
Regulatory Cues in Pulmonary Fibrosis—With Emphasis on the AIM2 Inflammasome

Abstract
:1. Introduction
2. Pulmonary Fibrosis
2.1. Fibroblast Activation
2.2. Epithelial-Mesenchymal Transition (EMT)
2.3. Epithelial Regeneration Impairment
2.4. Imbalanced Senescence
2.5. Immune-Cell Activation Deficiency
2.6. Inflammation and Oxidative Stress
3. Inflammasome
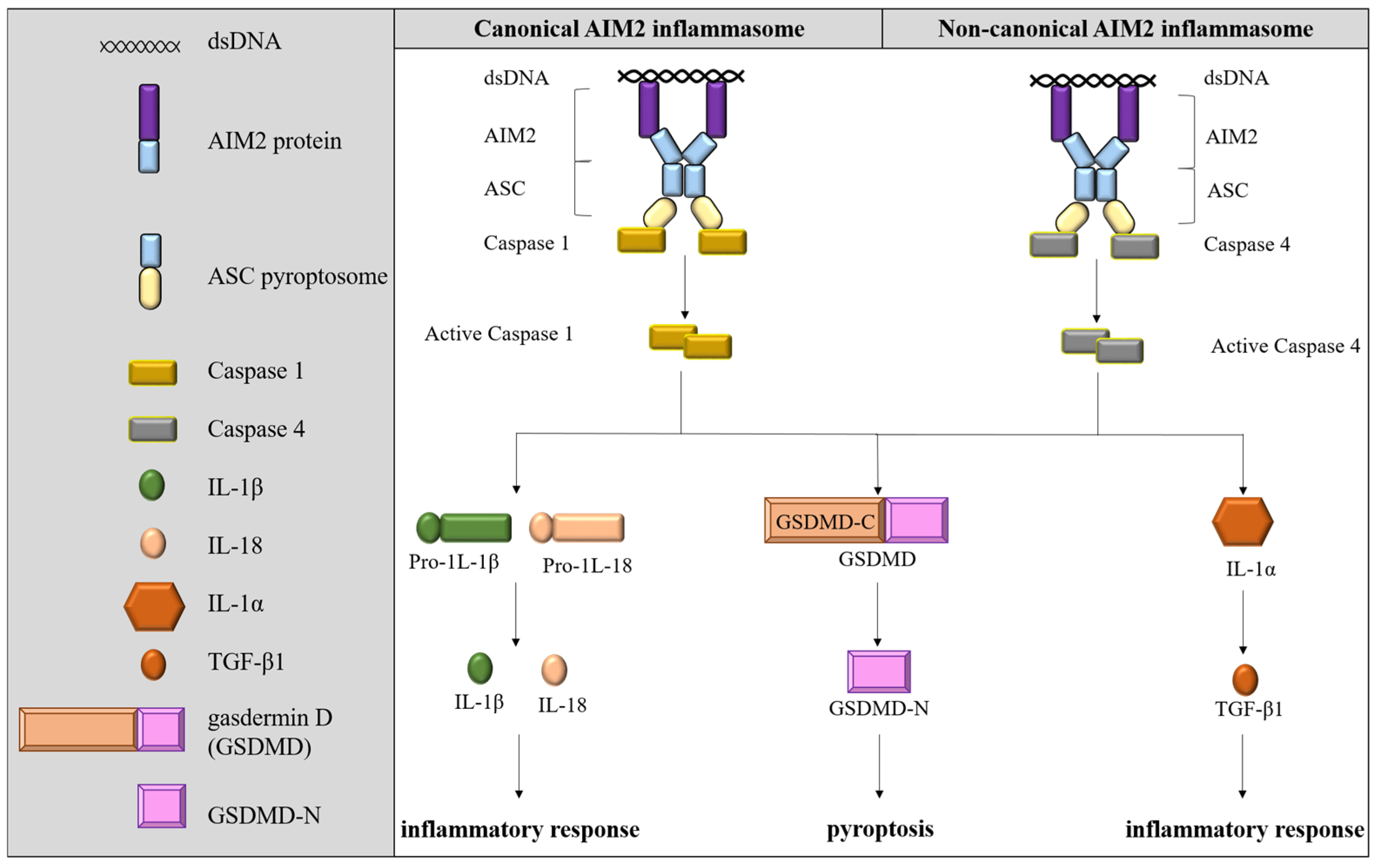
3.1. AIM2 Inflammasome
3.2. Role of the AIM2 Inflammasome in Pulmonary Fibrosis
3.3. Discovery of AIM2 Inflammasome Inhibitors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mattoo, H.; Pillai, S. Idiopathic pulmonary fibrosis and systemic sclerosis: Pathogenic mechanisms and therapeutic interventions. Cell. Mol. Life Sci. CMLS 2021, 78, 5527–5542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Duan, X.J.; Li, L.R.; Chen, Y.P. lncRNA NEAT1 promotes hypoxia-induced inflammation and fibrosis of alveolar epithelial cells via targeting miR-29a/NFATc3 axis. Kaohsiung J. Med. Sci. 2022, 38, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef] [Green Version]
- Inui, N.; Sakai, S.; Kitagawa, M. Molecular Pathogenesis of Pulmonary Fibrosis, with Focus on Pathways Related to TGF-β and the Ubiquitin-Proteasome Pathway. Int. J. Mol. Sci. 2021, 22, 6107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Chen, S.J.; Zhou, S.C.; Wu, S.Z.; Wang, H. Inflammasomes and Fibrosis. Front. Immunol. 2021, 12, 643149. [Google Scholar] [CrossRef]
- Meyer, K.C. Pulmonary fibrosis, part I: Epidemiology, pathogenesis, and diagnosis. Expert Rev. Respir. Med. 2017, 11, 343–359. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Kim, D.S. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Encycl. Respir. Med. 2022, 3, 199–217. [Google Scholar] [CrossRef]
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [Green Version]
- Min, L.; Shu-Li, Z.; Feng, Y.; Han, H.; Shao-Jun, L.; Sheng-Xiong, T.; Jia-Yu, T.; Xiang-Zhi, F.; Dan, F. NecroX-5 ameliorates bleomycin-induced pulmonary fibrosis via inhibiting NLRP3-mediated epithelial-mesenchymal transition. Respir. Res. 2022, 23, 128. [Google Scholar] [CrossRef]
- Chen, I.C.; Liu, Y.C.; Wu, Y.H.; Lo, S.H.; Dai, Z.K.; Hsu, J.H.; Tseng, Y.H. Evaluation of Proteasome Inhibitors in the Treatment of Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 1543. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.T.; Xiao, F.; Li, N.; Shan, S.; Qi, M.; Wang, Z.Y.; Zhang, S.N.; Wei, W.; Sun, W.Y. Inflammasome as an Effective Platform for Fibrosis Therapy. J. Inflamm. Res. 2021, 14, 1575–1590. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gonzalez, F.; Faner, R.; Rojas, M.; Agustí, A.; Serrano, M.; Sellarés, J. Cellular Senescence in Lung Fibrosis. Int. J. Mol. Sci. 2021, 22, 7012. [Google Scholar] [CrossRef] [PubMed]
- Bahri, S.; Mlika, M.; Nahdi, A.; Ben Ali, R.; Jameleddine, S. Thymus Vulgaris Inhibit Lung Fibrosis Progression and Oxidative Stress Induced by Bleomycin in Wistar Rats. Nutr. Cancer 2022, 74, 1420–1430. [Google Scholar] [CrossRef]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Tian, X.; Jia, Y.; Yang, M.; Li, Y.; Fernig, D.G. Functions of exogenous FGF signals in regulation of fibroblast to myofibroblast differentiation and extracellular matrix protein expression. Open Biol. 2022, 12, 210356. [Google Scholar] [CrossRef]
