
Structurally and Morphologically Distinct Pathological Tau Assemblies Differentially Affect GVB Accumulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
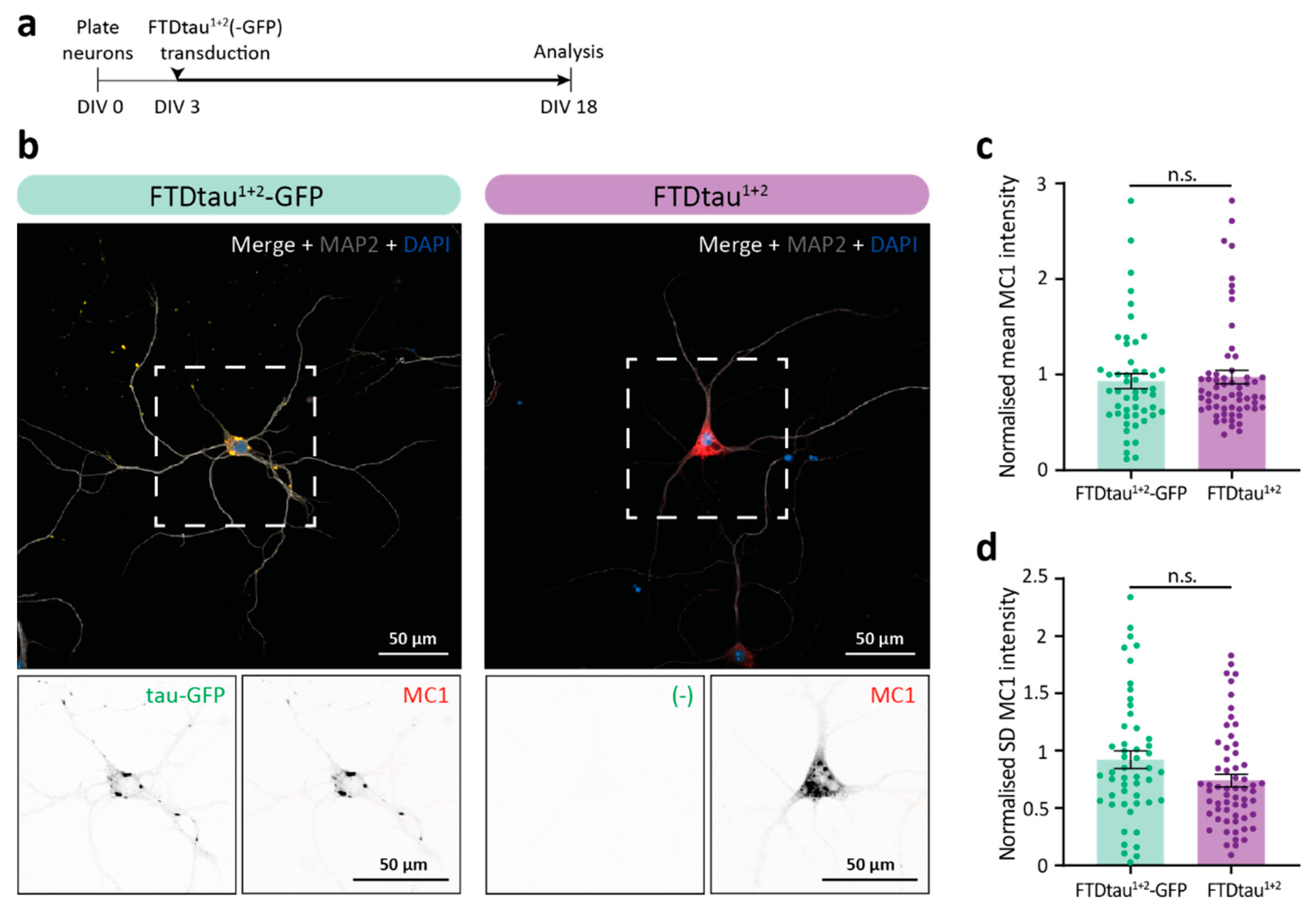
2.1. Pathological FTDtau1+2-GFP and FTDtau1+2 Assemblies Have Different Morphology and Subcellular Localization
2.2. FTDtau1+2-GFP and FTDtau1+2 Display Similar Progression of Pathological Tau Accumulation
2.3. FTDtau1+2-GFP and FTDtau1+2 Result in Different Filament Ultrastructure in Neurons
2.4. FTDtau1+2 Is Associated with a Higher GVB Load Than FTDtau1+2-GFP
3. Discussion
4. Materials and Methods
4.1. Primary Mouse Neuron Culture
4.2. Tau Pathology Induction
4.3. Fixation of Primary Neurons
4.4. Immunolabeling of Primary Neurons
4.5. Confocal Microscopy and Image Analysis
4.6. High-Content Microscopy and Automated Analysis
4.7. Transmission Electron Microscopy (TEM) Analysis
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Spillantini, M.G.; Goedert, M. Tau Pathology and Neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Dubey, H.; Gulati, K.; Ray, A. Recent Studies on Cellular and Molecular Mechanisms in Alzheimer’s Disease: Focus on Epigenetic Factors and Histone Deacetylase. Rev. Neurosci. 2018, 29, 241–260. [Google Scholar] [CrossRef]
- Carney, R.M.; Pericak-Vance, M.A.; Strang, K.H.; Golde, T.E.; Josephs, K.A. MAPT Mutations, Tauopathy, and Mechanisms of Neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Jack, C.R.; Wiste, H.J.; Schwarz, C.G.; Lowe, V.J.; Senjem, M.L.; Vemuri, P.; Weigand, S.D.; Therneau, T.M.; Knopman, D.S.; Gunter, J.L.; et al. Longitudinal Tau PET in Ageing and Alzheimer’s Disease. Brain 2018, 141, 1517–1528. [Google Scholar] [CrossRef] [Green Version]
- Van Bokhoven, P.; De Wilde, A.; Vermunt, L.; Leferink, P.S.; Heetveld, S.; Cummings, J.; Scheltens, P.; Vijverberg, E.G.B. The Alzheimer’s Disease Drug Development Landscape. Alzheimers Res. Ther. 2021, 13, 186. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Sigurdsson, E.M. Tau-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The Different Autophagy Degradation Pathways and Neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef]
- Wolozin, B.; Ivanov, P. Stress Granules and Neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of Cellular Proteostasis in Aging and Disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Walter, P. The Integrated Stress Response: From Mechanism to Disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- Köhler, C. Granulovacuolar Degeneration: A Neurodegenerative Change That Accompanies Tau Pathology. Acta Neuropathol. 2016, 132, 339–359. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, V.I.; Hoozemans, J.J.M.; Scheper, W. Untangling the Origin and Function of Granulovacuolar Degeneration Bodies in Neurodegenerative Proteinopathies. Acta Neuropathol. Commun. 2020, 8, 153. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Van Haastert, E.S.; Nijholt, D.A.T.; Rozemuller, A.J.M.; Eikelenboom, P.; Scheper, W. The Unfolded Protein Response Is Activated in Pretangle Neurons in Alzheimer’s Disease Hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [Green Version]
