–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, A.J.J.; King, J.; Scott, H.; Colman, P.; Yates, C.J. Insights into pituitary tumorigenesis: From Sanger sequencing to next-generation sequencing and beyond. Expert. Rev. Endocrinol. Metab. 2019, 14, 399–418. [Google Scholar] [CrossRef]
- Raverot, G.; Ilie, M.D.; Lasolle, H.; Amodru, V.; Trouillas, J.; Castinetti, F.; Brue, T. Aggressive Pituitary Tumours and Pituitary Carcinomas. Nat. Rev. Endocrinol. 2021, 17, 671–684. [Google Scholar] [CrossRef]
- Li, X. Molecular Network Basis of Invasive Pituitary Adenoma: A Review. Front. Endocrinol. 2019, 10, 10. [Google Scholar]
- Robertson, A.M.; Heaney, A.P. Molecular Markers in Pituitary Tumors. Curr. Opin. Endocrinol. Diabetes Obes. 2016, 23, 324–330. [Google Scholar] [CrossRef]
- Voellger, B.; Zhang, Z.; Benzel, J.; Wang, J.; Lei, T.; Nimsky, C.; Bartsch, J.-W. Targeting Aggressive Pituitary Adenomas at the Molecular Level—A Review. JCM 2021, 11, 124. [Google Scholar] [CrossRef]
- Jacoby, L.B.; Hedley-Whyte, E.T.; Pulaski, K.; Seizinger, B.R.; Martuza, R.L. Clonal Origin of Pituitary Adenomas. J. Neurosurg. 1990, 73, 731–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.M.; Biller, B.M.; Bikkal, H.; Zervas, N.T.; Arnold, A.; Klibanski, A. Clinically Nonfunctioning Pituitary Tumors Are Monoclonal in Origin. J. Clin. Investig. 1990, 86, 336–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsarrag, M.; Patel, P.D.; Chatrath, A.; Taylor, D.; Jane, J.A. Genomic and Molecular Characterization of Pituitary Adenoma Pathogenesis: Review and Translational Opportunities. Neurosurg. Focus 2020, 48, E11. [Google Scholar] [CrossRef] [PubMed]
- Clayton, R.N.; Farrell, W.E. Pituitary Tumour Clonality Revisited. Front. Horm. Res. 2004, 32, 186–204. [Google Scholar] [CrossRef]
- Lania, A.; Mantovani, G.; Spada, A. Genetics of pituitary tumors: Focus on G-protein mutations. Exp. Biol Med. 2003, 228, 1004–1017. [Google Scholar] [CrossRef]
- García-Guzmán, B.; Portocarrero-Ortiz, L.; Dorantes-Argandar, A.A.; Mercado, M. Hereditary Pituitary Tumor Syndromes: Genetic and Clinical Aspects. RIC 2020, 72, 3034. [Google Scholar] [CrossRef]
- Faltermeier, C.M.; Magill, S.T.; Blevins, L.S., Jr.; Aghi, M.K. Molecular Biology of Pituitary Adenomas. Neurosurg. Clin. N. Am. 2019, 30, 391–400. [Google Scholar] [CrossRef]
- Srirangam Nadhamuni, V.; Korbonits, M. Novel Insights into Pituitary Tumorigenesis: Genetic and Epigenetic Mechanisms. Endocr. Rev. 2020, 41, bnaa006. [Google Scholar] [CrossRef] [Green Version]
- Saeger, W. Pituitary tumors: Prognostic indicators. Endocrine 2005, 28, 57–66. [Google Scholar] [CrossRef]
- Sbiera, S.; Perez-Rivas, L.G.; Taranets, L.; Weigand, I.; Flitsch, J.; Graf, E.; Monoranu, C.-M.; Saeger, W.; Hagel, C.; Honegger, J.; et al. Driver Mutations in USP8 Wild-Type Cushing’s Disease. Neuro. Oncol. 2019, 21, 1273–1283. [Google Scholar] [CrossRef]
- Simon, J.; Theodoropoulou, M. Genetics of Cushing’s Disease. J. Neuroendocr. 2022, 34, e13148. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jian, X.; Deng, S.; Ma, Z.; Shou, X.; Shen, Y.; Zhang, Q.; Song, Z.; Li, Z.; Peng, H.; et al. Identification of Recurrent USP48 and BRAF Mu.u.utations in Cushing’s Disease. Nat. Commun. 2018, 9, 3171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murat, C.B.; Braga, P.B.S.; Fortes, M.A.H.Z.; Bronstein, M.D.; Corrêa-Giannella, M.L.C.; Giorgi, R.R. Mutation and Genomic Amplification of the PIK3CA Proto-Oncogene in Pituitary Adenomas. Braz. J. Med. Biol. Res. 2012, 45, 851–855. [Google Scholar] [CrossRef] [Green Version]
- Clayton, R.N.; Farrell, W.E. Clonality of Pituitary Tumours: More Complicated than Initially Envisaged? Brain Pathol. 2001, 11, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S. Pathogenesis of Pituitary Tumors. Nat. Rev. Endocrinol. 2011, 7, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Carreno, G.; Gonzalez-Meljem, J.M.; Haston, S.; Martinez-Barbera, J.P. Stem Cells and Their Role in Pituitary Tumorigenesis. Mol. Cell. Endocrinol. 2017, 445, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Ezzat, S.; Cheng, S.; Asa, S.L. Epigenetics of Pituitary Tumors: Pathogenetic and Therapeutic Implications. Mol. Cell. Endocrinol. 2018, 469, 70–76. [Google Scholar] [CrossRef]
