
Altered Tau Kinase Activity in rTg4510 Mice after a Single Interfaced CHIMERA Traumatic Brain Injury
 ,
,
Abstract
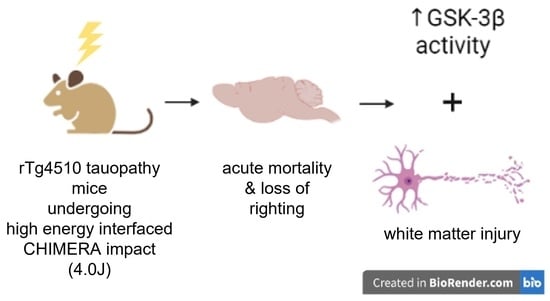
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. CHIMERA TBI with Interface at 4.0 J Induced High Mortality
2.2. TBI Induced Much Prolonged Loss of Righting Reflex and Chronic Axonal Injury and Microgliosis
2.3. TBI Induced Chronic Activation of GSK-3β
2.4. TBI Accelerated Increase in Plasma Total Tau in rTg4510 Mice
2.5. TBI Did Not Change Brain Tau Burden in rTg4510 Mice at 2-mo Post-Injury
2.6. TBI Did Not Change Levels of Autophagosomes and Lysosomes, Neurons, Astrocytes, Endothelial Cells, Synapses, or Size of Brain Regions
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Traumatic Brain Injury
4.3. Blood Collection and Euthanasia
4.4. Histology, Immunohistochemistry and Immunofluorescence
4.5. Image Quantification
4.6. Tissue Homogenization and Western Blot
4.7. Plasma Total Tau Analysis
4.8. Animal Genotyping
4.9. Quantitative Reverse Transcriptase-PCR (qRT-PCR)
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Omalu, B.I.; DeKosky, S.T.; Minster, R.L.; Kamboh, M.I.; Hamilton, R.L.; Wecht, C.H. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 2005, 57, 128–134; discussion 128–134. [Google Scholar] [CrossRef]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.; Budson, A.E.; Santini, V.E.; Lee, H.-S.; Kubilus, C.A.; Stern, R. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef]
- McKee, A.C.; Stein, T.; Nowinski, C.J.; Stern, R.; Daneshvar, D.; Alvarez, V.E.; Lee, H.-S.; Hall, G.; Wojtowicz, S.M.; Baugh, C.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef]
- Bieniek, K.F.; Cairns, N.J.; Crary, J.F.; Dickson, D.W.; Folkerth, R.D.; Keene, C.D.; Litvan, I.; Perl, D.P.; Stein, T.D.; Vonsattel, J.-P.; et al. The Second NINDS/NIBIB Consensus Meeting to Define Neuropathological Criteria for the Diagnosis of Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2021, 80, 210–219. [Google Scholar] [CrossRef]
- Smith, C.; Graham, D.I.; Murray, L.S.; Nicoll, J.A. Tau immunohistochemistry in acute brain injury. Neuropathol. Appl. Neurobiol. 2003, 29, 496–502. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012, 22, 142–149. [Google Scholar] [CrossRef]
- Kenney, K.; Iacono, D.; Edlow, B.L.; I Katz, D.; Diaz-Arrastia, R.; Dams-O’connor, K.; Daneshvar, D.; Stevens, A.; Moreau, A.; Tirrell, L.S.; et al. Dementia After Moderate-Severe Traumatic Brain Injury: Coexistence of Multiple Proteinopathies. J. Neuropathol. Exp. Neurol. 2018, 77, 50–63. [Google Scholar] [CrossRef]
- Tribett, T.; Erskine, B.; Bailey, K.; Brown, T.; Castellani, R.J. Chronic Traumatic Encephalopathy Pathology After Shotgun Injury to the Brain. J. Forensic Sci. 2019, 64, 1248–1252. [Google Scholar] [CrossRef]
- Gorgoraptis, N.; Li, L.M.; Whittington, A.; Zimmerman, K.A.; Maclean, L.M.; McLeod, C.; Ross, E.; Heslegrave, A.; Zetterberg, H.; Passchier, J.; et al. In vivo detection of cerebral tau pathology in long-term survivors of traumatic brain injury. Sci. Transl. Med. 2019, 11, eaaw1993. [Google Scholar] [CrossRef]
- Shively, S.B.; Edgerton, S.L.; Iacono, D.; Purohit, D.P.; Qu, B.-X.; Haroutunian, V.; Davis, K.L.; Diaz-Arrastia, R.; Perl, D.P. Localized cortical chronic traumatic encephalopathy pathology after single, severe axonal injury in human brain. Acta Neuropathol. 2017, 133, 353–366. [Google Scholar] [CrossRef]