- Nijholt, D.A.T.; Van Haastert, E.S.; Rozemuller, A.J.M.; Scheper, W.; Hoozemans, J.J.M. The Unfolded Protein Response Is Associated with Early Tau Pathology in the Hippocampus of Tauopathies. J. Pathol. 2012, 226, 693–702. [Google Scholar] [CrossRef]
- Ganz, A.B.; Beker, N.; Hulsman, M.; Sikkes, S.; Netherlands Brain Bank; Scheltens, P.; Smit, A.B.; Rozemuller, A.J.M.; Hoozemans, J.J.M.; Holstege, H. Neuropathology and Cognitive Performance in Self-Reported Cognitively Healthy Centenarians. Acta Neuropathol. Commun. 2018, 6, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiersma, V.I.; van Ziel, A.M.; Vazquez-Sanchez, S.; Nölle, A.; Berenjeno-Correa, E.; Bonaterra-Pastra, A.; Clavaguera, F.; Tolnay, M.; Musters, R.J.P.; van Weering, J.R.T.; et al. Granulovacuolar Degeneration Bodies Are Neuron-Selective Lysosomal Structures Induced by Intracellular Tau Pathology. Acta Neuropathol. 2019, 138, 943–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calafate, S.; Flavin, W.; Verstreken, P.; Moechars, D. Loss of Bin1 Promotes the Propagation of Tau Report Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep. 2016, 17, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.J.; Nathaniel, D.L.; Raghavan, P.; Nelson, M.; Tian, R.; Tse, E.; Hong, J.Y.; See, S.K.; Mok, S.A.; Hein, M.Y.; et al. Compromised Function of the ESCRT Pathway Promotes Endolysosomal Escape of Tau Seeds and Propagation of Tau Aggregation. J. Biol. Chem. 2019, 294, 18952–18966. [Google Scholar] [CrossRef] [Green Version]
- Flavin, W.P.; Bousset, L.; Green, Z.C.; Chu, Y.; Skarpathiotis, S.; Chaney, M.J.; Kordower, J.H.; Melki, R.; Campbell, E.M. Endocytic Vesicle Rupture Is a Conserved Mechanism of Cellular Invasion by Amyloid Proteins. Acta Neuropathol. 2017, 134, 629–653. [Google Scholar] [CrossRef]
- Jia, J.; Claude-Taupin, A.; Gu, Y.; Choi, S.W.; Peters, R.; Bissa, B.; Mudd, M.H.; Allers, L.; Pallikkuth, S.; Lidke, K.A.; et al. Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal. Dev. Cell 2020, 52, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, C.; Kirchner, P.; Bug, M.; Grum, D.; Koerver, L.; Schulze, N.; Poehler, R.; Dressler, A.; Fengler, S.; Arhzaouy, K.; et al. VCP/P97 Cooperates with YOD1, UBXD1 and PLAA to Drive Clearance of Ruptured Lysosomes by Autophagy. EMBO J. 2017, 36, 135–150. [Google Scholar] [CrossRef] [Green Version]
- Koller, E.J.; Gonzalez De La Cruz, E.; MacHula, T.; Ibanez, K.R.; Lin, W.L.; Williams, T.; Riffe, C.J.; Ryu, D.; Strang, K.H.; Liu, X.; et al. Combining P301L and S320F Tau Variants Produces a Novel Accelerated Model of Tauopathy. Hum. Mol. Genet. 2019, 28, 3255–3269. [Google Scholar] [CrossRef]
