
Dual-Warhead Conjugate Based on Fibroblast Growth Factor 2 Dimer Loaded with α-Amanitin and Monomethyl Auristatin E Exhibits Superior Cytotoxicity towards Cancer Cells Overproducing Fibroblast Growth Factor Receptor 1

Abstract
:1. Introduction
2. Results and Discussion
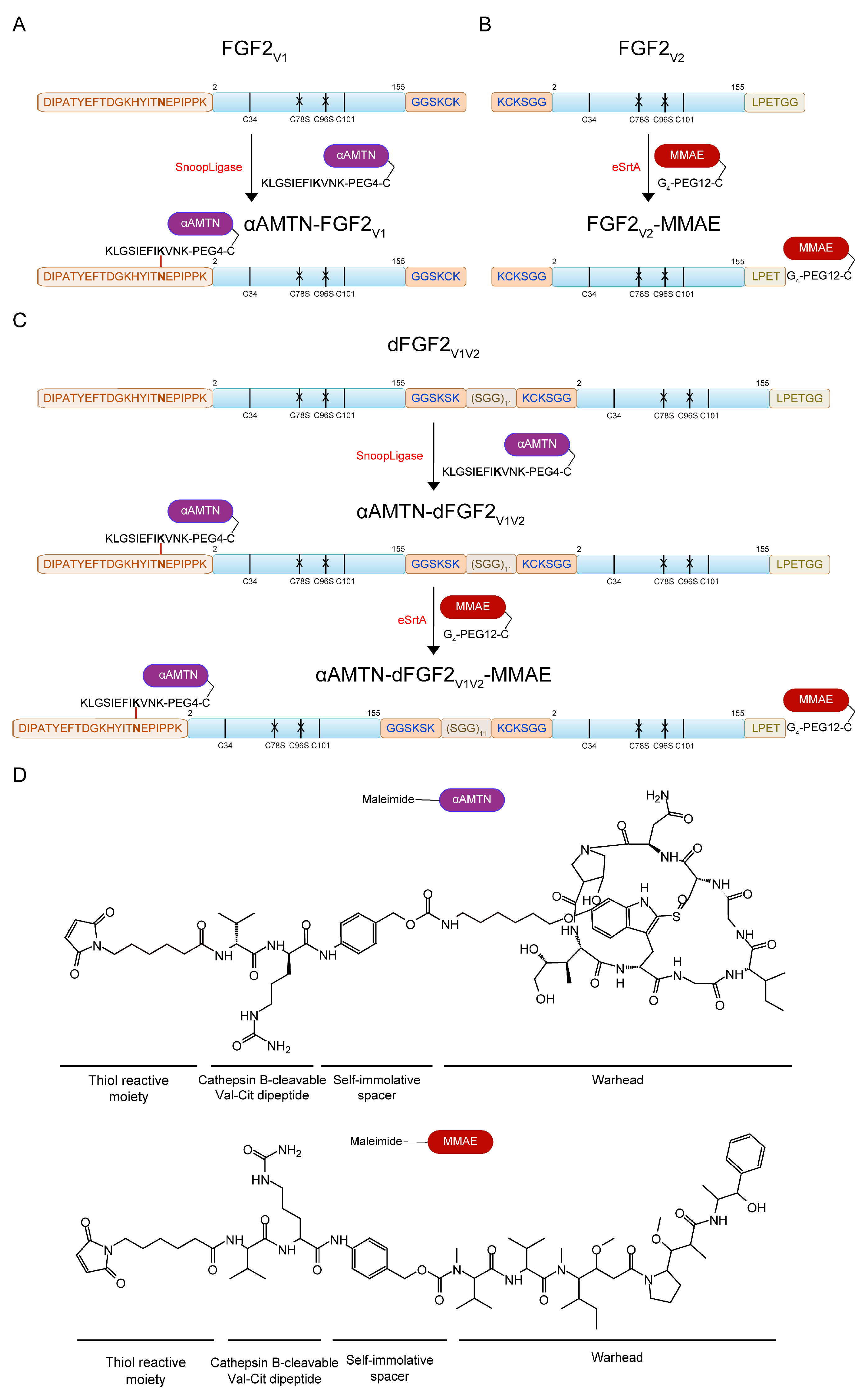
2.1. Engineering the Dimeric Dual-Warhead FGF2 Conjugate
2.1.1. Design Principle of the FGF2 Dimer (dFGF2V1V2)
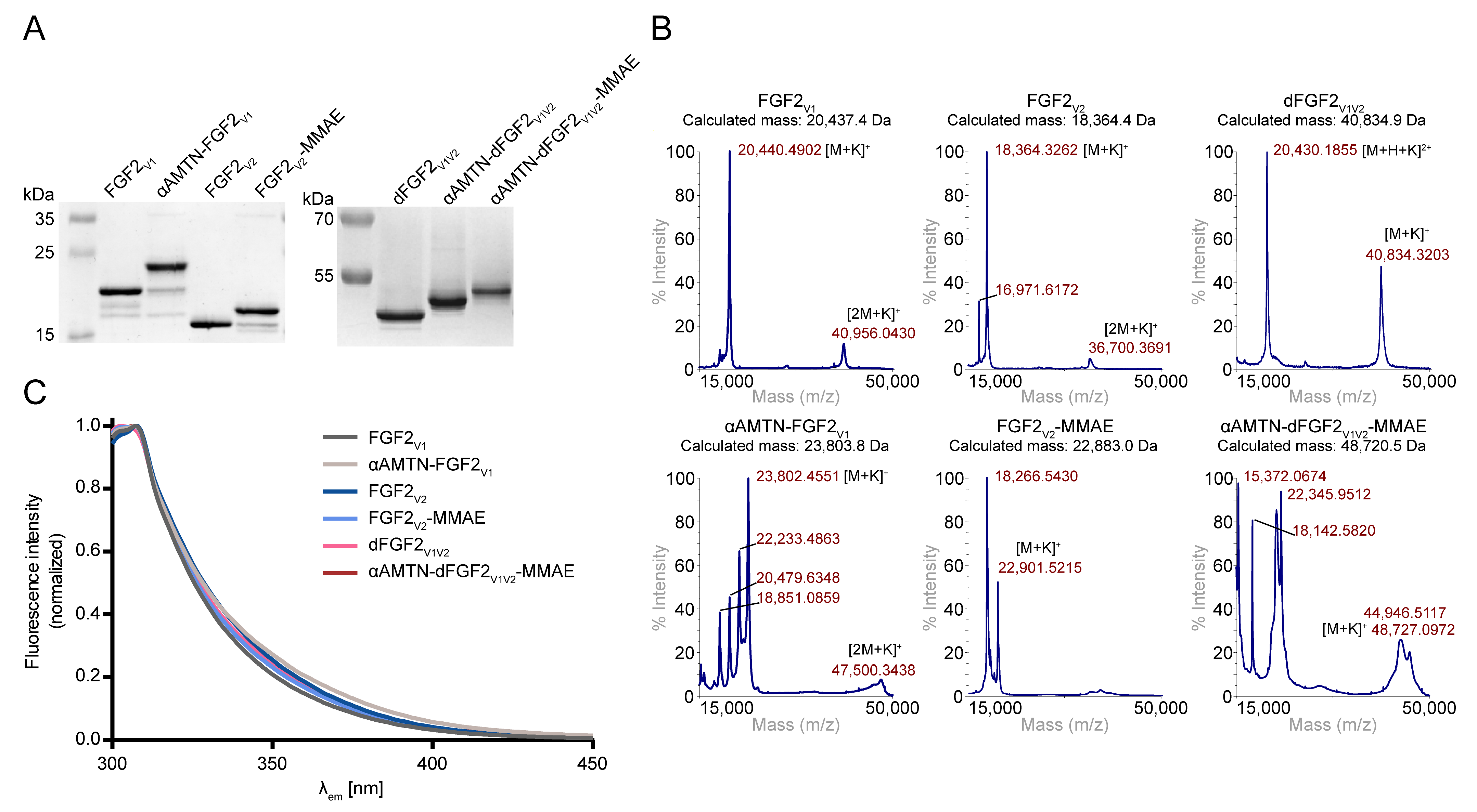
2.1.2. α-AMTN and MMAE Conjugation Reaction Scheme
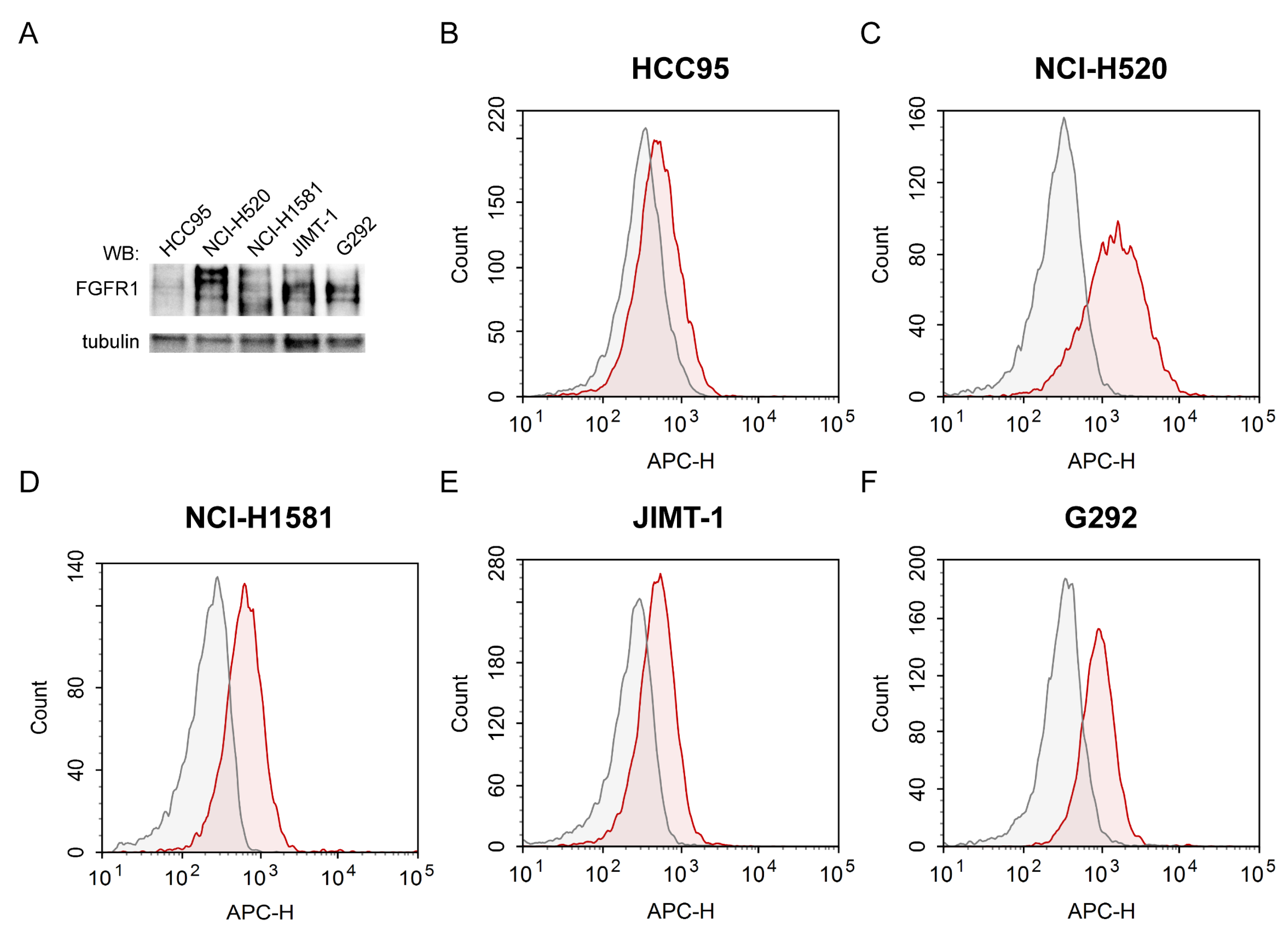
2.2. Evaluation of FGFR1 Expression Levels and Accessibility for FGF2 in Various Cancer Cell Lines
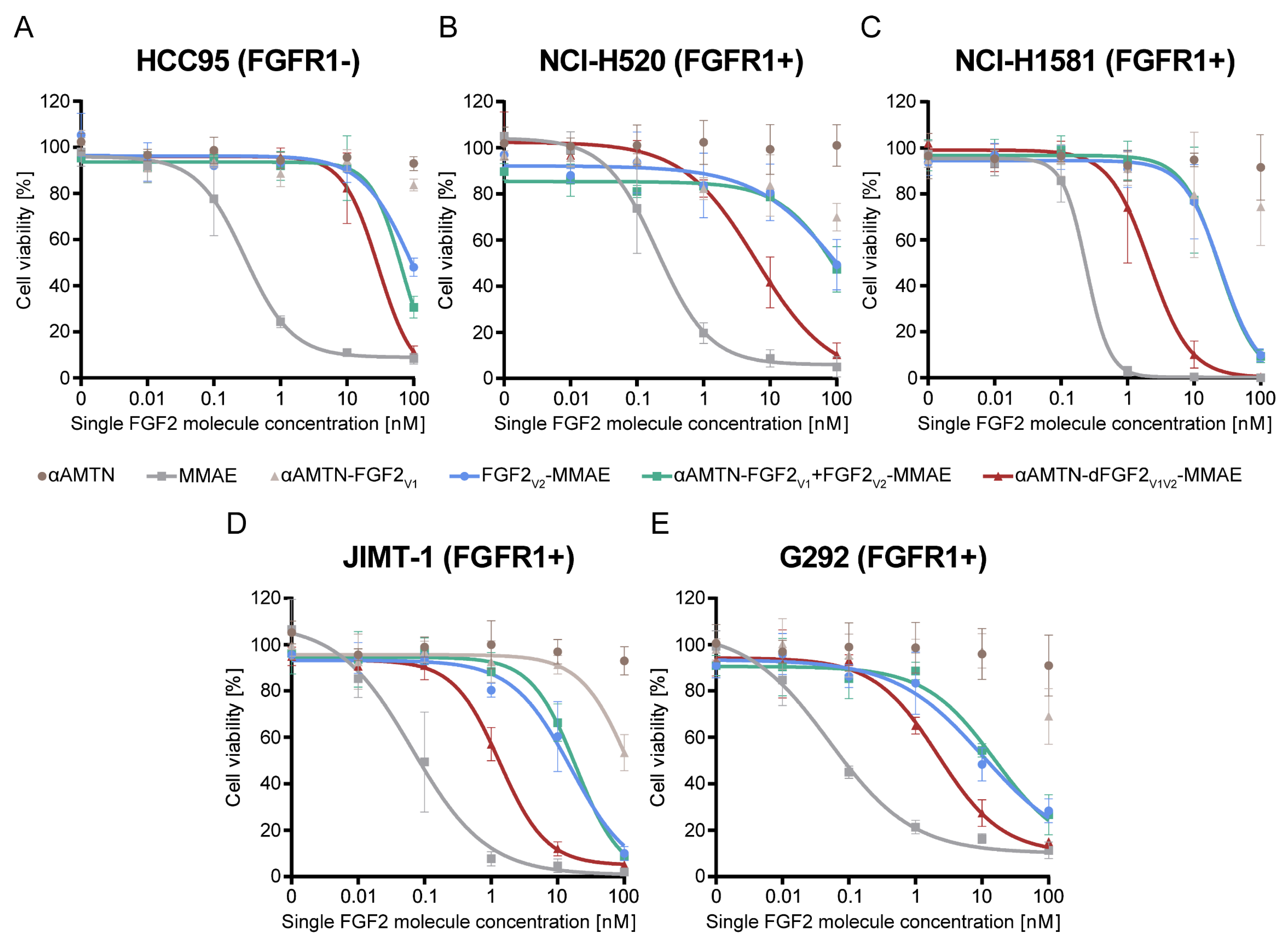
2.3. Superior Cytotoxicity of the Dimeric Dual-Warhead Conjugate
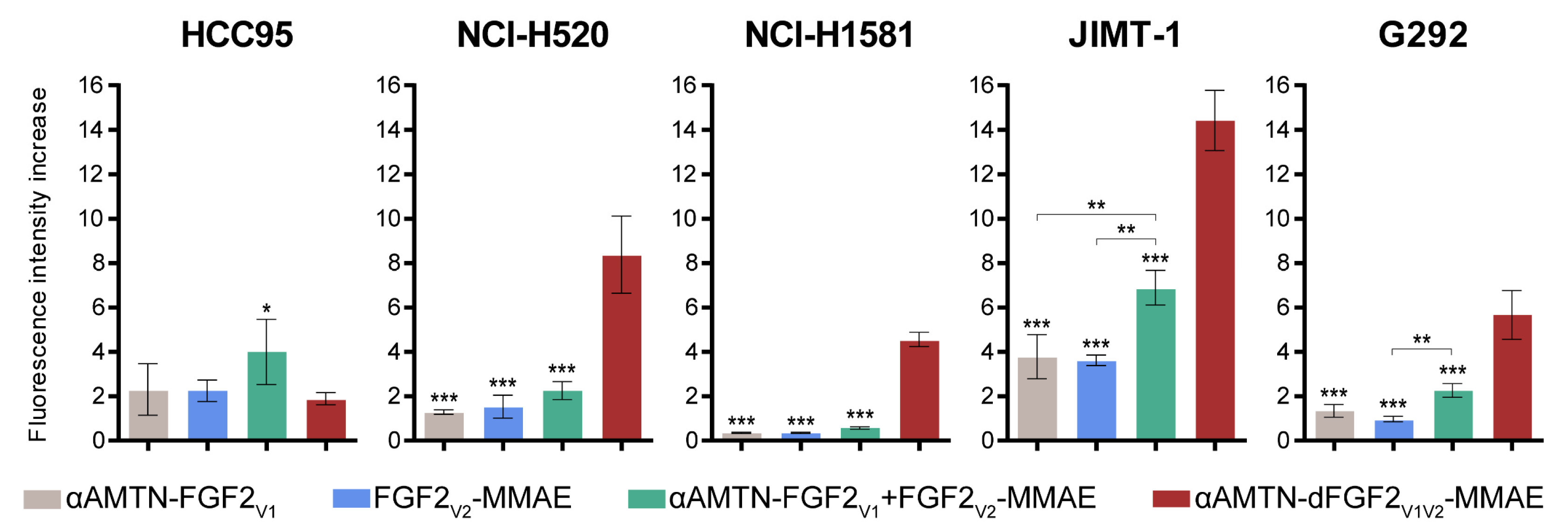
2.4. Selective and Efficient Internalization of the αAMTN-dFGF2V1V2-MMAE Conjugate into Cells Expressing FGFR1
3. Materials and Methods
3.1. Reagents
3.2. Cell Lines
3.3. Recombinant Proteins
3.4. Synthesis and Purification of KLGSIEFIKVNK-PEG4-C-vcαAMTN and GGGG-PEG12-C-vcMMAE
3.5. Synthesis of the Conjugates
3.5.1. Conjugation of the FGF2V1 and dFGF2V1V2 with αAMTN via SnoopLigase-Mediated Ligation
3.5.2. Double-Warhead Conjugate Synthesis. Conjugation of the αAMTN-dFGF2V1V2 and FGF2V2 with MMAE via eSrtA-Mediated Ligation
3.6. Mass Spectrometry
3.7. Spectrofluorimetry
3.8. Cytotoxicity Assay
3.9. Labeling of Conjugates and Wild-Type FGF2 with Fluorescent Probes
3.10. Flow Cytometric Evaluation of FGF2 Capacity to Bind to the Cells
3.11. Flow Cytometric Analysis of Conjugates’ Steady-State Internalization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADC | Antibody–drug conjugate. |
| ADCC | Antibody-dependent cellular cytotoxicity. |
| Ado | 8-amino-3,6-dioxaoctanoyl. |
| ATCC | American Type Culture Collection. |
| BSA | Bovine serum albumin. |
| CDC | Complement-dependent cytotoxicity. |
| DCM | Dichloromethane. |
| DIPEA | N,N-diisopropylethylamine. |
| DMAc | N,N-dimethylacetamide. |
| DMF | N,N′-dimethylformamide. |
| DTNB | 5,5-dithio-bis-(2-nitrobenzoic acid). |
| DTT | Dithiothreitol. |
| EC | Effective concentration. |
| EDTA | Ethylenediaminetetraacetic acid. |
| eSrtA | Evolved sortase A. |
| FA | Formic acid. |
| FGF | Fibroblast growth factor. |
| FGFR | Fibroblast growth factor receptor. |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid. |
| HPLC | High-performance liquid chromatography. |