- Simpson, D.J.; Hibberts, N.A.; McNicol, A.M.; Clayton, R.N.; Farrell, W.E. Loss of pRb Expression in Pituitary Adenomas Is Associated with Methylation of the RB1 CpG Island. Cancer Res. 2000, 60, 1211–1216. [Google Scholar]
- Yoshino, A.; Katayama, Y.; Ogino, A.; Watanabe, T.; Yachi, K.; Ohta, T.; Komine, C.; Yokoyama, T.; Fukushima, T. Promoter Hypermethylation Profile of Cell Cycle Regulator Genes in Pituitary Adenomas. J. Neurooncol. 2007, 83, 153–162. [Google Scholar] [CrossRef]
- Li, T.; Huang, H.; Huang, B.; Huang, B.; Lu, J. Histone Acetyltransferase P300 Regulates the Expression of Human Pituitary Tumor Transforming Gene (hPTTG). J. Genet. Genomics 2009, 36, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Cao, L.; Jia, Y.; Xiao, Y.; Zhang, X.; Gui, S. Emerging Roles of miRNA, lncRNA, circRNA, and Their Cross-Talk in Pituitary Adenoma. Cells 2022, 11, 2920. [Google Scholar] [CrossRef] [PubMed]
- Belaya, Z.; Khandaeva, P.; Nonn, L.; Nikitin, A.; Solodovnikov, A.; Sitkin, I.; Grigoriev, A.; Pikunov, M.; Lapshina, A.; Rozhinskaya, L.; et al. Circulating Plasma microRNA to Differentiate Cushing’s Disease From Ectopic ACTH Syndrome. Front. Endocrinol. 2020, 11, 331. [Google Scholar] [CrossRef]
- Beylerli, O.; Khasanov, D.; Gareev, I.; Valitov, E.; Sokhatskii, A.; Wang, C.; Pavlov, V.; Khasanova, G.; Ahmad, A. Differential Non-Coding RNAs Expression Profiles of Invasive and Non-Invasive Pituitary Adenomas. Non-Coding RNA Res. 2021, 6, 115–122. [Google Scholar] [CrossRef]
- Zhan, X.; Desiderio, D.M. Signaling Pathway Networks Mined from Human Pituitary Adenoma Proteomics Data. BMC Med. Genomics 2010, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Tanase, C.P.; Neagu, M.; Albulescu, R. Key Signaling Molecules in Pituitary Tumors. Expert. Rev. Mol. Diagn. 2009, 9, 859–877. [Google Scholar] [CrossRef]
- Muşat, M.; Vax, V.V.; Borboli, N.; Gueorguiev, M.; Bonner, S.; Korbonits, M.; Grossman, A.B. Cell Cycle Dysregulation in Pituitary Oncogenesis. Front. Horm. Res. 2004, 32, 34–62. [Google Scholar] [CrossRef]
- Neou, M.; Villa, C.; Armignacco, R.; Jouinot, A.; Raffin-Sanson, M.-L.; Septier, A.; Letourneur, F.; Diry, S.; Diedisheim, M.; Izac, B.; et al. Pangenomic Classification of Pituitary Neuroendocrine Tumors. Cancer Cell 2020, 37, 123–134.e5. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Q.; Zhu, J.; Yao, B.; Ma, C.; Qiao, N.; He, S.; Ye, Z.; Wang, Y.; Han, R.; et al. Integrated Proteogenomic Characterization across Major Histological Types of Pituitary Neuroendocrine Tumors. Cell Res. 2022, 32, 1047–1067. [Google Scholar] [CrossRef] [PubMed]
- Burman, P.; Trouillas, J.; Losa, M.; McCormack, A.; Petersenn, S.; Popovic, V.; Theodoropoulou, M.; Raverot, G.; Dekkers, O.M.; ESE Survey Collaborators. Aggressive Pituitary Tumours and Carcinomas, Characteristics and Management of 171 Patients. Eur. J. Endocrinol. 2022, 187, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Syro, L.V.; Rotondo, F.; Camargo, M.; Ortiz, L.D.; Serna, C.A.; Kovacs, K. Temozolomide and Pituitary Tumors: Current Understanding, Unresolved Issues, and Future Directions. Front. Endocrinol. 2018, 9, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gahete, M.D.; Jiménez-Vacas, J.M.; Alors-Pérez, E.; Herrero-Aguayo, V.; Fuentes-Fayos, A.C.; Pedraza-Arévalo, S.; Castaño, J.P.; Luque, R.M. Mouse Models of Endocrine Tumors. J. Endocrinol. 2019, 240, R73–R96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kano, M.; Suga, H.; Arima, H. Induction of Functional Hypothalamus and Pituitary Tissues From Pluripotent Stem Cells for Regenerative Medicine. J. Endocr. Soc. 2020, 5, bvaa188. [Google Scholar] [CrossRef] [PubMed]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR Signalling Pathway in Human Cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.-J.; Lai, Y.-H.; Manne, R.K.; Tsai, Y.-S.; Sarbassov, D.; Lin, H.-K. Akt: A Key Transducer in Cancer. J. Biomed. Sci. 2022, 29, 76. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Tschopp, O.; Baudry, A.; Dümmler, B.; Hynx, D.; Hemmings, B.A. Physiological Functions of Protein Kinase B/Akt. Biochem. Soc. Trans. 2004, 32, 350–354. [Google Scholar] [CrossRef]
