
Comparative Analysis of Cell Mixtures Deconvolution and Gene Signatures Generated for Blood, Immune and Cancer Cells

 ,
,  ,
,  , , and
, , and
Abstract
:1. Introduction
1.1. Cell Heterogeneity
1.2. Deconvolution to Decompose Mixtures
1.3. Formulation of Deconvolution
2. Results
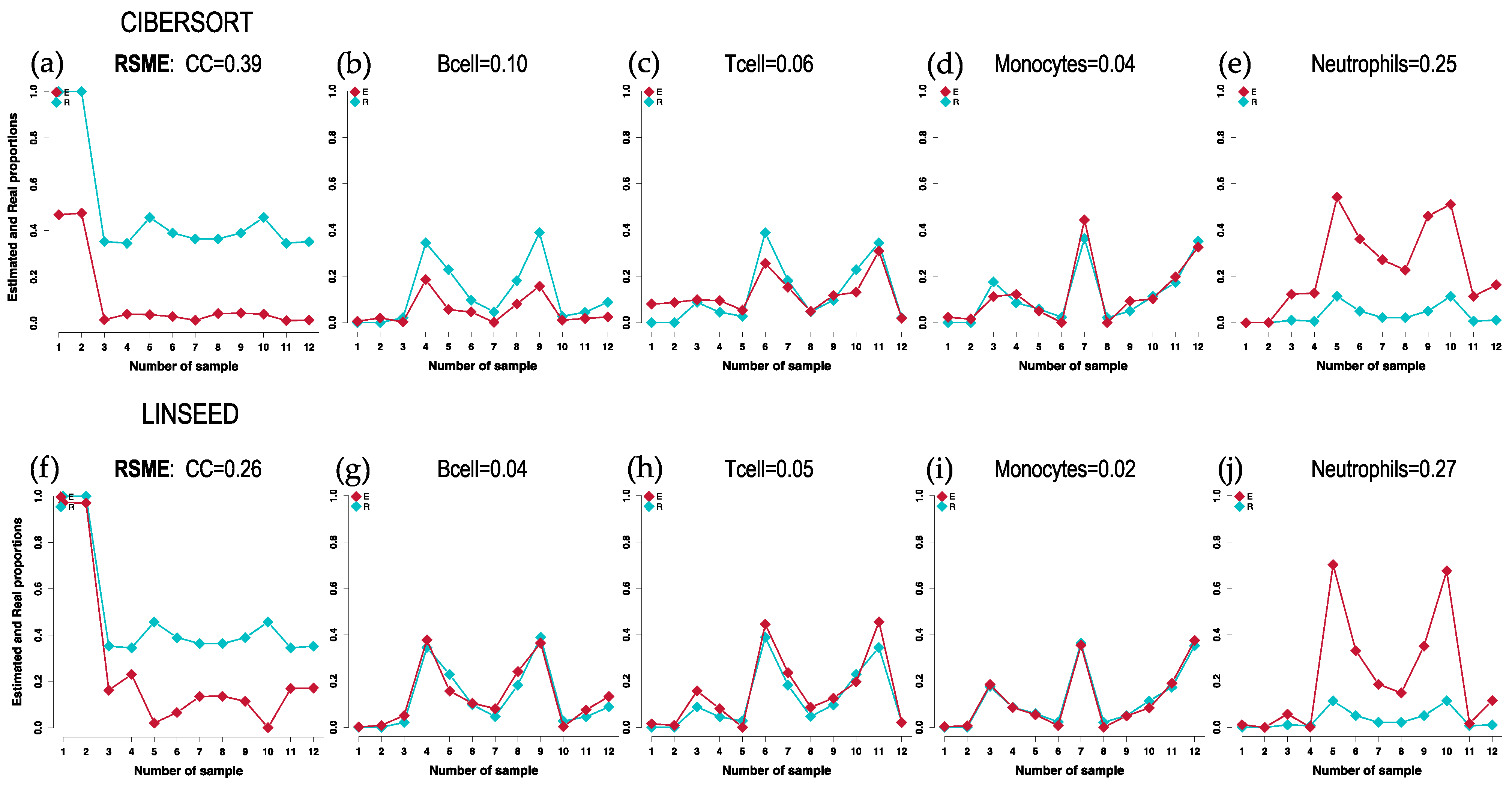
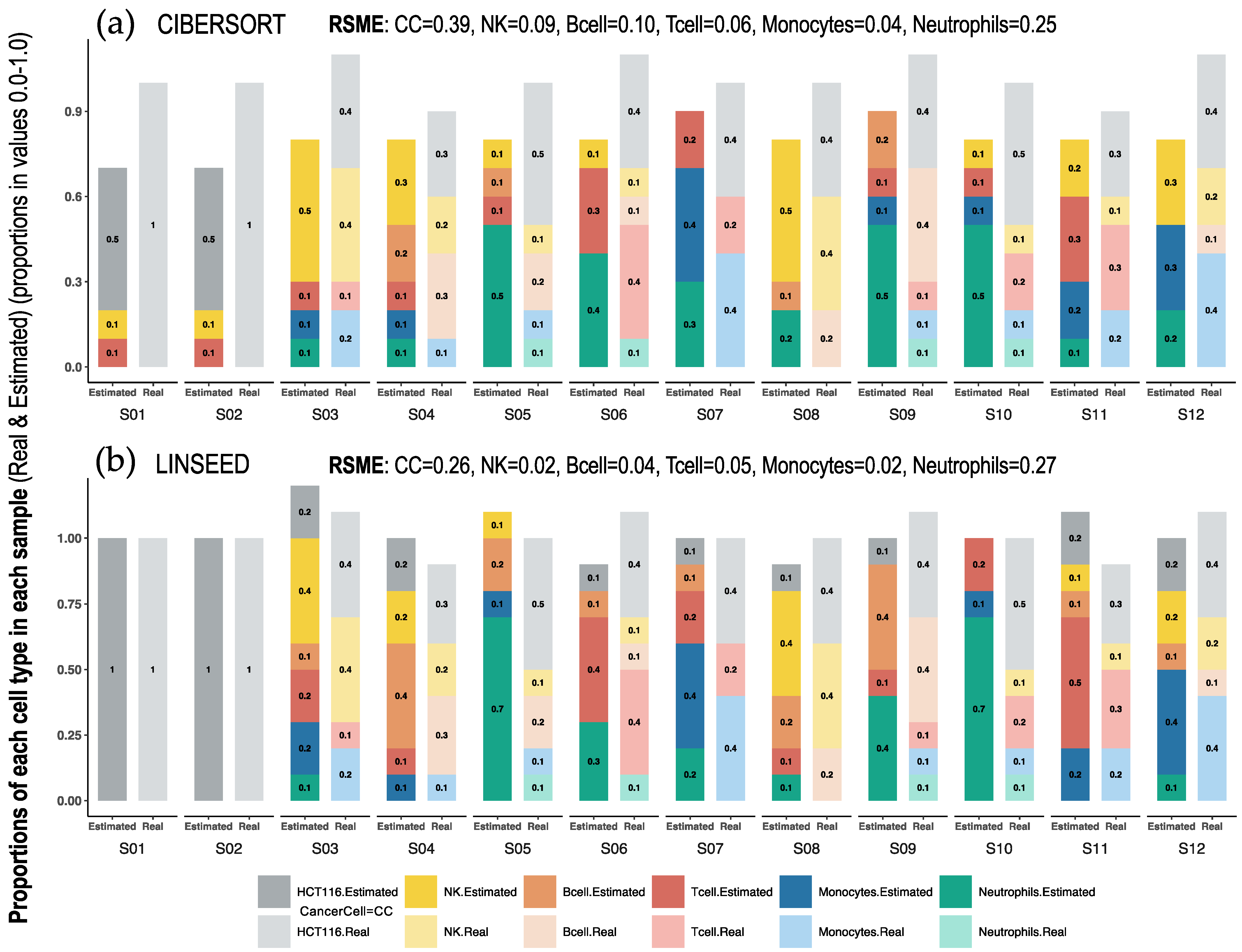