| HRP | Horseradish peroxidase. |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside. |
| MALDI | Matrix-assisted laser desorption/ionization. |
| MMAE | Monomethyl auristatin E. |
| MS | Mass spectrometry. |
| TCEP | Tris(2-carboxyethyl)phosphine. |
| OD | Optical density. |
| PABC | Para-aminobenzylcarbamate. |
| PBS | Phosphate-buffered saline. |
| PEG | Poly(ethylene glycol). |
| PMSF | Phenylmethylsulfonyl fluoride. |
| PVDF | Polyvinylidene difluoride. |
| RP | Reversed phase. |
| SD | Standard deviation. |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis. |
| SPPS | Solid phase peptide synthesis. |
| TB | Terrific broth. |
| TCA | Trichloroacetic acid. |
| TCE | 2,2,2-trichloroethan-1-ol. |
| TFA | Trifluoroacetic acid. |
| TIS | Triisopropylsilane. |
| TOF | Time of flight. |
| WT | Wild-type. |
| αAMTN | α-amanitin. |
References
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Elsheikh, S.E.; Green, A.R.; Lambros, M.B.K.; Turner, N.C.; Grainge, M.J.; Powe, D.; Ellis, I.O.; Reis-Filho, J.S. FGFR1 amplification in breast carcinomas: A chromogenic in situ hybridisation analysis. Breast Cancer Res. 2007, 9, R23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Servetto, A.; Formisano, L.; Arteaga, C.L. FGFR signaling and endocrine resistance in breast cancer: Challenges for the clinical development of FGFR inhibitors. Biochim. Biophys. Acta-Rev. Cancer 2021, 1876, 188595. [Google Scholar] [CrossRef]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Bange, J.; Prechtl, D.; Cheburkin, Y.; Specht, K.; Harbeck, N.; Schmitt, M.; Knyazeva, T.; Müller, S.; Gärtner, S.; Sures, I.; et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg388 allele. Cancer Res. 2002, 62, 840–847. [Google Scholar]
- Kommalapati, A.; Tella, S.H.; Borad, M.; Javle, M.; Mahipal, A. Fgfr inhibitors in oncology: Insight on the management of toxicities in clinical practice. Cancers 2021, 13, 2968. [Google Scholar] [CrossRef] [PubMed]
- Pacini, L.; Jenks, A.D.; Lima, N.C.; Huang, P.H. Targeting the fibroblast growth factor receptor (Fgfr) family in lung cancer. Cells 2021, 10, 1154. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Lowery, M.; Shroff, R.T.; Heinz Weiss, K.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Zhou, Z.; Chen, Z.; Xu, G.; Chen, Y. Fibroblast growth factor receptors (Fgfrs): Structures and small molecule inhibitors. Cells 2019, 8, 614. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.P.; Hung, T.H.; Hsiao, S.H.; Huang, Y.H.; Hung, L.C.; Yu, Y.J.; Chang, Y.T.; Wang, S.P.; Wu, Y.S. Erdafitinib resensitizes ABCB1-overexpressing multidrug-resistant cancer cells to cytotoxic anticancer drugs. Cancers 2020, 12, 1366. [Google Scholar] [CrossRef]