- Chen, W.S.; Xu, P.-Z.; Gottlob, K.; Chen, M.-L.; Sokol, K.; Shiyanova, T.; Roninson, I.; Weng, W.; Suzuki, R.; Tobe, K.; et al. Growth Retardation and Increased Apoptosis in Mice with Homozygous Disruption of the Akt1 Gene. Genes Dev. 2001, 15, 2203–2208. [Google Scholar] [CrossRef] [Green Version]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a Protein Target of the FKBP12-Rapamycin Complex in Mammalian Cells (∗). J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.J.; Albers, M.W.; Bum Shin, T.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A Mammalian Protein Targeted by G1-Arresting Rapamycin–Receptor Complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef]
- Akbarzadeh, M.; Mihanfar, A.; Akbarzadeh, S.; Yousefi, B.; Majidinia, M. Crosstalk between miRNA and PI3K/AKT/mTOR Signaling Pathway in Cancer. Life Sci. 2021, 285. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. mTOR Signaling in Metabolism and Cancer. Cells 2020, 9, 2278. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and Downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR Signalling and Cellular Metabolism Are Mutual Determinants in Cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in Cancer and the Tumor Microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The Translational Landscape of mTOR Signalling Steers Cancer Initiation and Metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the mTORC1 Protein Kinase. Mol. Cell. 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a Binding Partner of Target of Rapamycin (TOR), Mediates TOR Action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The Mammalian Target of Rapamycin (mTOR) Partner, Raptor, Binds the mTOR Substrates P70 S6 Kinase and 4E-BP1 through Their TOR Signaling (TOS) Motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, L.P.; Yue, W.; Santen, R.J.; Lawrence, J.C. Farnesylthiosalicylic Acid Inhibits Mammalian Target of Rapamycin (mTOR) Activity Both in Cells and in Vitro by Promoting Dissociation of the mTOR-Raptor Complex. Mol. Endocrinol. 2005, 19, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex That Signals to the Cell Growth Machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holz, M.K.; Blenis, J. Identification of S6 Kinase 1 as a Novel Mammalian Target of Rapamycin (mTOR)-Phosphorylating Kinase. J. Biol. Chem. 2005, 280, 26089–26093. [Google Scholar] [CrossRef] [Green Version]
- Pullen, N.; Thomas, G. The Modular Phosphorylation and Activation of P70s6k. FEBS Lett. 1997, 410, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of mTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway That Regulates the Cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 Is Necessary for Akt/PKB Phosphorylation, and Its Isoforms Define Three Distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [Green Version]
- Oh, W.J.; Jacinto, E. mTOR Complex 2 Signaling and Functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabilii, S.; Ricciardi, M.R.; Piedimonte, M.; Gianfelici, V.; Bianchi, M.P.; Tafuri, A. Biological Aspects of mTOR in Leukemia. Int. J. Mol. Sci. 2018, 19, 2396. [Google Scholar] [CrossRef] [Green Version]
- Marquard, F.E.; Jücker, M. PI3K/AKT/mTOR Signaling as a Molecular Target in Head and Neck Cancer. Biochem. Pharm. 2020, 172, 113729. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.-I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Karagianni, F.; Pavlidis, A.; Malakou, L.S.; Piperi, C.; Papadavid, E. Predominant Role of mTOR Signaling in Skin Diseases with Therapeutic Potential. Int. J. Mol. Sci. 2022, 23, 1693. [Google Scholar] [CrossRef] [PubMed]
- Simioni, C.; Martelli, A.M.; Zauli, G.; Melloni, E.; Neri, L.M. Targeting mTOR in Acute Lymphoblastic Leukemia. Cells 2019, 8, 190. [Google Scholar] [CrossRef] [Green Version]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR Signaling Pathway in Ovarian Cancer: Biological and Therapeutic Significance. Semin. Cancer. Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Armengol, G.; Rojo, F.; Castellví, J.; Iglesias, C.; Cuatrecasas, M.; Pons, B.; Baselga, J.; Ramón y Cajal, S. 4E-Binding Protein 1: A Key Molecular “Funnel Factor” in Human Cancer with Clinical Implications. Cancer Res. 2007, 67, 7551–7555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivanco, I.; Sawyers, C.L. The Phosphatidylinositol 3-Kinase AKT Pathway in Human Cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Kouvaraki, M.A.; Liakou, C.; Paraschi, A.; Dimas, K.; Patsouris, E.; Tseleni-Balafouta, S.; Rassidakis, G.Z.; Moraitis, D. Activation of mTOR Signaling in Medullary and Aggressive Papillary Thyroid Carcinomas. Surgery 2011, 150, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Gild, M.L.; Landa, I.; Ryder, M.; Ghossein, R.A.; Knauf, J.A.; Fagin, J.A. Targeting mTOR in RET Mutant Medullary and Differentiated Thyroid Cancer Cells. Endocr. Relat. Cancer 2013, 20, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Lyra, J.; Vinagre, J.; Batista, R.; Pinto, V.; Prazeres, H.; Rodrigues, F.; Eloy, C.; Sobrinho-Simões, M.; Soares, P. mTOR Activation in Medullary Thyroid Carcinoma with RAS Mutation. Eur. J. Endocrinol. 