- Kis, K.; Liu, X.; Hagood, J.S. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev. Mol. Med. 2011, 13, e27. [Google Scholar] [CrossRef]
- Lasky, J.A.; Brody, A.R. Interstitial fibrosis and growth factors. Environ. Health Perspect. 2000, 108 (Suppl. S4), 751–762. [Google Scholar] [CrossRef] [Green Version]
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587. [Google Scholar] [CrossRef]
- Moore, M.W.; Herzog, E.L. Regulation and Relevance of Myofibroblast Responses in Idiopathic Pulmonary Fibrosis. Curr. Pathobiol. Rep. 2013, 1, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2018, 27, 180033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Wen, L.; Shi, Q.F.; Gao, F.; Huang, B.; Meng, J.; Hu, C.P.; Wang, C.M. Scutellarin ameliorates pulmonary fibrosis through inhibiting NF-κB/NLRP3-mediated epithelial-mesenchymal transition and inflammation. Cell Death Dis. 2020, 11, 978. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. CMLS 2021, 78, 2031–2057. [Google Scholar] [CrossRef]
- Zoz, D.F.; Lawson, W.E.; Blackwell, T.S. Idiopathic pulmonary fibrosis: A disorder of epithelial cell dysfunction. Am. J. Med. Sci. 2011, 341, 435–438. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Ward, C.; Eapen, M.S.; Myers, S.; Hallgren, O.; Levine, H.; Sohal, S.S. Epithelial-mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2018, 247, 346–358. [Google Scholar] [CrossRef] [Green Version]
- Pain, M.; Bermudez, O.; Lacoste, P.; Royer, P.J.; Botturi, K.; Tissot, A.; Brouard, S.; Eickelberg, O.; Magnan, A. Tissue remodelling in chronic bronchial diseases: From the epithelial to mesenchymal phenotype. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2014, 23, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Salton, F.; Volpe, M.C.; Confalonieri, M. Epithelial⁻Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina 2019, 55, 83. [Google Scholar] [CrossRef] [Green Version]
- Chuliá-Peris, L.; Carreres-Rey, C.; Gabasa, M.; Alcaraz, J.; Carretero, J.; Pereda, J. Matrix Metalloproteinases and Their Inhibitors in Pulmonary Fibrosis: EMMPRIN/CD147 Comes into Play. Int. J. Mol. Sci. 2022, 23, 6894. [Google Scholar] [CrossRef]
- Kim, K.H.; Burkhart, K.; Chen, P.; Frevert, C.W.; Randolph-Habecker, J.; Hackman, R.C.; Soloway, P.D.; Madtes, D.K. Tissue inhibitor of metalloproteinase-1 deficiency amplifies acute lung injury in bleomycin-exposed mice. Am. J. Respir. Cell Mol. Biol. 2005, 33, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.E.; Huizar, I.; Bench, E.M.; Sussman, S.W.; Wang, Y.; Khokha, R.; Parks, W.C. Tissue inhibitor of metalloproteinases 3 regulates resolution of inflammation following acute lung injury. Am. J. Pathol. 2010, 176, 64–73. [Google Scholar] [CrossRef] [PubMed]