- Tabernero, J.; Bahleda, R.; Dienstmann, R.; Infante, J.R.; Mita, A.; Italiano, A.; Calvo, E.; Moreno, V.; Adamo, B.; Gazzah, A.; et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 3401–3408. [Google Scholar] [CrossRef]
- Siefker-Radtke, A.O.; Mellado, B.; Decaestecker, K.; Burke, J.M.; O’Hagan, A.; Avadhani, A.N.; Zhong, B.; Santiago-Walker, A.E.; De Porre, P.; Brookman-May, S.; et al. A phase 2 study of JNJ-42756493, a pan-FGFR tyrosine kinase inhibitor, in patients (pts) with metastatic or unresectable urothelial cancer (UC) harboring FGFR gene alterations. J. Clin. Oncol. 2016, 34, TPS4575. [Google Scholar] [CrossRef]
- Perera, T.P.S.; Jovcheva, E.; Mevellec, L.; Vialard, J.; De Lange, D.; Verhulst, T.; Paulussen, C.; Van De Ven, K.; King, P.; Freyne, E.; et al. Discovery & pharmacological characterization of JNJ-42756493 (Erdafitinib), a functionally selective small-molecule FGFR family inhibitor. Mol. Cancer Ther. 2017, 16, 1010–1020. [Google Scholar] [CrossRef] [Green Version]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR landscape in cancer: Analysis of 4,853 tumors by next-generation sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Javle, M.; Roychowdhury, S.; Kelley, R.K.; Sadeghi, S.; Macarulla, T.; Weiss, K.H.; Waldschmidt, D.T.; Goyal, L.; Borbath, I.; El-Khoueiry, A.; et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: Mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol. Hepatol. 2021, 6, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Chan, A.G.; Ahene, A.; Bellovin, D.I.; Deng, R.; Hsu, A.W.; Jeffry, U.; Palencia, S.; Powers, J.; Zanghi, J.; et al. Preclinical characterization of bemarituzumab, an anti-FGFR2b antibody for the treatment of cancer. MAbs 2021, 13, 1981202. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Tesfaye, A.; Tejani, M.; Cheung, E.; Eisenberg, P.; Scott, A.J.; Eng, C.; Hnatyszyn, J.; Marina, N.; Powers, J.; et al. Bemarituzumab with modified FOLFOX6 for advanced FGFR2-positive gastroesophageal cancer: FIGHT Phase III study design. Future Oncol. 2019, 15, 2073–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, H.; Liu, L.; Gao, Y.; Ahene, A.; Collins, H. Covariate effects and population pharmacokinetic analysis of the anti-FGFR2b antibody bemarituzumab in patients from phase 1 to phase 2 trials. Cancer Chemother. Pharmacol. 