2014, 171, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Zining, J.; Lu, X.; Caiyun, H.; Yuan, Y. Genetic Polymorphisms of mTOR and Cancer Risk: A Systematic Review and Updated Meta-Analysis. Oncotarget 2016, 7, 57464–57480. [Google Scholar] [CrossRef] [Green Version]
- Mirabilii, S.; Ricciardi, M.R.; Tafuri, A. mTOR Regulation of Metabolism in Hematologic Malignancies. Cells 2020, 9, 404. [Google Scholar] [CrossRef] [Green Version]
- Chamcheu, J.C.; Roy, T.; Uddin, M.B.; Banang-Mbeumi, S.; Chamcheu, R.-C.N.; Walker, A.L.; Liu, Y.-Y.; Huang, S. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells 2019, 8, 803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derwich, A.; Sykutera, M.; Bromińska, B.; Andrusiewicz, M.; Ruchała, M.; Sawicka-Gutaj, N. Clinical Implications of mTOR Expression in Papillary Thyroid Cancer—A Systematic Review. Cancers 2023, 15, 1665. [Google Scholar] [CrossRef] [PubMed]
- Stirewalt, D.L.; Kopecky, K.J.; Meshinchi, S.; Appelbaum, F.R.; Slovak, M.L.; Willman, C.L.; Radich, J.P. FLT3, RAS, and TP53 Mutations in Elderly Patients with Acute Myeloid Leukemia. Blood 2001, 97, 3589–3595. [Google Scholar] [CrossRef] [Green Version]
- Ullah, R.; Yin, Q.; Snell, A.H.; Wan, L. RAF-MEK-ERK Pathway in Cancer Evolution and Treatment. Semin. Cancer Biol. 2022, 85, 123–154. [Google Scholar] [CrossRef] [PubMed]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK Signaling Pathways Inhibitors as Anticancer Agents: Structural and Pharmacological Perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK Pathway in Cell Growth, Malignant Transformation and Drug Resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK Mitogen-Activated Protein Kinase Cascade for the Treatment of Cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [Green Version]
- Howe, L.R.; Leevers, S.J.; Gómez, N.; Nakielny, S.; Cohen, P.; Marshall, C.J. Activation of the MAP Kinase Pathway by the Protein Kinase Raf. Cell 1992, 71, 335–342. [Google Scholar] [CrossRef]
- Cuevas, B.D.; Abell, A.N.; Johnson, G.L. Role of Mitogen-Activated Protein Kinase Kinase Kinases in Signal Integration. Oncogene 2007, 26, 3159–3171. [Google Scholar] [CrossRef] [Green Version]
- Yap, J.L.; Worlikar, S.; MacKerell, A.D., Jr.; Shapiro, P.; Fletcher, S. Small-Molecule Inhibitors of the ERK Signaling Pathway: Towards Novel Anticancer Therapeutics. ChemMedChem 2011, 6, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Chong, H.; Vikis, H.G.; Guan, K.-L. Mechanisms of Regulating the Raf Kinase Family. Cell. Signal. 2003, 15, 463–469. [Google Scholar] [CrossRef]
- Lewis, T.S.; Shapiro, P.S.; Ahn, N.G. Signal Transduction through MAP Kinase Cascades. Adv. Cancer Res. 1998, 74, 49–139. [Google Scholar] [CrossRef]
- Younis, E. Oncogenesis of Thyroid Cancer. Asian Pac. J. Cancer Prev. 2017, 18, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK–RAS–RAF Signaling Pathway in Cancer Therapy. Expert. Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazieres, J.; Cropet, C.; Montané, L.; Barlesi, F.; Souquet, P.J.; Quantin, X.; Dubos-Arvis, C.; Otto, J.; Favier, L.; Avrillon, V.; et al. Vemurafenib in Non-Small-Cell Lung Cancer Patients with BRAFV600 and BRAFnonV600 Mutations. Ann. Oncol. 2020, 31, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Crippen, M.M.; Kılıç, S.; Eloy, J.A. Updates in the Management of Sinonasal Mucosal Melanoma. Curr. Opin. Otolaryngol. Head Neck Surg. 2018, 26, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF Kinases for Cancer Therapy: BRAF-Mutated Melanoma and Beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [Green Version]
- The PRISMA. A 2020 Statement: An Updated Guideline for Reporting Systematic Reviews|The BMJ. Available online: https://www.bmj.com/content/372/bmj.n71 (accessed on 27 January 2023).