- García-Alvarez, J.; Ramirez, R.; Checa, M.; Nuttall, R.K.; Sampieri, C.L.; Edwards, D.R.; Selman, M.; Pardo, A. Tissue inhibitor of metalloproteinase-3 is up-regulated by transforming growth factor-beta1 in vitro and expressed in fibroblastic foci in vivo in idiopathic pulmonary fibrosis. Exp. Lung Res. 2006, 32, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Ruiz, V.; Cabrera, S.; Segura, L.; Ramírez, R.; Barrios, R.; Pardo, A. TIMP-1, -2, -3, and -4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lung microenvironment? Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L562-74. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.; Fu, M.; Wang, M.; Wei, Y.; Wei, X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 2022, 21, 104. [Google Scholar] [CrossRef]
- Nam, M.W.; Kim, C.W.; Choi, K.C. Epithelial-Mesenchymal Transition-Inducing Factors Involved in the Progression of Lung Cancers. Biomol. Ther. 2022, 30, 213–220. [Google Scholar] [CrossRef]
- Wang, L.; Li, S.; Yao, Y.; Yin, W.; Ye, T. The role of natural products in the prevention and treatment of pulmonary fibrosis: A review. Food Funct. 2021, 12, 990–1007. [Google Scholar] [CrossRef]
- Willis, B.C.; Borok, Z. TGF-beta-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhu, X.; Hua, Y.; Zhao, Q.; Wang, K.; Zhen, L.; Wang, G.; Lü, J.; Luo, A.; Cho, W.C.; et al. YY1 mediates TGF-β1-induced EMT and pro-fibrogenesis in alveolar epithelial cells. Respir. Res. 2019, 20, 249. [Google Scholar] [CrossRef]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Fu, X.; Chen, X.; Han, X.; Dong, P. M2 macrophages induce EMT through the TGF-β/Smad2 signaling pathway. Cell Biol. Int. 2017, 41, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Fang, Y.; Jiang, F.; Li, L.; Zhou, H.; Xu, X.; Ning, W. Neohesperidin inhibits TGF-β1/Smad3 signaling and alleviates bleomycin-induced pulmonary fibrosis in mice. Eur. J. Pharmacol. 2019, 864, 172712. [Google Scholar] [CrossRef]
- Lv, Q.; Wang, J.; Xu, C.; Huang, X.; Ruan, Z.; Dai, Y. Pirfenidone alleviates pulmonary fibrosis in vitro and in vivo through regulating Wnt/GSK-3β/β-catenin and TGF-β1/Smad2/3 signaling pathways. Mol. Med. (Camb. Mass.) 2020, 26, 49. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C.; Mercer, P.F. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S1), S16–S20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.K.; Wu, T.J.; Hong, J.H.; Chen, F.H.; Yu, J.; Wang, C.C. Radiation Induces Pulmonary Fibrosis by Promoting the Fibrogenic Differentiation of Alveolar Stem Cells. Stem Cells Int. 2020, 2020, 6312053. [Google Scholar] [CrossRef] [PubMed]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Hadjicharalambous, M.R.; Lindsay, M.A. Idiopathic Pulmonary Fibrosis: Pathogenesis and the Emerging Role of Long Non-Coding RNAs. Int. J. Mol. Sci. 2020, 21, 524. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Luo, Z.; Zhou, Y. Regeneration-Associated Transitional State Cells in Pulmonary Fibrosis. Int. J. Mol. Sci. 2022, 23, 6757. [Google Scholar] [CrossRef]