2021, 88, 899–910. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Enzinger, P.C.; Kang, Y.K.; Qin, S.; Yamaguchi, K.; Kim, I.H.; Saeed, A.; Oh, S.C.; Li, J.; Turk, H.M.; et al. Bemarituzumab in patients with FGFR2b-selected gastric or gastro-oesophageal junction adenocarcinoma (FIGHT): A randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2022, 23, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Bahleda, R.; Hierro, C.; Sanson, M.; Bridgewater, J.; Arkenau, H.T.; Tran, B.; Kelley, R.K.; Park, J.O.; Javle, M.; et al. Futibatinib, an Irreversible FGFR1–4 Inhibitor, in Patients with Advanced Solid Tumors Harboring FGF/FGFR Aberrations: A Phase I Dose-Expansion Study. Cancer Discov. 2022, 12, 402–415. [Google Scholar] [CrossRef]
- Bahleda, R.; Meric-Bernstam, F.; Goyal, L.; Tran, B.; He, Y.; Yamamiya, I.; Benhadji, K.A.; Matos, I.; Arkenau, H.T. Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1–4 inhibitor in patients with advanced solid tumors. Ann. Oncol. 2020, 31, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Sootome, H.; Fujita, H.; Ito, K.; Ochiiwa, H.; Fujioka, Y.; Ito, K.; Miura, A.; Sagara, T.; Ito, S.; Ohsawa, H.; et al. Futibatinib is a novel irreversible FGFR 1-4 inhibitor that shows selective antitumor activity against FGFR-deregulated tumors. Cancer Res. 2020, 80, 4986–4997. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Shitara, K.; Kojima, T.; Kuboki, Y.; Matsubara, N.; Bando, H.; Yoh, K.; Naito, Y.; Hirai, H.; Kurokawa, Y.; et al. Phase I study of the irreversible fibroblast growth factor receptor 1–4 inhibitor futibatinib in Japanese patients with advanced solid tumors. Cancer Sci. 2023, 114, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, H.; Yun, M.R.; Kang, H.N.; Pyo, K.H.; Park, H.J.; Lee, J.M.; Choi, H.M.; Ellinghaus, P.; Ocker, M.; et al. Activation of the Met kinase confers acquired drug resistance in FGFR-targeted lung cancer therapy. Oncogenesis 2016, 5, e241. [Google Scholar] [CrossRef] [Green Version]
- Yue, S.; Li, Y.; Chen, X.; Wang, J.; Li, M.; Chen, Y.; Wu, D. FGFR-TKI resistance in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 23. [Google Scholar] [CrossRef]
- Szlachcic, A.; Zakrzewska, M.; Lobocki, M.; Jakimowicz, P.; Otlewski, J. Design and characteristics of cytotoxic fibroblast growth factor 1 conjugate for fibroblast growth factor receptor-targeted cancer therapy. Drug. Des. Devel. Ther. 2016, 10, 2547–2560. [Google Scholar] [CrossRef] [Green Version]