- Monsalves, E.; Juraschka, K.; Tateno, T.; Agnihotri, S.; Asa, S.L.; Ezzat, S.; Zadeh, G. The PI3K/AKT/mTOR Pathway in the Pathophysiology and Treatment of Pituitary Adenomas. Endocr. Relat. Cancer 2014, 21, R331–R344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, W.; Sanders, A.J.; Jia, G.; Liu, X.; Lu, R.; Jiang, W.G. Expression of the mTOR Pathway Regulators in Human Pituitary Adenomas Indicates the Clinical Course. Anticancer Res. 2013, 33, 3123–3131. [Google Scholar]
- Zatelli, M.C.; Minoia, M.; Filieri, C.; Tagliati, F.; Buratto, M.; Ambrosio, M.R.; Lapparelli, M.; Scanarini, M.; degli Uberti, E.C. Effect of Everolimus on Cell Viability in Nonfunctioning Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2010, 95, 968–976. [Google Scholar] [CrossRef] [Green Version]
- Voronel, O.; Fedorchak, K.; Ali, S.M.; Elvin, J.A.; Vergilio, J.-A.; Suh, J.; Ramkissoon, S.H.; Miller, V.A.; Stephens, P.J.; Ross, J.S. Recurrent and Aggressive Pituitary Adenomas and Carcinomas: A Comprehensive Genomic Profiling Study. Lab. Investig. 2016, 96, 156A. [Google Scholar] [CrossRef] [Green Version]
- Sajjad, E.A.; Zieliński, G.; Maksymowicz, M.; Hutnik, Ł.; Bednarczuk, T.; Włodarski, P. mTOR Is Frequently Active in GH-Secreting Pituitary Adenomas without Influencing Their Morphopathological Features. Endocr. Pathol. 2013, 24, 11–19. [Google Scholar] [CrossRef]
- Maiter, D. Management of Dopamine Agonist-Resistant Prolactinoma. Neuroendocrinology 2019, 109, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Sun, R.; Wen, G.; Zhong, C.; Yang, J.; Zhu, J.; Cong, Z.; Luo, X.; Ma, C. Bromocriptine and Cabergoline Induce Cell Death in Prolactinoma Cells via the ERK/EGR1 and AKT/mTOR Pathway Respectively. Cell Death Dis. 2019, 10, 335. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Tang, C.; Cong, Z.; Yuan, F.; Cai, X.; Yang, J.; Ma, C. ACT001 Reverses Resistance of Prolactinomas via AMPK-Mediated EGR1 and mTOR Pathways. Endocr. Relat. Cancer 2021, 29, 33–46. [Google Scholar] [CrossRef]
- Yamamoto, M.; Nakao, T.; Ogawa, W.; Fukuoka, H. Aggressive Cushing’s Disease: Molecular Pathology and Its Therapeutic Approach. Front. Endocrinol. 2021, 12, 650791. [Google Scholar] [CrossRef]
- Di Pasquale, C.; Gentilin, E.; Falletta, S.; Bellio, M.; Buratto, M.; degli Uberti, E.; Chiara Zatelli, M. PI3K/Akt/mTOR Pathway Involvement in Regulating Growth Hormone Secretion in a Rat Pituitary Adenoma Cell Line. Endocrine 2018, 60, 308–316. [Google Scholar] [CrossRef]
- Li, J.; Li, C.; Wang, J.; Song, G.; Zhao, Z.; Wang, H.; Wang, W.; Li, H.; Li, Z.; Miao, Y.; et al. Genome-Wide Analysis of Differentially Expressed lncRNAs and mRNAs in Primary Gonadotrophin Adenomas by RNA-Seq. Oncotarget 2017, 8, 4585–4606. [Google Scholar] [CrossRef] [Green Version]
- Mangili, F.; Esposito, E.; Treppiedi, D.; Catalano, R.; Marra, G.; Di Muro, G.; Barbieri, A.M.; Locatelli, M.; Lania, A.G.; Mangone, A.; et al. DRD2 Agonist Cabergoline Abolished the Escape Mechanism Induced by mTOR Inhibitor Everolimus in Tumoral Pituitary Cells. Front. Endocrinol. 2022, 13, 867822. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, J.; Gao, H.; Yuan, T.; Kang, J.; Jin, L.; Gui, S.; Zhang, Y. Role of EGFL7/EGFR-Signaling Pathway in Migration and Invasion of Growth Hormone-Producing Pituitary Adenomas. Sci. China Life Sci. 2018, 61, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Feng, M.; Dai, C.; Bao, X.; Deng, K.; Yao, Y.; Wang, R. Expression of EGFR in Pituitary Corticotroph Adenomas and Its Relationship With Tumor Behavior. Front. Endocrinol. 2019, 10, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roof, A.K. Consider the Context: Ras/ERK and PI3K/AKT/mTOR Signaling Outcomes Are Pituitary Cell Type-Specific. Mol. Cell. Endocrinol. 2018, 10, 87–96. [Google Scholar] [CrossRef]
- De Dios, N.; Orrillo, S.; Irizarri, M.; Theas, M.S.; Boutillon, F.; Candolfi, M.; Seilicovich, A.; Goffin, V.; Pisera, D.; Ferraris, J. JAK2/STAT5 Pathway Mediates Prolactin-Induced Apoptosis of Lactotropes. Neuroendocrinology 2019, 108, 84–97. [Google Scholar] [CrossRef]
- Booth, A.; Trudeau, T.; Gomez, C.; Lucia, M.S.; Gutierrez-Hartmann, A. Persistent ERK/MAPK Activation Promotes Lactotrope Differentiation and Diminishes Tumorigenic Phenotype. Mol. Endocrinol. 2014, 28, 1999–2011. [Google Scholar] [CrossRef] [Green Version]