2.1. Comparison of Cell Type Proportions Correlations Using Four Deconvolution Methods
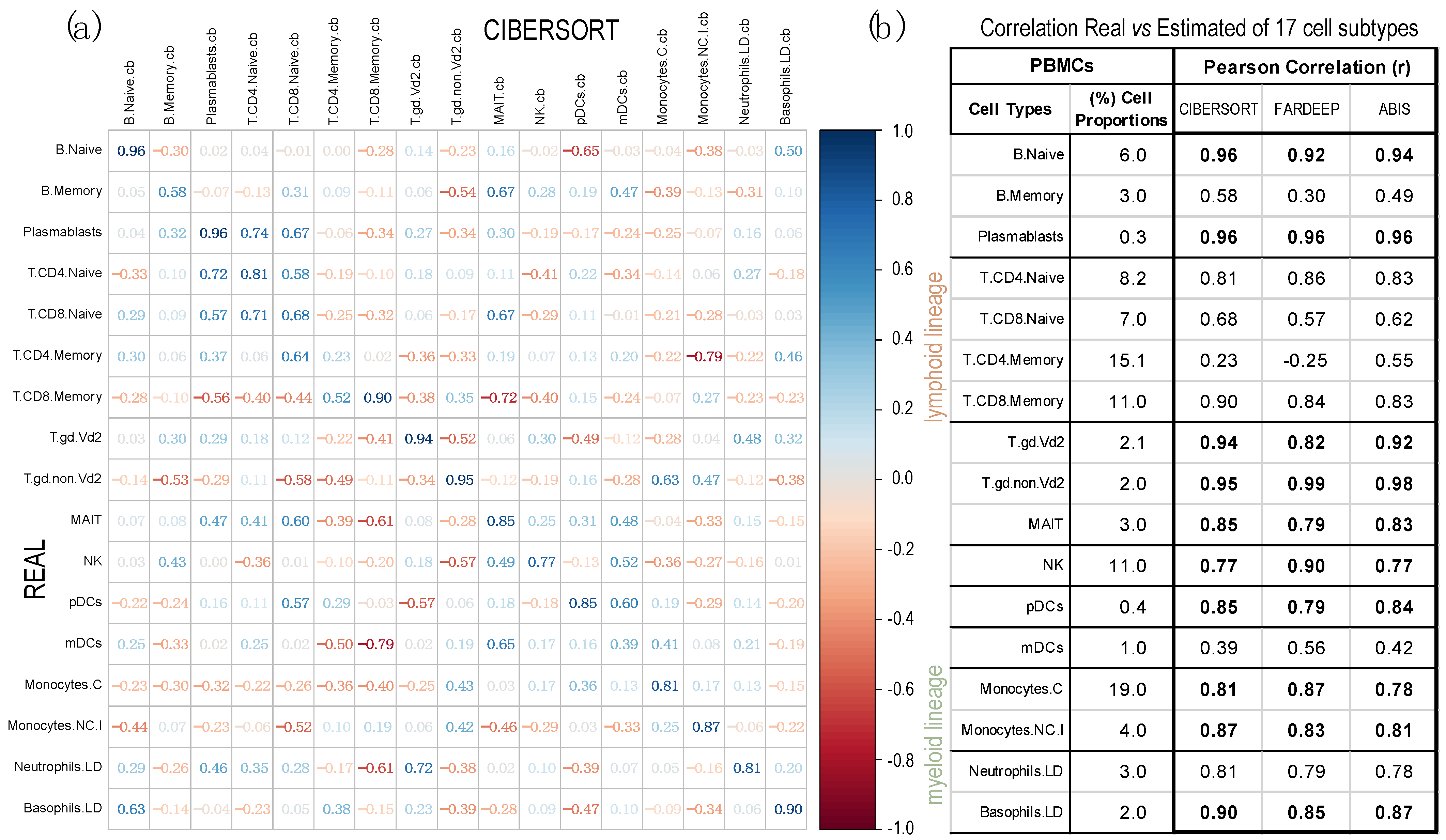
2.2. Comparison of Proportions of 17 Cell Types, Identified in PBMCs, Calculated Using Different Deconvolution Methods against the Proportions Experimentally Determined
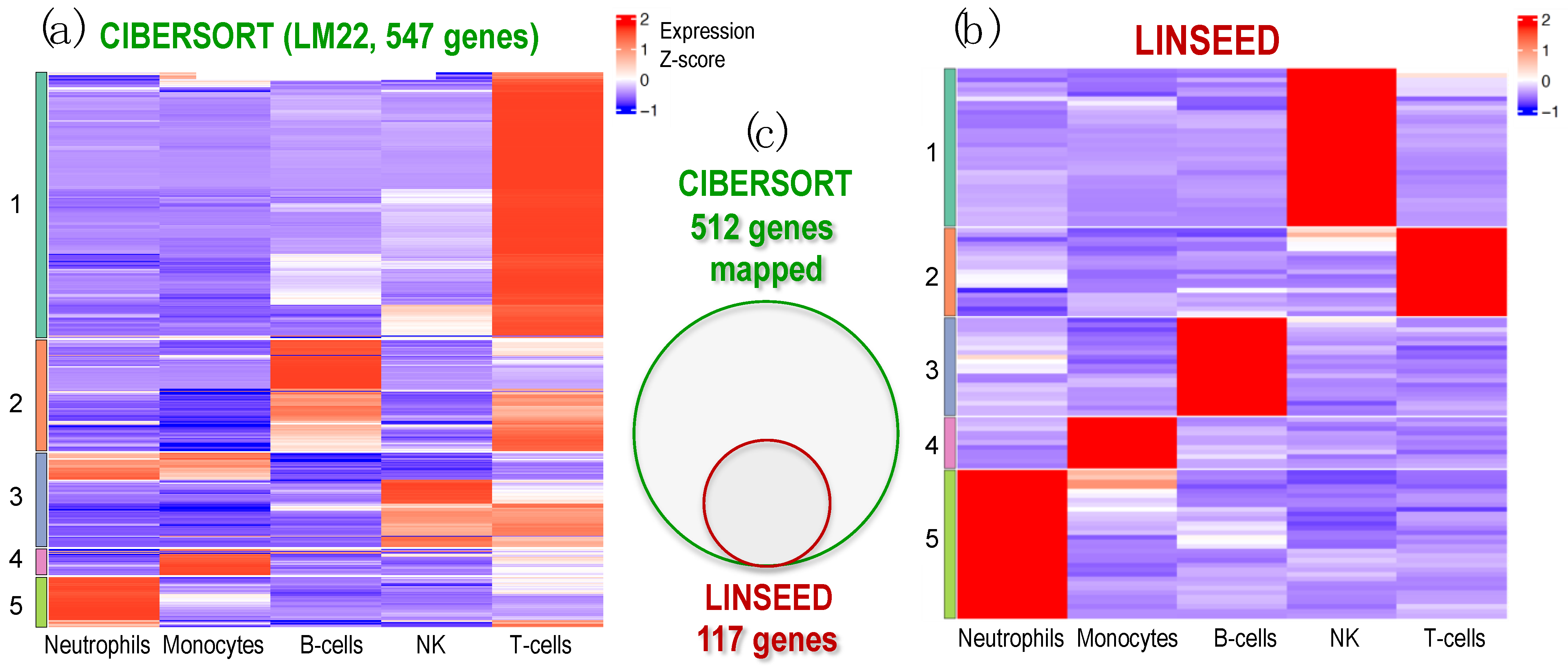
2.3. Identification of Cell-Specific Gene Signatures Obtained by the Combination of Two Deconvolution Methods
3. Discussion
4. Materials and Methods
4.1. Datasets
- (i)
- LM22: Signature matrix composed of 22 immune cell types and 547 genes, designed by CIBERSORT authors [10]. We used it to decompose the mixture samples (bulk expression data) of dataset GSE64385.
- (ii)
- ‘sigmatrixMicro.txt’: Matrix consisting of 819 genes characterizing 11 immune cell types in complex cell mixtures. Signal expression was obtained with Illumina microarrays [47]. We applied this matrix to decompose the bulk in GSE106898.
- (iii)
- ‘sigmatrixRNAseq.txt’: Signature matrix composed of 1296 gene biomarkers to identify 17 immune cell populations. Signal expression was obtained with Illumina RNA-seq [48]. We applied this matrix to deconvolute the bulk in GSE107011.