- Lobocki, M.; Zakrzewska, M.; Szlachcic, A.; Krzyscik, M.A.; Sokolowska-Wedzina, A.; Otlewski, J. High-Yield Site-Specific Conjugation of Fibroblast Growth Factor 1 with Monomethylauristatin E via Cysteine Flanked by Basic Residues. Bioconjug. Chem. 2017, 28, 1850–1858. [Google Scholar] [CrossRef]
- Krzyscik, M.A.; Zakrzewska, M.; Sørensen, V.; Sokolowska-Wedzina, A.; Lobocki, M.; Swiderska, K.W.; Krowarsch, D.; Wiedlocha, A.; Otlewski, J. Cytotoxic Conjugates of Fibroblast Growth Factor 2 (FGF2) with Monomethyl Auristatin E for Effective Killing of Cells Expressing FGF Receptors. ACS Omega 2017, 2, 3792–3805. [Google Scholar] [CrossRef] [Green Version]
- Swiderska, K.W.; Szlachcic, A.; Czyrek, A.; Zakrzewska, M.; Otlewski, J. Site-specific conjugation of fibroblast growth factor 2 (FGF2) based on incorporation of alkyne-reactive unnatural amino acid. Bioorg. Med. Chem. 2017, 25, 3685–3693. [Google Scholar] [CrossRef]
- Świderska, K.W.; Szlachcic, A.; Opaliński, Ł.; Zakrzewska, M.; Otlewski, J. FGF2 dual warhead conjugate with monomethyl auristatin E and α-amanitin displays a cytotoxic effect towards cancer cells overproducing FGF receptor 1. Int. J. Mol. Sci. 2018, 19, 2098. [Google Scholar] [CrossRef] [Green Version]
- Krzyscik, M.A.; Opaliński, Ł.; Otlewski, J. Novel Method for Preparation of Site-Specific, Stoichiometric-Controlled Dual Warhead Conjugate of FGF2 via Dimerization Employing Sortase A-Mediated Ligation. Mol. Pharm. 2019, 16, 3588–3599. [Google Scholar] [CrossRef] [PubMed]
- Nawrocka, D.; Krzyscik, M.A.; Opaliński, Ł.; Zakrzewska, M.; Otlewski, J. Stable fibroblast growth factor 2 dimers with high pro-survival and mitogenic potential. Int. J. Mol. Sci. 2020, 21, 4108. [Google Scholar] [CrossRef] [PubMed]
- Ducry, L.; Stump, B. Antibody−Drug Conjugates: Linking Cytotoxic Payloads to Monoclonal Antibodies. Bioconjug. Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef]
- Reddy Chichili, V.P.; Kumar, V.; Sivaraman, J. Linkers in the structural biology of protein-protein interactions. Protein Sci. 2013, 22, 153–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug. Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Abdollahpour-Alitappeh, M.; Lotfinia, M.; Gharibi, T.; Mardaneh, J.; Farhadihosseinabadi, B.; Larki, P.; Faghfourian, B.; Sepehr, K.S.; Abbaszadeh-Goudarzi, K.; Abbaszadeh-Goudarzi, G.; et al. Antibody–drug conjugates (ADCs) for cancer therapy: Strategies, challenges, and successes. J. Cell. Physiol. 2019, 234, 5628–5642. [Google Scholar] [CrossRef]