- Croft, C.L.; Cruz, P.E.; Ryu, D.H.; Ceballos-Diaz, C.; Strang, K.H.; Woody, B.M.; Lin, W.L.; Deture, M.; Rodríguez-Lebrón, E.; Dickson, D.W.; et al. RAAV-Based Brain Slice Culture Models of Alzheimer’s and Parkinson’s Disease Inclusion Pathologies. J. Exp. Med. 2019, 216, 539–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorge-Oliva, M.; Smits, J.F.M.; Wiersma, V.I.; Hoozemans, J.J.M.; Scheper, W. Granulovacuolar Degeneration Bodies Are Independently Induced by Tau and α-Synuclein Pathology. Alzheimer’s Res. Ther. 2022, 14, 187. [Google Scholar] [CrossRef] [PubMed]
- Kaniyappan, S.; Tepper, K.; Biernat, J.; Chandupatla, R.R.; Hübschmann, S.; Irsen, S.; Bicher, S.; Klatt, C.; Mandelkow, E.M.; Mandelkow, E. FRET-Based Tau Seeding Assay Does Not Represent Prion-like Templated Assembly of Tau Filaments. Mol. Neurodegener. 2020, 15, 39. [Google Scholar] [CrossRef]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Van Slegtenhorst, M.; Gwinn-Hardy, K.; Murphy, M.P.; Baker, M.; Yu, X.; et al. Neurofibrillary Tangles, Amyotrophy and Progressive Motor Disturbance in Mice Expressing Mutant (P301L) Tau Protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef]
- Lin, W.L.; Lewis, J.; Yen, S.H.; Hutton, M.; Dickson, D.W. Ultrastructural Neuronal Pathology in Transgenic Mice Expressing Mutant (P301L) Human Tau. J. Neurocytol. 2003, 32, 1091–1105. [Google Scholar] [CrossRef]
- Lin, W.L.; Lewis, J.; Yen, S.H.; Hutton, M.; Dickson, D.W. Filamentous Tau in Oligodendrocytes and Astrocytes of Transgenic Mice Expressing the Human Tau Isoform with the P301L Mutation. Am. J. Pathol. 2003, 162, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Terry, R.D.; Gonatas, N.K.; Weiss, M. Ultrastructural Studies in Alzheimer’S Presenile Dementia. Am. J. Pathol. 1964, 44, 269–297. [Google Scholar]
- Iqbal, K.; Wisniewski, H.M.; Grundke-Iqbal, I.; Korthals, J.K.; Terry, R.D. Chemical Pathology of Neurofibrils: Neurofibrillary Tangles of Alzheimer’s Presenile-Senile Dementia. J. Histochem. Cytochem. 1975, 23, 563–569. [Google Scholar] [CrossRef]
- Waxman, E.A.; Giasson, B.I. Induction of Intracellular Tau Aggregation Is Promoted by α-Synuclein Seeds and Provides Novel Insights into the Hyperphosphorylation of Tau. J. Neurosci. 2011, 31, 7604–7618. [Google Scholar] [CrossRef] [Green Version]
- Köhler, C.; Dinekov, M.; Götz, J. Granulovacuolar Degeneration and Unfolded Protein Response in Mouse Models of Tauopathy and Aβ Amyloidosis. Neurobiol. Dis. 2014, 71, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Stutzbach, L.D.; Xie, S.X.; Naj, A.C.; Albin, R.; Gilman, S.; Lee, V.M.Y.; Trojanowski, J.Q.; Devlin, B.; Schellenberg, G.D. The Unfolded Protein Response Is Activated in Disease-Affected Brain Regions in Progressive Supranuclear Palsy and Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, Y.; Matsubara, T.; Takahashi, T.; Kurashige, T.; Dohi, E.; Hiji, M.; Nagano, Y.; Yamawaki, T.; Matsumoto, M. Granulovacuolar Degenerations Appear in Relation to Hippocampal Phosphorylated Tau Accumulation in Various Neurodegenerative Disorders. PLoS ONE 2011, 6, e26996. [Google Scholar] [CrossRef]