- Huang, Y.; Zheng, Y.; Yin, J.; Lu, T.; Li, M.; Liang, J.; Hu, Z.; Bi, G.; Zhan, C.; Xue, L.; et al. Reconstructing the Developmental Trajectories of Multiple Subtypes in Pulmonary Parenchymal Epithelial Cells by Single-Cell RNA-seq. Front. Genet. 2020, 11, 573429. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.N.; Tang, X.X. New Perspectives on the Aberrant Alveolar Repair of Idiopathic Pulmonary Fibrosis. Front. Cell Dev. Biol. 2020, 8, 580026. [Google Scholar] [CrossRef] [PubMed]
- Venosa, A. Senescence in Pulmonary Fibrosis: Between Aging and Exposure. Front. Med. 2020, 7, 606462. [Google Scholar] [CrossRef] [PubMed]
- Ozsvari, B.; Nuttall, J.R.; Sotgia, F.; Lisanti, M.P. Azithromycin and Roxithromycin define a new family of “senolytic” drugs that target senescent human fibroblasts. Aging 2018, 10, 3294–3307. [Google Scholar] [CrossRef]
- Parimon, T.; Hohmann, M.S.; Yao, C. Cellular Senescence: Pathogenic Mechanisms in Lung Fibrosis. Int. J. Mol. Sci. 2021, 22, 6214. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [Green Version]
- Blokland, K.E.C.; Waters, D.W.; Schuliga, M.; Read, J.; Pouwels, S.D.; Grainge, C.L.; Jaffar, J.; Westall, G.; Mutsaers, S.E.; Prêle, C.M.; et al. Senescence of IPF Lung Fibroblasts Disrupt Alveolar Epithelial Cell Proliferation and Promote Migration in Wound Healing. Pharmaceutics 2020, 12, 389. [Google Scholar] [CrossRef]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [Green Version]
- Shenderov, K.; Collins, S.L.; Powell, J.D.; Horton, M.R. Immune dysregulation as a driver of idiopathic pulmonary fibrosis. J. Clin. Investig. 2021, 131, e143226. [Google Scholar] [CrossRef]
- Panda, A.; Arjona, A.; Sapey, E.; Bai, F.; Fikrig, E.; Montgomery, R.R.; Lord, J.M.; Shaw, A.C. Human innate immunosenescence: Causes and consequences for immunity in old age. Trends Immunol. 2009, 30, 325–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanouchi, H.; Ohtsuki, Y.; Fujita, J.; Bandoh, S.; Yoshinouchi, T.; Ishida, T. The distribution and number of Leu-7 (CD57) positive cells in lung tissue from patients with pulmonary fibrosis. Acta Med. Okayama 2002, 56, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, D.; Wang, L.; Wang, S.; Roden, A.C.; Zhao, H.; Li, X.; Prakash, Y.S.; Matteson, E.L.; Tschumperlin, D.J.; et al. Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL-17RA in human fibrotic interstitial lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L487–L497. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Kishore, A.; Petrek, M. Roles of Macrophage Polarization and Macrophage-Derived miRNAs in Pulmonary Fibrosis. Front. Immunol. 2021, 12, 678457. [Google Scholar] [CrossRef]
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436. [Google Scholar] [CrossRef]
- Kolahian, S.; Fernandez, I.E.; Eickelberg, O.; Hartl, D. Immune Mechanisms in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef] [Green Version]
- Lis-López, L.; Bauset, C.; Seco-Cervera, M.; Cosín-Roger, J. Is the Macrophage Phenotype Determinant for Fibrosis Development? Biomedicines 2021, 9, 1747. [Google Scholar] [CrossRef]
- Jegal, Y. The role of neutrophils in the pathogenesis of IPF. Korean J. Intern. Med. 2022, 37, 945–946. [Google Scholar] [CrossRef]