- Bushnell, D.A.; Cramer, P.; Kornberg, R.D. Structural basis of transcription: α-amanitin-RNA polymerase II cocrystal at 2.8 Å resolution. Proc. Natl. Acad. Sci. USA 2002, 99, 1218–1222. [Google Scholar] [CrossRef] [Green Version]
- Moldenhauer, G.; Salnikov, A.V.; Lüttgau, S.; Herr, I.; Anderl, J.; Faulstich, H. Therapeutic potential of amanitin-conjugated anti-epithelial cell adhesion molecule monoclonal antibody against pancreatic carcinoma. J. Natl. Cancer Inst. 2012, 104, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chemie-Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [Green Version]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Wang, L.; Amphlett, G.; Blättler, W.A.; Lambert, J.M.; Zhang, W. Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci. 2005, 14, 2436–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug. Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Baalmann, M.; Ziegler, M.J.; Werther, P.; Wilhelm, J.; Wombacher, R. Enzymatic and site-specific ligation of minimal-size tetrazines and triazines to proteins for bioconjugation and live-cell imaging. Bioconjug. Chem. 2019, 30, 1405–1414. [Google Scholar] [CrossRef]
- Liu, Y.; Tian, F.; Shi, S.; Deng, Y.; Zheng, P. Enzymatic Protein-Protein Conjugation through Internal Site Verified at the Single-Molecule Level. J. Phys. Chem. Lett. 2021, 12, 10914–10919. [Google Scholar] [CrossRef] [PubMed]
- Buldun, C.M.; Jean, J.X.; Bedford, M.R.; Howarth, M. SnoopLigase Catalyzes Peptide–Peptide Locking and Enables Solid-Phase Conjugate Isolation. J. Am. Chem. Soc. 2018, 140, 3008–3018. [Google Scholar] [CrossRef]
- Fierer, J.O.; Veggiani, G.; Howarth, M. SpyLigase peptide–peptide ligation polymerizes affibodies to enhance magnetic cancer cell capture. Proc. Natl. Acad. Sci. USA 2014, 111, E1176–E1181. [Google Scholar] [CrossRef] [Green Version]
- Morgan, H.E.; Turnbull, W.B.; Webb, M.E. Challenges in the use of sortase and other peptide ligases for site-specific protein modification. Chem. Soc. Rev. 2022, 51, 4121–4145. [Google Scholar] [CrossRef]
- Chuprakov, S.; Ogunkoya, A.O.; Barfield, R.M.; Bauzon, M.; Hickle, C.; Kim, Y.C.; Yeo, D.; Zhang, F.; Rabuka, D.; Drake, P.M. Tandem-Cleavage Linkers Improve the in Vivo Stability and Tolerability of Antibody-Drug Conjugates. Bioconjug. Chem. 2021, 32, 746–754. [Google Scholar] [CrossRef]