4.2. Brief Description of the Cell Mixture Deconvolution Methods Used
4.2.1. DECONICA: Deconvolution through Immune Component Analysis
4.2.2. LINSEED: Linear Subspace Identification for Gene Expression
4.2.3. ABIS: ABsolute Immune Signal Deconvolution
4.2.4. FARDEEP: Fast and Robust Deconvolution of Expression Profiles
4.2.5. CIBERSORT: Estimation of Cell Types Abundances in a Mixed Cell Population Using Gene Expression Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.F.; Wei, W.; Smithy, J.W.; Acs, B.; Toki, M.I.; Blenman, K.R.M.; Zelterman, D.; Kluger, H.M.; Rimm, D.L. Multiplex Quantitative Analysis of Tumor-Infiltrating Lymphocytes and Immunotherapy Outcome in Metastatic Melanoma. Clin. Cancer Res. 2019, 25, 2442–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uryvaev, A.; Passhak, M.; Hershkovits, D.; Sabo, E.; Bar-Sela, G. The role of tumor-infiltrating lymphocytes (TILs) as a predictive biomarker of response to anti-PD1 therapy in patients with metastatic non-small cell lung cancer or metastatic melanoma. Med. Oncol. 2018, 35, 25. [Google Scholar] [CrossRef] [PubMed]
- Mami-Chouaib, F.; Blanc, C.; Corgnac, S.; Hans, S.; Malenica, I.; Granier, C.; Tihy, I.; Tartour, E. Resident memory T cells, critical components in tumor immunology. J. Immunother. Cancer 2018, 6, 87. [Google Scholar] [CrossRef] [Green Version]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.-J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef] [Green Version]
- Pascual-García, M.; Bonfill-Teixidor, E.; Planas-Rigol, E.; Rubio-Perez, C.; Iurlaro, R.; Arias, A.; Cuartas, I.; Sala-Hojman, A.; Escudero, L.; Martinez-Ricarte, F.; et al. LIF regulates CXCL9 in tumor-associated macrophages and prevents CD8+ T cell tumor-infiltration impairing anti-PD1 therapy. Nat. Commun. 2019, 10, 2416. [Google Scholar] [CrossRef] [Green Version]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein–α. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Shen-Orr, S.S.; Gaujoux, R. Computational deconvolution: Extracting cell type-specific information from heterogeneous samples. Curr. Opin. Immunol. 2013, 25, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Cherry, E.C. Some Experiments on the Recognition of Speech, with One and with Two Ears. J. Acoust. Soc. Am. 1953, 25, 975–979. [Google Scholar] [CrossRef]
- Shinn-Cunningham, B.G. Object-based auditory and visual attention. Trends Cogn. Sci. 2008, 12, 182–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czerwińska, U. Unsupervised Deconvolution of Bulk Omics Profiles: Methodology and Application to Characterize the Im-mune Landscape in Tumors. Ph.D. Thesis, Sorbonne University, París, France, 2018. Available online: https://urszulaczerwinska.github.io/UCzPhDThesis/ (accessed on 30 June 2021).