- Ding, L.; Yang, J.; Zhang, C.; Zhang, X.; Gao, P. Neutrophils Modulate Fibrogenesis in Chronic Pulmonary Diseases. Front. Med. 2021, 8, 616200. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.; Kim, J.; Ko, Y.G.; Kim, H.Y. The Dynamic Contribution of Neutrophils in the Chronic Respiratory Diseases. Allergy Asthma Immunol. Res. 2022, 14, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Achaiah, A.; Rathnapala, A.; Pereira, A.; Bothwell, H.; Dwivedi, K.; Barker, R.; Iotchkova, V.; Benamore, R.; Hoyles, R.K.; Ho, L.P. Neutrophil lymphocyte ratio as an indicator for disease progression in Idiopathic Pulmonary Fibrosis. BMJ Open Respir. Res. 2022, 9, e001202. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, G.; Liu, A.; Herzog, E.L. Evolving Perspectives on Innate Immune Mechanisms of IPF. Front. Mol. Biosci. 2021, 8, 676569. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.D.; Kliment, C.R.; Metz, H.E.; Kim, K.H.; Kargl, J.; Agostini, B.A.; Crum, L.T.; Oczypok, E.A.; Oury, T.A.; Houghton, A.M. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J. Leukoc. Biol. 2015, 98, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Takemasa, A.; Ishii, Y.; Fukuda, T. A neutrophil elastase inhibitor prevents bleomycin-induced pulmonary fibrosis in mice. Eur. Respir. J. 2012, 40, 1475–1482. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.H.; Li, M.; Ma, H.F.; Li, W.; Liu, L.; Yin, Y.; Zhou, X.M.; Hou, G. Protective effects of GHK-Cu in bleomycin-induced pulmonary fibrosis via anti-oxidative stress and anti-inflammation pathways. Life Sci. 2020, 241, 117139. [Google Scholar] [CrossRef]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef] [Green Version]
- Trachalaki, A.; Tsitoura, E.; Mastrodimou, S.; Invernizzi, R.; Vasarmidi, E.; Bibaki, E.; Tzanakis, N.; Molyneaux, P.L.; Maher, T.M.; Antoniou, K. Enhanced IL-1β Release Following NLRP3 and AIM2 Inflammasome Stimulation Is Linked to mtROS in Airway Macrophages in Pulmonary Fibrosis. Front. Immunol. 2021, 12, 661811. [Google Scholar] [CrossRef]
- Zuo, L.; Wijegunawardana, D. Redox Role of ROS and Inflammation in Pulmonary Diseases. Adv. Exp. Med. Biol. 2021, 1304, 187–204. [Google Scholar] [CrossRef]
- Barnes, J.L.; Gorin, Y. Myofibroblast differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney Int. 2011, 79, 944–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Liu, G.S.; Dusting, G.J.; Chan, E.C. NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox Biol. 2014, 2, 267–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnula, V.L.; Fattman, C.L.; Tan, R.J.; Oury, T.D. Oxidative stress in pulmonary fibrosis: A possible role for redox modulatory therapy. Am. J. Respir. Crit. Care Med. 2005, 172, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terlizzi, M.; Molino, A.; Colarusso, C.; Donovan, C.; Imitazione, P.; Somma, P.; Aquino, R.P.; Hansbro, P.M.; Pinto, A.; Sorrentino, R. Activation of the Absent in Melanoma 2 Inflammasome in Peripheral Blood Mononuclear Cells From Idiopathic Pulmonary Fibrosis Patients Leads to the Release of Pro-Fibrotic Mediators. Front. Immunol. 2018, 9, 670. [Google Scholar] [CrossRef] [Green Version]