- Treppiedi, D.; Barbieri, A.M.; Di Muro, G.; Marra, G.; Mangili, F.; Catalano, R.; Esposito, E.; Ferrante, E.; Serban, A.L.; Locatelli, M.; et al. Genetic Profiling of a Cohort of Italian Patients with Acth-Secreting Pituitary Tumors and Characterization of a Novel Usp8 Gene Variant. Cancers 2021, 13, 4022. [Google Scholar] [CrossRef]
- Chanson, P.; Dormoy, A.; Dekkers, O.M. Use of Radiotherapy after Pituitary Surgery for Non-Functioning Pituitary Adenomas. Eur. J. Endocrinol. 2019, 181, D1–D13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekkers, O.M.; Karavitaki, N.; Pereira, A.M. The Epidemiology of Aggressive Pituitary Tumors (and Its Challenges). Rev. Endocr. Metab. Disord. 2020, 21, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Way, J.S.; Zhang, X.; Sergey, M.; Bergsneider, M.; Wang, M.B.; Yong, W.H.; Heaney, A.P. Effect of Everolimus in Treatment of Aggressive Prolactin-Secreting Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2019, 104, 1929–1936. [Google Scholar] [CrossRef]
- Donovan, L.E.; Arnal, A.V.; Wang, S.-H.; Odia, Y. Widely Metastatic Atypical Pituitary Adenoma with mTOR Pathway STK11(F298L) Mutation Treated with Everolimus Therapy. CNS Oncol. 2016, 5, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Jouanneau, E.; Wierinckx, A.; Ducray, F.; Favrel, V.; Borson-Chazot, F.; Honnorat, J.; Trouillas, J.; Raverot, G. New Targeted Therapies in Pituitary Carcinoma Resistant to Temozolomide. Pituitary 2012, 15, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Alshaikh, O.M.; Asa, S.L.; Mete, O.; Ezzat, S. An Institutional Experience of Tumor Progression to Pituitary Carcinoma in a 15-Year Cohort of 1055 Consecutive Pituitary Neuroendocrine Tumors. Endocr. Pathol. 2019, 30, 118–127. [Google Scholar] [CrossRef]
- Nogami, H.; Koshida, R.; Omori, H.; Shibata, M.; Harigaya, T.; Takei, Y. Inhibition of Epidermal Growth Factor Receptor Stimulates Prolactin Expression in Primary Culture of the Mouse Pituitary Gland. J. Neuroendocr. 2019, 31, e12764. [Google Scholar] [CrossRef]
- Hubina, E.; Nanzer, A.M.; Hanson, M.R.; Ciccarelli, E.; Losa, M.; Gaia, D.; Papotti, M.; Terreni, M.R.; Khalaf, S.; Jordan, S.; et al. Somatostatin Analogues Stimulate P27 Expression and Inhibit the MAP Kinase Pathway in Pituitary Tumours. Eur. J. Endocrinol. 2006, 155, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Radl, D.; De Mei, C.; Chen, E.; Lee, H.; Borrelli, E. Each Individual Isoform of the Dopamine D2 Receptor Protects from Lactotroph Hyperplasia. Mol. Endocrinol. 2013, 27, 953–965. [Google Scholar] [CrossRef] [Green Version]
- Cai, F.; Chen, S.; Yu, X.; Zhang, J.; Liang, W.; Zhang, Y.; Chen, Y.; Chen, S.; Hong, Y.; Yan, W.; et al. Transcription Factor GTF2B Regulates AIP Protein Expression in Growth Hormone-Secreting Pituitary Adenomas and Influences Tumor Phenotypes. Neuro. Oncol. 2022, 24, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Chanal, M.; Chevallier, P.; Raverot, V.; Fonteneau, G.; Lucia, K.; Monteserin Garcia, J.L.; Rachwan, A.; Jouanneau, E.; Trouillas, J.; Honnorat, J.; et al. Differential Effects of PI3K and Dual PI3K/mTOR Inhibition in Rat Prolactin-Secreting Pituitary Tumors. Mol. Cancer Ther. 2016, 15, 1261–1270. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Ruan, L.; Pu, J.; Zhong, A.; Wang, F.; Tan, S.; Huang, H.; Mu, J.; Yang, G. Metformin Suppresses Growth and Adrenocorticotrophic Hormone Secretion in Mouse Pituitary Corticotroph Tumor AtT20 cells. Mol. Cell. Endocrinol. 2018, 478, 53–61. [Google Scholar] [CrossRef]