- Nakamori, M.; Takahashi, T.; Yamazaki, Y.; Kurashige, T.; Yamawaki, T.; Matsumoto, M. Cyclin-Dependent Kinase 5 Immunoreactivity for Granulovacuolar Degeneration. Neuroreport 2012, 23, 867–872. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; Mackenzie, D.; Kamath, T.; Commins, C.; et al. Tau Protein Liquid–Liquid Phase Separation Can Initiate Tau Aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, W.; Yang, Y.; Murzin, A.G.; Falcon, B.; Kotecha, A.; Van Beers, M.; Tarutani, A.; Kametani, F.; Garringer, H.J.; et al. Structure-Based Classification of Tauopathies. Nature 2021, 598, 359–363. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Stadelmann, C. Problems of Cell Death in Neurodegeneration and Alzheimer’s Disease. J. Alzheimer’s Dis. 2001, 3, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadelmann, C.; Deckwerth, T.L.; Srinivasan, A.; Bancher, C.; Jellinger, K.; Lassmann, H.; Brück, W.; Jellinger, K.; Lassmann, H. Activation of Caspase-3 in Single Neurons and Autophagic Granules of Granulovacuolar Degeneration in Alzheimer’s Disease. Evidence for Apoptotic Cell Death. Am. J. Pathol. 1999, 155, 1459–1466. [Google Scholar] [CrossRef]
- Koper, M.J.; Van Schoor, E.; Ospitalieri, S.; Vandenberghe, R.; Vandenbulcke, M.; von Arnim, C.A.F.; Tousseyn, T.; Balusu, S.; De Strooper, B.; Thal, D.R. Necrosome Complex Detected in Granulovacuolar Degeneration Is Associated with Neuronal Loss in Alzheimer’s Disease. Acta Neuropathol. 2020, 139, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M.Y. Seeding of Normal Tau by Pathological Tau Conformers Drives Pathogenesis of Alzheimer-like Tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jorge-Oliva, M.; van Weering, J.R.T.; Scheper, W. Structurally and Morphologically Distinct Pathological Tau Assemblies Differentially Affect GVB Accumulation. Int. J. Mol. Sci. 2023, 24, 10865. https://doi.org/10.3390/ijms241310865
Jorge-Oliva M, van Weering JRT, Scheper W. Structurally and Morphologically Distinct Pathological Tau Assemblies Differentially Affect GVB Accumulation. International Journal of Molecular Sciences. 2023; 24(13):10865. https://doi.org/10.3390/ijms241310865
Chicago/Turabian StyleJorge-Oliva, Marta, Jan R. T. van Weering, and Wiep Scheper. 2023. "Structurally and Morphologically Distinct Pathological Tau Assemblies Differentially Affect GVB Accumulation" International Journal of Molecular Sciences 24, no. 13: 10865. https://doi.org/10.3390/ijms241310865