- Lasithiotaki, I.; Giannarakis, I.; Tsitoura, E.; Samara, K.D.; Margaritopoulos, G.A.; Choulaki, C.; Vasarmidi, E.; Tzanakis, N.; Voloudaki, A.; Sidiropoulos, P.; et al. NLRP3 inflammasome expression in idiopathic pulmonary fibrosis and rheumatoid lung. Eur. Respir. J. 2016, 47, 910–918. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.J.; Lee, M.; Stout-Delgado, H.W.; Moon, J.S. DROSHA-Dependent miRNA and AIM2 Inflammasome Activation in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 1668. [Google Scholar] [CrossRef] [Green Version]
- Platnich, J.M.; Muruve, D.A. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch. Biochem. Biophys. 2019, 670, 4–14. [Google Scholar] [CrossRef]
- Lang, T.; Lee, J.P.W.; Elgass, K.; Pinar, A.A.; Tate, M.D.; Aitken, E.H.; Fan, H.; Creed, S.J.; Deen, N.S.; Traore, D.A.K.; et al. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat. Commun. 2018, 9, 2223. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Loughran, P.; Shapiro, R.; Shrivastava, I.H.; Antoine, D.J.; Li, T.; Yan, Z.; Fan, J.; Billiar, T.R.; Scott, M.J. Redox-dependent regulation of hepatocyte absent in melanoma 2 inflammasome activation in sterile liver injury in mice. Hepatology 2017, 65, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Planès, R.; Pinilla, M.; Santoni, K.; Hessel, A.; Passemar, C.; Lay, K.; Paillette, P.; Valadão, A.C.; Robinson, K.S.; Bastard, P.; et al. Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol. Cell 2022, 82, 2385–2400. [Google Scholar] [CrossRef]
- Sharma, M.; de Alba, E. Structure, Activation and Regulation of NLRP3 and AIM2 Inflammasomes. Int. J. Mol. Sci. 2021, 22, 872. [Google Scholar] [CrossRef]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Asp. Med. 2020, 76, 100924. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colunga Biancatelli, R.M.L.; Solopov, P.A.; Catravas, J.D. The Inflammasome NLR Family Pyrin Domain-Containing Protein 3 (NLRP3) as a Novel Therapeutic Target for Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2022, 192, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Hosseinian, N.; Cho, Y.; Lockey, R.F.; Kolliputi, N. The role of the NLRP3 inflammasome in pulmonary diseases. Ther. Adv. Respir. Dis. 2015, 9, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, C.; Swartzwelter, B.; Koenders, M.I.; Azam, T.; Tengesdal, I.W.; Powers, N.; de Graaf, D.M.; Dinarello, C.A.; Joosten, L.A.B. NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Res. Ther. 2018, 20, 169. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Bhattacharya, M.; Roy, S.; Tian, Y.; Yin, Q. Immunobiology and structural biology of AIM2 inflammasome. Mol. Asp. Med. 2020, 76, 100869. [Google Scholar] [CrossRef]