- Okamura, Y.; Kawakami, I.; Watanabe, K.; Oshima, K.; Niizato, K.; Ikeda, K.; Akiyama, H.; Hasegawa, M. Tau progression in single severe frontal traumatic brain injury in human brains. J. Neurol. Sci. 2019, 407, 116495. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Uryu, K.; Higuchi, M.; Longhi, L.; Hoover, R.; Fujimoto, S.; McIntosh, T.; Lee, V.M.-Y.; Trojanowski, J.Q.; Petraglia, A.L.; et al. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. J. Neurotrauma 2005, 22, 1134–1141. [Google Scholar] [CrossRef]
- Mouzon, B.; Saltiel, N.; Ferguson, S.; Ojo, J.; Lungmus, C.; Lynch, C.; Algamal, M.; Morin, A.; Carper, B.; Bieler, G.; et al. Impact of age on acute post-TBI neuropathology in mice expressing humanized tau: A Chronic Effects of Neurotrauma Consortium Study. Brain Inj. 2018, 32, 1285–1294. [Google Scholar] [CrossRef]
- Gangolli, M.; Benetatos, J.; Esparza, T.J.; Fountain, E.M.; Seneviratne, S.; Brody, D.L. Repetitive Concussive and Subconcussive Injury in a Human Tau Mouse Model Results in Chronic Cognitive Dysfunction and Disruption of White Matter Tracts, But Not Tau Pathology. J. Neurotrauma 2019, 36, 735–755. [Google Scholar] [CrossRef]
- Ojo, J.O.; Mouzon, B.; Algamal, M.; Leary, P.; Lynch, C.; Abdullah, L.; Evans, J.; Mullan, M.; Bachmeier, C.; Stewart, W.; et al. Chronic Repetitive Mild Traumatic Brain Injury Results in Reduced Cerebral Blood Flow, Axonal Injury, Gliosis, and Increased T-Tau and Tau Oligomers. J. Neuropathol. Exp. Neurol. 2016, 75, 636–655. [Google Scholar] [CrossRef]
- Tran, H.T.; LaFerla, F.M.; Holtzman, D.M.; Brody, D.L. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J. Neurosci. 2011, 31, 9513–9525. [Google Scholar] [CrossRef]
- Xu, L.; Ryu, J.; Nguyen, J.V.; Arena, J.; Rha, E.; Vranis, P.; Hitt, D.; Marsh-Armstrong, N.; Koliatsos, V.E. Evidence for accelerated tauopathy in the retina of transgenic P301S tau mice exposed to repetitive mild traumatic brain injury. Exp. Neurol. 2015, 273, 168–176. [Google Scholar] [CrossRef]
- Edwards, G., 3rd; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic Brain Injury Induces Tau Aggregation and Spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef]
- Cheng, H.; Deaton, L.M.; Qiu, M.; Ha, S.; Pacoma, R.; Lao, J.; Tolley, V.; Moran, R.; Keeton, A.; Lamb, J.R.; et al. Tau overexpression exacerbates neuropathology after repeated mild head impacts in male mice. Neurobiol. Dis. 2020, 134, 104683. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Morganti, J.M.; Bodnar, C.N.; Webster, S.J.; Higgins, E.K.; Roberts, K.N.; Snider, H.; Meier, S.E.; Nation, G.K.; Goulding, D.S.; et al. The effects of mild closed head injuries on tauopathy and cognitive deficits in rodents: Primary results in wild type and rTg4510 mice, and a systematic review. Exp. Neurol. 2020, 326, 113180. [Google Scholar] [CrossRef]
- Namjoshi, D.R.; Cheng, W.H.; McInnes, K.A.; Martens, K.M.; Carr, M.; Wilkinson, A.; Fan, J.; Robert, J.; Hayat, A.; Cripton, P.A.; et al. Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): A novel, surgery-free model of traumatic brain injury. Mol. Neurodegener. 2014, 9, 55. [Google Scholar] [CrossRef]
- Bashir, A.; Abebe, Z.A.; McInnes, K.A.; Button, E.B.; Tatarnikov, I.; Cheng, W.H.; Haber, M.; Wilkinson, A.; Barron, C.; Diaz-Arrastia, R.; et al. Increased severity of the CHIMERA model induces acute vascular injury, sub-acute deficits in memory recall, and chronic white matter gliosis. Exp. Neurol. 2020, 324, 113116. [Google Scholar] [CrossRef]
- Cheung, H.; Cheng, W.H.; Button, E.B.; Bashir, A.; Barron, C.J.; Wilkinson, A.; Wellington, C.L. ApoA-I deficiency has a subtle effect on acute inflammatory responses after experimental Traumatic Brain Injury. Can. J. Undergrad. Res. 2021, 6. Available online: https://ojs.library.ubc.ca/index.php/cjur/article/view/193944 (accessed on 25 May 2023).