- Chen, I.; Dorr, B.M.; Liu, D.R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. USA 2011, 108, 11399–11404. [Google Scholar] [CrossRef] [Green Version]
- Pishesha, N.; Ingram, J.R.; Ploegh, H.L. Sortase A: A Model for Transpeptidation and Its Biological Applications. Annu. Rev. Cell. Dev. Biol. 2018, 34, 163–188. [Google Scholar] [CrossRef]
- Mao, H.; Hart, S.A.; Schink, A.; Pollok, B.A. Sortase-Mediated Protein Ligation: A New Method for Protein Engineering. J. Am. Chem. Soc. 2004, 126, 2670–2671. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Karas, M.; Rosu, F.; Gabelica, V. Influence of the Matrix on Analyte Fragmentation in Atmospheric Pressure MALDI. J. Am. Soc. Mass. Spectrom. 2006, 17, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynes, M.W.; Hinz, T.K.; Gao, D.; Martini, M.; Marek, L.A.; Ware, K.E.; Edwards, M.G.; Böhm, D.; Perner, S.; Helfrich, B.A.; et al. FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI sensitivity across all lung cancer histologies. Clin. Cancer Res. 2014, 20, 3299–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jendryczko, K.; Rzeszotko, J.; Krzyscik, M.A.; Szymczyk, J.; Otlewski, J.; Szlachcic, A. Peptibody Based on FGFR1-Binding Peptides From the FGF4 Sequence as a Cancer-Targeting Agent. Front. Pharmacol. 2021, 12, 2811. [Google Scholar] [CrossRef]
- Jendryczko, K.; Rzeszotko, J.; Krzyscik, M.A.; Kocyła, A.; Szymczyk, J.; Otlewski, J.; Szlachcic, A. Drug Conjugation via Maleimide-Thiol Chemistry Does Not Affect Targeting Properties of Cysteine-Containing Anti-FGFR1 Peptibodies. Mol. Pharm. 2022, 19, 1422–1433. [Google Scholar] [CrossRef]
- Dutt, A.; Ramos, A.H.; Hammerman, P.S.; Mermel, C.; Cho, J.; Sharifnia, T.; Chande, A.; Tanaka, K.E.; Stransky, N.; Greulich, H.; et al. Inhibitor-sensitive fgfr1 amplification in human non-small cell lung cancer. PLoS ONE 2011, 6, e20351. [Google Scholar] [CrossRef] [Green Version]
- Pahl, A.; Lutz, C.; Hechler, T. Amanitins and their development as a payload for antibody-drug conjugates. Drug. Discov. Today Technol. 2018, 30, 85–89. [Google Scholar] [CrossRef]
- Lutz, C.; Simon, W.; Werner-Simon, S.; Pahl, A.; Müller, C. Total Synthesis of α- and β-Amanitin. Angew. Chemie-Int. Ed. 2020, 59, 11390–11393. [Google Scholar] [CrossRef]
- Chen, H.; Lin, Z.; Arnst, K.E.; Miller, D.D.; Li, W. Tubulin inhibitor-based antibody-drug conjugates for cancer therapy. Molecules 2017, 22, 1281. [Google Scholar] [CrossRef] [Green Version]