- Ruwanpura, S.M.; McLeod, L.; Dousha, L.F.; Seow, H.J.; West, A.C.; West, A.J.; Weng, T.; Alanazi, M.; MacDonald, M.; King, P.T.; et al. Cross-talk between IL-6 trans-signaling and AIM2 inflammasome/IL-1β axes bridge innate immunity and epithelial apoptosis to promote emphysema. Proc. Natl. Acad. Sci. USA 2022, 119, e2201494119. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016, 46, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Yin, Q. AIM2 inflammasome activation and regulation: A structural perspective. J. Struct. Biol. 2017, 200, 279–282. [Google Scholar] [CrossRef]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef] [Green Version]
- Kumari, P.; Russo, A.J.; Shivcharan, S.; Rathinam, V.A. AIM2 in health and disease: Inflammasome and beyond. Immunol. Rev. 2020, 297, 83–95. [Google Scholar] [CrossRef]
- Sharma, B.R.; Karki, R.; Kanneganti, T.D. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019, 49, 1998–2011. [Google Scholar] [CrossRef] [Green Version]
- Komada, T.; Chung, H.; Lau, A.; Platnich, J.M.; Beck, P.L.; Benediktsson, H.; Duff, H.J.; Jenne, C.N.; Muruve, D.A. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J. Am. Soc. Nephrol. JASN 2018, 29, 1165–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viganò, E.; Mortellaro, A. Caspase-11: The driving factor for noncanonical inflammasomes. Eur. J. Immunol. 2013, 43, 2240–2245. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, C.; Li, H.B.; Tong, J.; Ouyang, X.; Cetinbas, N.M.; Zhu, S.; Strowig, T.; Lam, F.C.; Zhao, C.; et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 2016, 354, 765–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Peng, S.; Shan, X.; Deng, G.; Shen, L.; Sun, J.; Jiang, C.; Yang, X.; Chang, Z.; Sun, X.; et al. Inhibition of AIM2 inflammasome-mediated pyroptosis by Andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis. Cell Death Dis. 2019, 10, 957. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.J.; Hong, K.S.; Jeong, J.H.; Lee, M.; Choi, A.M.K.; Stout-Delgado, H.W.; Moon, J.S. DROSHA-Dependent AIM2 Inflammasome Activation Contributes to Lung Inflammation during Idiopathic Pulmonary Fibrosis. Cells 2019, 8, 938. [Google Scholar] [CrossRef] [Green Version]
- Colarusso, C.; Terlizzi, M.; Molino, A.; Imitazione, P.; Somma, P.; Rega, R.; Saccomanno, A.; Aquino, R.P.; Pinto, A.; Sorrentino, R. AIM2 Inflammasome Activation Leads to IL-1α and TGF-β Release From Exacerbated Chronic Obstructive Pulmonary Disease-Derived Peripheral Blood Mononuclear Cells. Front. Pharmacol. 2019, 10, 257. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, R.; Banerjee, S.; Amin, S.A.; Adhikari, N.; Jha, T. Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: A review. Eur. J. Med. Chem. 2020, 192, 112171. [Google Scholar] [CrossRef]
- Zheng, Q.; Lei, Y.; Hui, S.; Tong, M.; Liang, L. HDAC3 promotes pulmonary fibrosis by activating NOTCH1 and STAT1 signaling and up-regulating inflammasome components AIM2 and ASC. Cytokine 2022, 153, 155842. [Google Scholar] [CrossRef]
- Finnerty, J.P.; Ponnuswamy, A.; Dutta, P.; Abdelaziz, A.; Kamil, H. Efficacy of antifibrotic drugs, nintedanib and pirfenidone, in treatment of progressive pulmonary fibrosis in both idiopathic pulmonary fibrosis (IPF) and non-IPF: A systematic review and meta-analysis. BMC Pulm. Med. 2021, 21, 411. [Google Scholar] [CrossRef]