- Cheng, W.H.; Martens, K.M.; Bashir, A.; Cheung, H.; Stukas, S.; Gibbs, E.; Namjoshi, D.R.; Button, E.B.; Wilkinson, A.; Barron, C.J.; et al. CHIMERA repetitive mild traumatic brain injury induces chronic behavioural and neuropathological phenotypes in wild-type and APP/PS1 mice. Alzheimer’s Res. Ther. 2019, 11, 6. [Google Scholar] [CrossRef]
- Namjoshi, D.R.; Cheng, W.H.; Carr, M.; Martens, K.M.; Zareyan, S.; Wilkinson, A.; McInnes, K.; Cripton, P.; Wellington, C.L. Chronic Exposure to Androgenic-Anabolic Steroids Exacerbates Axonal Injury and Microgliosis in the CHIMERA Mouse Model of Repetitive Concussion. PLoS ONE 2016, 11, e0146540. [Google Scholar] [CrossRef]
- Haber, M.; Hutchinson, E.B.; Sadeghi, N.; Cheng, W.H.; Namjoshi, D.; Cripton, P.; Irfanoglu, M.O.; Wellington, C.; Diaz-Arrastia, R.; Pierpaoli, C. Defining an Analytic Framework to Evaluate Quantitative MRI Markers of Traumatic Axonal Injury: Preliminary Results in a Mouse Closed Head Injury Model. eNeuro 2017, 4, ENEURO.0164-17.2017. [Google Scholar] [CrossRef]
- Namjoshi, D.R.; Cheng, W.H.; Bashir, A.; Wilkinson, A.; Stukas, S.; Martens, K.M.; Whyte, T.; Abebe, Z.A.; McInnes, K.A.; Cripton, P.A.; et al. Defining the biomechanical and biological threshold of murine mild traumatic brain injury using CHIMERA (Closed Head Impact Model of Engineered Rotational Acceleration). Exp. Neurol. 2017, 292, 80–91. [Google Scholar] [CrossRef]
- Cheng, W.H.; Stukas, S.; Martens, K.M.; Namjoshi, D.R.; Button, E.B.; Wilkinson, A.; Bashir, A.; Robert, J.; Cripton, P.; Wellington, C.L. Age at injury and genotype modify acute inflammatory and neurofilament-light responses to mild CHIMERA traumatic brain injury in wild-type and APP/PS1 mice. Exp. Neurol. 2018, 301, 26–38. [Google Scholar] [CrossRef]
- Andriessen, T.M.; Jacobs, B.; Vos, P.E. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J. Cell. Mol. Med. 2010, 14, 2381–2392. [Google Scholar] [CrossRef]
- Salottolo, K.; Carrick, M.; Levy, A.S.; Morgan, B.C.; Slone, D.S.; Bar-Or, D. The epidemiology, prognosis, and trends of severe traumatic brain injury with presenting Glasgow Coma Scale of 3. J. Crit. Care 2017, 38, 197–201. [Google Scholar] [CrossRef]
- Roozenbeek, B.; Maas, A.I.; Menon, D.K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 231–236. [Google Scholar] [CrossRef]
- Avila, J.; León-Espinosa, G.; García, E.; García-Escudero, V.; Hernández, F.; DeFelipe, J. Tau Phosphorylation by GSK3 in Different Conditions. Int. J. Alzheimer’s Dis. 2012, 2012, 578373. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Cabezas-Opazo, F.; Deaton, C.A.; Vergara, E.H.; Johnson, G.V.W.; Quintanilla, R.A. It’s all about tau. Prog. Neurobiol. 2019, 175, 54–76. [Google Scholar] [CrossRef]
- Pei, J.-J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J. Neuropathol. Exp. Neurol. 1999, 58, 1010–1019. [Google Scholar] [CrossRef]
- Llorens-Marã tin, M.; Jurado, J.; Hernã¡Ndez, F.; Ãvila, J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [CrossRef]
- Zhao, S.; Fu, J.; Liu, X.; Wang, T.; Zhang, J.; Zhao, Y. Activation of Akt/GSK-3beta/beta-catenin signaling pathway is involved in survival of neurons after traumatic brain injury in rats. Neurol. Res. 2012, 34, 400–407. [Google Scholar] [CrossRef]