- Lee, M.; Theodoropoulou, M.; Graw, J.; Roncaroli, F.; Zatelli, M.C.; Pellegata, N.S. Levels of P27 Sensitize to Dual PI3K/mTOR Inhibition. Mol. Cancer Ther. 2011, 10, 1450–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.; Zhang, B.; Liu, X.; Ma, S.; Yang, Y.; Yao, Y.; Feng, M.; Bao, X.; Li, G.; Wang, J.; et al. Inhibition of PI3K/AKT/mTOR Pathway Enhances Temozolomide-Induced Cytotoxicity in Pituitary Adenoma Cell Lines in Vitro and Xenografted Pituitary Adenoma in Female Nude Mice. Endocrinology 2013, 154, 1247–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; See, A.P.; Aziz, K.; Thiyagarajan, S.; Salih, T.; Gajula, R.P.; Armour, M.; Phallen, J.; Terezakis, S.; Kleinberg, L.; et al. Nelfinavir Induces Radiation Sensitization in Pituitary Adenoma Cells. Cancer Biol. Ther. 2011, 12, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Peverelli, E.; Olgiati, L.; Locatelli, M.; Magni, P.; Fustini, M.F.; Frank, G.; Mantovani, G.; Beck-Peccoz, P.; Spada, A.; Lania, A. The Dopamine-Somatostatin Chimeric Compound BIM-23A760 Exerts Antiproliferative and Cytotoxic Effects in Human Non-Functioning Pituitary Tumors by Activating ERK1/2 and P38 Pathways. Cancer Lett. 2010, 288, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Xue, Y.; Cao, L.; Liu, Q.; Liu, C.; Shan, X.; Wang, H.; Gu, Y.; Zhang, Y. ESR1 and Its Antagonist Fulvestrant in Pituitary Adenomas. Mol. Cell. Endocrinol. 2017, 443, 32–41. [Google Scholar] [CrossRef]
- Cooper, O.; Bonert, V.; Rudnick, J.; Pressman, B.; Melmed, S. SUN-442 EGFR/ErbB2 Targeted Therapy for Aggressive Prolactinomas. J. Endocr. Soc. 2019, 3, SUN-442. [Google Scholar] [CrossRef]
- Cooper, O.; Bonert, V.S.; Rudnick, J.; Pressman, B.D.; Lo, J.; Salvatori, R.; Yuen, K.C.J.; Fleseriu, M.; Melmed, S. EGFR/ErbB2-Targeting Lapatinib Therapy for Aggressive Prolactinomas. J. Clin. Endocrinol. Metab. 2021, 106, e917–e925. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; He, Q.; Meng, X.; Zhou, S.; Zhu, Y.; Xu, J.; Tao, R. Apatinib (YN968D1) and Temozolomide in Recurrent Invasive Pituitary Adenoma: Case Report and Literature Review. World Neurosurg. 2019, 124, 319–322. [Google Scholar] [CrossRef]
- Burman, P.; Casar-Borota, O.; Perez-Rivas, L.G.; Dekkers, O.M. Aggressive Pituitary Tumors and Pituitary Carcinomas: From Pathology to Treatment. J. Clin. Endocrinol. Metab. 2023, 108, 1585–1601. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, J.M.; Lee, E.J. Functional Expression of CXCR4 in Somatotrophs: CXCL12 Activates GH Gene, GH Production and Secretion, and Cellular Proliferation. J. Endocrinol. 2008, 199, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Lin, S.; Lin, Y.; Wu, S.; Zhuo, M.; Zhang, A.; Zheng, J.; You, Z. BRAF-Activated WT1 Contributes to Cancer Growth and Regulates Autophagy and Apoptosis in Papillary Thyroid Carcinoma. J. Transl. Med. 2022, 20, 79. [Google Scholar] [CrossRef] [PubMed]
- De Martino, I.; Fedele, M.; Palmieri, D.; Visone, R.; Cappabianca, P.; Wierinckx, A.; Trouillas, J.; Fusco, A. B-RAF Mutations Are a Rare Event in Pituitary Adenomas. J. Endocrinol. Investig. 2007, 30, RC1–RC3. [Google Scholar] [CrossRef]
- Ewing, I.; Pedder-Smith, S.; Franchi, G.; Ruscica, M.; Emery, M.; Vax, V.; Garcia, E.; Czirják, S.; Hanzély, Z.; Kola, B.; et al. A Mutation and Expression Analysis of the Oncogene BRAF in Pituitary Adenomas. Clin. Endocrinol. 2007, 66, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Cooper, O.; Vlotides, G.; Fukuoka, H.; Greene, M.I.; Melmed, S. Expression and Function of ErbB Receptors and Ligands in the Pituitary. Endocr. -Relat. Cancer 2011, 18, R197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Shlomo, A.; Cooper, O. The Role of Tyrosine Kinase Inhibitors in the Treatment of Pituitary Tumors: From Bench to Bedside. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 24, 301. [Google Scholar] [CrossRef]
- Theodoropoulou, M.; Arzberger, T.; Gruebler, Y.; Jaffrain-Rea, M.L.; Schlegel, J.; Schaaf, L.; Petrangeli, E.; Losa, M.; Stalla, G.K.; Pagotto, U. Expression of Epidermal Growth Factor Receptor in Neoplastic Pituitary Cells: Evidence for a Role in Corticotropinoma Cells. J. Endocrinol. 2004, 183, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Kano, M.; Araki, T.; Cooper, O.; Fukuoka, H.; Tone, Y.; Tone, M.; Melmed, S. ErbB Receptor-Driven Prolactinomas Respond to Targeted Lapatinib Treatment in Female Transgenic Mice. Endocrinology 2015, 156, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Vlotides, G.; Siegel, E.; Donangelo, I.; Gutman, S.; Ren, S.-G.; Melmed, S. Rat Prolactinoma Cell Growth Regulation by Epidermal Growth Factor Receptor Ligands. Cancer Res. 2008, 68, 6377–6386. [Google Scholar] [CrossRef] [Green Version]
- Kwatra, M.M. A Rational Approach to Target the Epidermal Growth Factor Receptor in Glioblastoma. Current Cancer Drug Targets 2017, 17, 290–296. [Google Scholar] [CrossRef]