- Lu, P.; Nakorchevskiy, A.; Marcotte, E.M. Expression deconvolution: A reinterpretation of DNA microarray data reveals dynamic changes in cell populations. Proc. Natl. Acad. Sci. USA 2003, 100, 10370–10375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, A.; Quon, G.; Rosenberg, A.M.; Yeung, R.S.M.; Morris, Q.; BBOP Study Consortium. Gene Expression Deconvolution for Uncovering Molecular Signatures in Response to Therapy in Juvenile Idiopathic Arthritis. PLoS ONE 2016, 11, e0156055. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.; Thu, D.; Waldvogel, H.; Faull, R.; Luthi-Carter, R. Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat. Methods 2011, 8, 945–947. [Google Scholar] [CrossRef]
- Qiao, W.; Quon, G.; Csaszar, E.; Yu, M.; Morris, Q.; Zandstra, P.W. PERT: A Method for Expression Deconvolution of Human Blood Samples from Varied Microenvironmental and Developmental Conditions. PLoS Comput. Biol. 2012, 8, e1002838. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, S.; Zuckerman, N.; Goldsmith, A.; Grama, A. A Critical Survey of Deconvolution Methods for Separating Cell Types in Complex Tissues. Proc. IEEE 2016, 105, 340–366. [Google Scholar] [CrossRef]
- Gong, T.; Szustakowski, J.D. DeconRNASeq: A statistical framework for deconvolution of heterogeneous tissue samples based on mRNA-Seq data. Bioinformatics 2013, 29, 1083–1085. [Google Scholar] [CrossRef] [Green Version]
- Cappello, V.; Marchetti, L.; Parlanti, P.; Landi, S.; Tonazzini, I.; Cecchini, M.; Piazza, V.; Gemmi, M. Ultrastructural Characterization of the Lower Motor System in a Mouse Model of Krabbe Disease. Sci. Rep. 2016, 6, 1. [Google Scholar] [CrossRef]
- Gaujoux, R.; Seoighe, C. Semi-supervised Nonnegative Matrix Factorization for gene expression deconvolution: A case study. Infect. Genet. Evol. 2012, 12, 913–921. [Google Scholar] [CrossRef]
- Sturm, G.; Finotello, F.; List, M. In Silico Cell-Type Deconvolution Methods in Cancer Immunotherapy. Methods Mol. Biol. 2020, 2120, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumour–host interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y. Adipocyte and lipid metabolism in cancer drug resistance. J. Clin. Investig. 2019, 129, 3006–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2018; Volume 1711, pp. 243–259. [Google Scholar] [CrossRef]
- Liu, C.C.; Steen, C.B.; Newman, A.M. Computational approaches for characterizing the tumor immune microenvironment. Immunology 2019, 158, 70–84. [Google Scholar] [CrossRef] [Green Version]
- Craven, K.E.; Gökmen-Polar, Y.; Badve, S.S. CIBERSORT analysis of TCGA and METABRIC identifies subgroups with better outcomes in triple negative breast cancer. Sci. Rep. 2021, 11, 4691. [Google Scholar] [CrossRef]
- Kawada, J.-I.; Takeuchi, S.; Imai, H.; Okumura, T.; Horiba, K.; Suzuki, T.; Torii, Y.; Yasuda, K.; Imanaka-Yoshida, K.; Ito, Y. Immune cell infiltration landscapes in pediatric acute myocarditis analyzed by CIBERSORT. J. Cardiol. 2020, 77, 174–178. [Google Scholar] [CrossRef]
- Chen, T.; Hua, W.; Xu, B.; Chen, H.; Xie, M.; Sun, X.; Ge, X. Robust rank aggregation and cibersort algorithm applied to the identification of key genes in head and neck squamous cell cancer. Math. Biosci. Eng. 2021, 18, 4491–4507. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; An, Y.; Xu, B.; Wang, N.; Sun, X.; Sun, M. Potential Impact of ALKBH5 and YTHDF1 on Tumor Immunity in Colon Adenocarcinoma. Front. Oncol. 2021, 11, 670490. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Yan, M.; Heath, B.R.; Lei, Y.L.; Xie, Y. Fast and robust deconvolution of tumor infiltrating lymphocyte from expression profiles using least trimmed squares. PLoS Comput. Biol. 2019, 15, e1006976. [Google Scholar] [CrossRef] [Green Version]
- Sturm, G.; Finotello, F.; Petitprez, F.; Zhang, J.D.; Baumbach, J.; Fridman, W.H.; List, M.; Aneichyk, T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019, 35, i436–i445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobos, F.A.; Alquicira-Hernandez, J.; Powell, J.E.; Mestdagh, P.; De Preter, K. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat. Commun. 2020, 11, 5650. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Liu, Z. A benchmark for RNA-seq deconvolution analysis under dynamic testing environments. Genome Biol. 2021, 22, 101. [Google Scholar] [CrossRef]