- Shapira, M.; Licht, A.; Milman, A.; Pick, C.G.; Shohami, E.; Eldar-Finkelman, H. Role of glycogen synthase kinase-3β in early depressive behavior induced by mild traumatic brain injury. Mol. Cell. Neurosci. 2007, 34, 571–577. [Google Scholar] [CrossRef]
- Dash, P.K.; Johnson, D.; Clark, J.; Orsi, S.A.; Zhang, M.; Zhao, J.; Grill, R.J.; Moore, A.N.; Pati, S. Involvement of the glycogen synthase kinase-3 signaling pathway in TBI pathology and neurocognitive outcome. PLoS ONE 2011, 6, e24648. [Google Scholar] [CrossRef]
- Farr, S.A.; Niehoff, M.L.; Kumar, V.B.; Roby, D.A.; Morley, J.E. Inhibition of Glycogen Synthase Kinase 3beta as a Treatment for the Prevention of Cognitive Deficits after a Traumatic Brain Injury. J. Neurotrauma 2019, 36, 1869–1875. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of Microglia in the Central Nervous System Diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Degradation of tau protein by autophagy and proteasomal pathways. Biochem. Soc. Trans. 2012, 40, 644–652. [Google Scholar] [CrossRef]
- Chesser, A.S.; Pritchard, S.M.; Johnson, G.V. Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front. Neurol. 2013, 4, 122. [Google Scholar] [CrossRef]
- Jiang, S.; Bhaskar, K. Degradation and Transmission of Tau by Autophagic-Endolysosomal Networks and Potential Therapeutic Targets for Tauopathy. Front. Mol. Neurosci. 2020, 13, 586731. [Google Scholar] [CrossRef]
- Wu, J.; Lipinski, M.M. Autophagy in Neurotrauma: Good, Bad, or Dysregulated. Cells 2019, 8, 693. [Google Scholar] [CrossRef]
- Kuninaka, N.; Kawaguchi, M.; Ogawa, M.; Sato, A.; Arima, K.; Murayama, S.; Saito, Y. Simplification of the modified G allyas method. Neuropathology 2015, 35, 10–15. [Google Scholar] [CrossRef]
- Gallyas Silver Stain. 2012. Available online: https://www.protocolsonline.com/histology/dyes-and-stains/neurohistology/gallyas-silver-stain/ (accessed on 1 June 2019).
- Yanamandra, K.; Kfoury, N.; Jiang, H.; Mahan, T.E.; Ma, S.; Maloney, S.E.; Wozniak, D.F.; Diamond, M.I.; Holtzman, D.M. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 2013, 80, 402–414. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, W.H.; Cheung, H.; Kang, A.; Fan, J.; Cooper, J.; Anwer, M.; Barron, C.; Wilkinson, A.; Hu, G.; Yue, J.; et al. Altered Tau Kinase Activity in rTg4510 Mice after a Single Interfaced CHIMERA Traumatic Brain Injury. Int. J. Mol. Sci. 2023, 24, 9439. https://doi.org/10.3390/ijms24119439
Cheng WH, Cheung H, Kang A, Fan J, Cooper J, Anwer M, Barron C, Wilkinson A, Hu G, Yue J, et al. Altered Tau Kinase Activity in rTg4510 Mice after a Single Interfaced CHIMERA Traumatic Brain Injury. International Journal of Molecular Sciences. 2023; 24(11):9439. https://doi.org/10.3390/ijms24119439
Chicago/Turabian StyleCheng, Wai Hang, Honor Cheung, Amy Kang, Jianjia Fan, Jennifer Cooper, Mehwish Anwer, Carlos Barron, Anna Wilkinson, Grace Hu, Jefferey Yue, and et al. 2023. "Altered Tau Kinase Activity in rTg4510 Mice after a Single Interfaced CHIMERA Traumatic Brain Injury" International Journal of Molecular Sciences 24, no. 11: 9439. https://doi.org/10.3390/ijms24119439