- Feng, K.; Guo, Y.; Dai, H.; Wang, Y.; Li, X.; Jia, H.; Han, W. Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of Patients with EGFR-Expressing Advanced Relapsed/Refractory Non-Small Cell Lung Cancer. Sci. China Life Sci. 2016, 59, 468–479. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, X.; Zhou, Y.; Luo, Y.; Li, C.; Yuan, X. Expression and Clinical Significance of EGFL7 in Malignant Glioma. J. Cancer Res. Clin. Oncol. 2010, 136, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, J.; Yang, H.; Gao, H.; Li, C.; Lan, X.; Zhang, Y. Attenuation of EGFL7 Expression Inhibits Growth Hormone–Producing Pituitary Adenomas Growth and Invasion. Hum. Gene Ther. 2018, 29, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Q.; Gao, H.; Wan, D.; Li, C.; Li, Z.; Zhang, Y. EGFL7 Participates in Regulating Biological Behavior of Growth Hormone-Secreting Pituitary Adenomas via Notch2/DLL3 Signaling Pathway. Tumour Biol. 2017, 39, 1010428317706203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for Cancer Therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Dworakowska, D.; Wlodek, E.; Leontiou, C.A.; Igreja, S.; Cakir, M.; Teng, M.; Prodromou, N.; Góth, M.I.; Grozinsky-Glasberg, S.; Gueorguiev, M.; et al. Activation of RAF/MEK/ERK and PI3K/AKT/mTOR Pathways in Pituitary Adenomas and Their Effects on Downstream Effectors. Endocr. Relat. Cancer 2009, 16, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Effect of Combined Treatment with a Pan-PI3K Inhibitor or an Isoform-Specific PI3K Inhibitor and Everolimus on Cell Proliferation in GH-Secreting Pituitary Tumour in an Experimental Setting|SpringerLink. Available online: https://link.springer.com/article/10.1007/s12020-018-1677-2 (accessed on 29 April 2022).
- Lee, M.; Wiedemann, T.; Gross, C.; Leinhäuser, I.; Roncaroli, F.; Braren, R.; Pellegata, N.S. Targeting PI3K/mTOR Signaling Displays Potent Antitumor Efficacy against Nonfunctioning Pituitary Adenomas. Clin. Cancer Res. 2015, 21, 3204–3215. [Google Scholar] [CrossRef] [Green Version]
- Lamb, L.S.; Sim, H.-W.; McCormack, A.I. Exploring the Role of Novel Medical Therapies for Aggressive Pituitary Tumors: A Review of the Literature—“Are We There Yet?”. Cancers 2020, 12, 308. [Google Scholar] [CrossRef] [Green Version]



| Drug Name | Publication | Drug Mechanism | Type of Tumor | Outcome |
|---|---|---|---|---|
| lapatinib | McCormack et al. (2018) [11], Cooper et al. (2019) [147], Cooper et al. (2021) [148], | dual EGFR and HER2/Neu inhibitor | 10 APT-PRL; 2 APT unspecified | 6 SD, 1 PR, 5 PD |
| erlotinib | McCormack et al. (2018) [11] | EGFR inhibitor | APT-ACTH | 1 PD |
| gefitinib | McCormack et al. (2018) [11] | EGFR inhibitor | APT-PRL/GH | 1 PR |
| sunitinib | McCormack et al. (2018) [11], Alshaikh et al. (2019) [135], Burman et al. (2022) [50] | oral multireceptor TK inhibitor | 2 APT unspecified; 1 APT-ACTH | 3 PD |
| apatinib plus temozolomide | Wang et al. (2019) [149] | VEGF inhibitor | APT-GH | 1 CR |
| bevacizumab | Burman et al. (2023) [150] | recombinant monoclonal antibody blocking VEGF | 8 APT and 4 PC | 2 PR, 6 SD, 4 PD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Derwich, A.; Sykutera, M.; Bromińska, B.; Rubiś, B.; Ruchała, M.; Sawicka-Gutaj, N. The Role of Activation of PI3K/AKT/mTOR and RAF/MEK/ERK Pathways in Aggressive Pituitary Adenomas—New Potential Therapeutic Approach—A Systematic Review. Int. J. Mol. Sci. 2023, 24, 10952. https://doi.org/10.3390/ijms241310952
Derwich A, Sykutera M, Bromińska B, Rubiś B, Ruchała M, Sawicka-Gutaj N. The Role of Activation of PI3K/AKT/mTOR and RAF/MEK/ERK Pathways in Aggressive Pituitary Adenomas—New Potential Therapeutic Approach—A Systematic Review. International Journal of Molecular Sciences. 2023; 24(13):10952. https://doi.org/10.3390/ijms241310952
Chicago/Turabian StyleDerwich, Aleksandra, Monika Sykutera, Barbara Bromińska, Błażej Rubiś, Marek Ruchała, and Nadia Sawicka-Gutaj. 2023. "The Role of Activation of PI3K/AKT/mTOR and RAF/MEK/ERK Pathways in Aggressive Pituitary Adenomas—New Potential Therapeutic Approach—A Systematic Review" International Journal of Molecular Sciences 24, no. 13: 10952. https://doi.org/10.3390/ijms241310952