- Sutton, G.J.; Poppe, D.; Simmons, R.K.; Walsh, K.; Nawaz, U.; Lister, R.; Gagnon-Bartsch, J.A.; Voineagu, I. Comprehensive evaluation of deconvolution methods for human brain gene expression. Nat. Commun. 2022, 13, 1358. [Google Scholar] [CrossRef]
- Im, Y.; Kim, Y. A comprehensive overview of RNA deconvolution methods and their application. Mol. Cells 2023, 46, 99–105. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Wang, X.; Park, J.; Susztak, K.; Zhang, N.R.; Li, M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 2019, 10, 380. [Google Scholar] [CrossRef] [Green Version]
- Tsoucas, D.; Dong, R.; Chen, H.; Zhu, Q.; Guo, G.; Yuan, G.-C. Accurate estimation of cell-type composition from gene expression data. Nat. Commun. 2019, 10, 2975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jew, B.; Alvarez, M.; Rahmani, E.; Miao, Z.; Ko, A.; Garske, K.M.; Sul, J.H.; Pietiläinen, K.H.; Pajukanta, P.; Halperin, E. Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat. Commun. 2020, 11, 1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimnezhad, A. More accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Bioinform. Adv. 2022, 2, vbac049. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Monaco, G.; Lee, B.; Xu, W.; Mustafah, S.; Hwang, Y.Y.; Carré, C.; Burdin, N.; Visan, L.; Ceccarelli, M.; Poidinger, M.; et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 2019, 26, 1627–1640.e7. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Monaco, G.; Wong, E.H.; Tan, W.L.W.; Kared, H.; Simoni, Y.; Tan, S.W.; How, W.Z.Y.; Tan, C.T.Y.; Lee, B.; et al. Mapping of γ/δ T cells reveals Vδ2+ T cells resistance to senescence. Ebiomedicine 2018, 39, 44–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyvarinen, A. Fast ICA for noisy data using gaussian moments. In Proceedings of the IEEE International Symposium on Circuits and Systems, Orlando, FL, USA, 30 May–2 June 1999; p. 5. [Google Scholar] [CrossRef]
- Zaitsev, K.; Bambouskova, M.; Swain, A.; Artyomov, M.N. Complete deconvolution of cellular mixtures based on linearity of transcriptional signatures. Nat. Commun. 2019, 10, 2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantzig, G.B. Origins of the simplex method. In A History of Scientific Computing; ACM: New York, NY, USA, 1990; pp. 141–151. [Google Scholar] [CrossRef]
- Xu, Q.; Yan, M.; Huang, C.; Xiong, J.; Huang, Q.; Yao, Y. Exploring Outliers in Crowdsourced Ranking for QoE. In Proceedings of the 25th ACM International Conference on Multimedia, Mountain View, CA, USA, 23–27 October 2017; p. 17. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number | Gene Expression Platform | Samples | Genes | Biological Source | Cell Types | Reference |
|---|---|---|---|---|---|---|
| GSE64385 | Microarray HGU133 Plus 2.0—Affymetrix | 12 | 54,675 | PBMCs 1, PMNs 2, and Cancer Cells (HCT116) | 5 | [10] |
| GSE107011 | RNA-seq HiSeq 2000—Illumina | 13 | 17,487 | PBMCs | 17 | [48] |
| GSE106898 | Microarray Human HT-12 V4.0—Illumina | 13 | 17,487 | PBMCs | 11 | [47] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso-Moreda, N.; Berral-González, A.; De La Rosa, E.; González-Velasco, O.; Sánchez-Santos, J.M.; De Las Rivas, J. Comparative Analysis of Cell Mixtures Deconvolution and Gene Signatures Generated for Blood, Immune and Cancer Cells. Int. J. Mol. Sci. 2023, 24, 10765. https://doi.org/10.3390/ijms241310765
Alonso-Moreda N, Berral-González A, De La Rosa E, González-Velasco O, Sánchez-Santos JM, De Las Rivas J. Comparative Analysis of Cell Mixtures Deconvolution and Gene Signatures Generated for Blood, Immune and Cancer Cells. International Journal of Molecular Sciences. 2023; 24(13):10765. https://doi.org/10.3390/ijms241310765
Chicago/Turabian StyleAlonso-Moreda, Natalia, Alberto Berral-González, Enrique De La Rosa, Oscar González-Velasco, José Manuel Sánchez-Santos, and Javier De Las Rivas. 2023. "Comparative Analysis of Cell Mixtures Deconvolution and Gene Signatures Generated for Blood, Immune and Cancer Cells" International Journal of Molecular Sciences 24, no. 13: 10765. https://doi.org/10.3390/ijms241310765