- Mckertish, C.M.; Kayser, V. Advances and limitations of antibody drug conjugates for cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef]
- Li, N.; Hill, K.S.; Elferink, L.A. Analysis of Receptor Tyrosine Kinase Internalization Using Flow Cytometry. Membr. Traffick. 2008, 21, 305–317. [Google Scholar]
- Krzyscik, M.A.; Zakrzewska, M.; Sørensen, V.; Øy, G.F.; Brunheim, S.; Haugsten, E.M.; Mælandsmo, G.M.; Wiedlocha, A.; Otlewski, J. Fibroblast Growth Factor 2 Conjugated with Monomethyl Auristatin e Inhibits Tumor Growth in a Mouse Model. Biomacromolecules 2021, 22, 4169–4180. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preparation | Cell Line | ||||
|---|---|---|---|---|---|
| HCC95 | NCI-H520 | NCI-H1581 | JIMT-1 | G292 | |
| EC50 [nM] | |||||
| Non-conjugated αAMTN | 1238 ** | n.t. a | 998.6 ** | 1278 ** | 967.1 ** |
| Non-conjugated MMAE | 0.3398 ** | 0.2761 ** | 0.2288 ** | 0.09179 * | 0.09103 ** |
| αAMTN-FGF2V1 | 487.7 *** | 205.5 *** | 233.2 ** | 112.5 ** | 225.6 ** |
| FGF2V2-MMAE | 91.53 ** | 82.67 *** | 22.60 ** | 12.83 * | 10.77 * |
| αAMTN-FGF2V1 + FGF2V2-MMAE | 52.29 | 73.46 *** | 23.28 *** | 16.40 ** | 14.45 ** |
| αAMTN-FGF2V1V2-MMAE | 32.45 | 6.803 | 2.195 | 1.304 | 2.510 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nawrocka, D.; Krzyscik, M.A.; Sluzalska, K.D.; Otlewski, J. Dual-Warhead Conjugate Based on Fibroblast Growth Factor 2 Dimer Loaded with α-Amanitin and Monomethyl Auristatin E Exhibits Superior Cytotoxicity towards Cancer Cells Overproducing Fibroblast Growth Factor Receptor 1. Int. J. Mol. Sci. 2023, 24, 10143. https://doi.org/10.3390/ijms241210143
Nawrocka D, Krzyscik MA, Sluzalska KD, Otlewski J. Dual-Warhead Conjugate Based on Fibroblast Growth Factor 2 Dimer Loaded with α-Amanitin and Monomethyl Auristatin E Exhibits Superior Cytotoxicity towards Cancer Cells Overproducing Fibroblast Growth Factor Receptor 1. International Journal of Molecular Sciences. 2023; 24(12):10143. https://doi.org/10.3390/ijms241210143
Chicago/Turabian StyleNawrocka, Daria, Mateusz Adam Krzyscik, Katarzyna Dominika Sluzalska, and Jacek Otlewski. 2023. "Dual-Warhead Conjugate Based on Fibroblast Growth Factor 2 Dimer Loaded with α-Amanitin and Monomethyl Auristatin E Exhibits Superior Cytotoxicity towards Cancer Cells Overproducing Fibroblast Growth Factor Receptor 1" International Journal of Molecular Sciences 24, no. 12: 10143. https://doi.org/10.3390/ijms241210143