- Yang, C.; Song, C.; Wang, Y.; Zhou, W.; Zheng, W.; Zhou, H.; Deng, G.; Li, H.; Xiao, W.; Yang, Z.; et al. Re-Du-Ning injection ameliorates radiation-induced pneumonitis and fibrosis by inhibiting AIM2 inflammasome and epithelial-mesenchymal transition. Phytomed. Int. J. Phytother. Phytopharm. 2022, 102, 154184. [Google Scholar] [CrossRef]
- Wu, C.F.; Bi, X.L.; Yang, J.Y.; Zhan, J.Y.; Dong, Y.X.; Wang, J.H.; Wang, J.M.; Zhang, R.; Li, X. Differential effects of ginsenosides on NO and TNF-alpha production by LPS-activated N9 microglia. Int. Immunopharmacol. 2007, 7, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.; Han, B.C.; Lee, S.H.; Lee, G.S. Fructose-arginine, a non-saponin molecule of Korean Red Ginseng, attenuates AIM2 inflammasome activation. J. Ginseng Res. 2020, 44, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Li, C.; Qian, F.; Zhao, Y.; Shi, X.; Hong, D.; Ai, Q.; Zhong, L. Ginsenoside Rg1 Inhibits Microglia Pyroptosis Induced by Lipopolysaccharide Through Regulating STAT3 Signaling. J. Inflamm. Res. 2021, 14, 6619–6632. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Nakahata, S.; Kondo, Y.; Izumi, A.; Yamamoto, K.; Ichikawa, T.; Tamura, T.; Noumi, K.; Yamashita, Y.; Morishita, K. Overexpression of absent in melanoma 2 in oral squamous cell carcinoma contributes to tumor progression. Biochem. Biophys. Res. Commun. 2019, 509, 82–88. [Google Scholar] [CrossRef]
- Zhang, M.; Jin, C.; Yang, Y.; Wang, K.; Zhou, Y.; Zhou, Y.; Wang, R.; Li, T.; Hu, R. AIM2 promotes non-small-cell lung cancer cell growth through inflammasome-dependent pathway. J. Cell. Physiol. 2019, 234, 20161–20173. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhang, M.; Ying, Q.; Xie, X.; Yue, S.; Tong, B.; Wei, Q.; Bai, Z.; Ma, L. Decrease of AIM2 mediated by luteolin contributes to non-small cell lung cancer treatment. Cell Death Dis. 2019, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug | Pulmonary Fibrosis Model | Effect | Ref |
|---|---|---|---|
| RDN | (1) RILI in Female C57BL/6 mice |
| [120] |
| ANDRO | (1) RILI in Female C57BL/6 mice (1) 8 Gy of X-ray radiation-stimulated BMDMs |
| [114] |
| RGFP966 | (1) A bleomycin-induced pulmonary fibrosis mouse model (2) TGF-β1-challenged MRC-5 cells |
| [118] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, Y.-H.; Chen, I.-C.; Li, W.-C.; Hsu, J.-H. Regulatory Cues in Pulmonary Fibrosis—With Emphasis on the AIM2 Inflammasome. Int. J. Mol. Sci. 2023, 24, 10876. https://doi.org/10.3390/ijms241310876
Tseng Y-H, Chen I-C, Li W-C, Hsu J-H. Regulatory Cues in Pulmonary Fibrosis—With Emphasis on the AIM2 Inflammasome. International Journal of Molecular Sciences. 2023; 24(13):10876. https://doi.org/10.3390/ijms241310876
Chicago/Turabian StyleTseng, Yu-Hsin, I-Chen Chen, Wan-Chun Li, and Jong-Hau Hsu. 2023. "Regulatory Cues in Pulmonary Fibrosis—With Emphasis on the AIM2 Inflammasome" International Journal of Molecular Sciences 24, no. 13: 10876. https://doi.org/10.3390/ijms241310876