

Natural Cyclophilin A Inhibitors Suppress the Growth of Cancer Stem Cells in Non-Small Cell Lung Cancer by Disrupting Crosstalk between CypA/CD147 and EGFR


Abstract
:1. Introduction
2. Results
2.1. C9 and CsA Suppress the Growth of NSCLC CSCs
2.2. C9 and CsA Suppress NSCLC CSC-Derived Tumor Growth In Vivo
2.3. C9 and CsA Promote Apoptosis in NSCLC CSCs
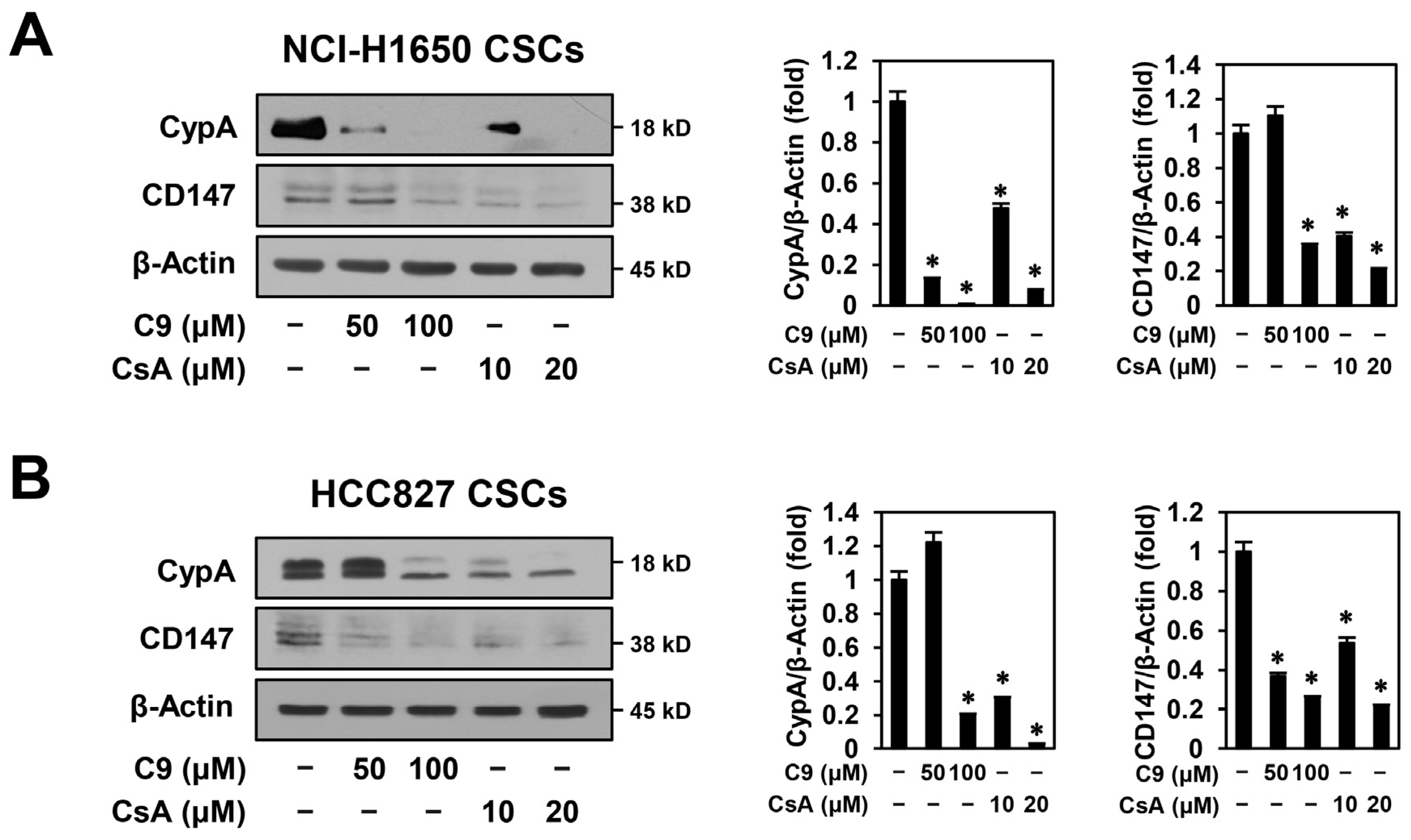
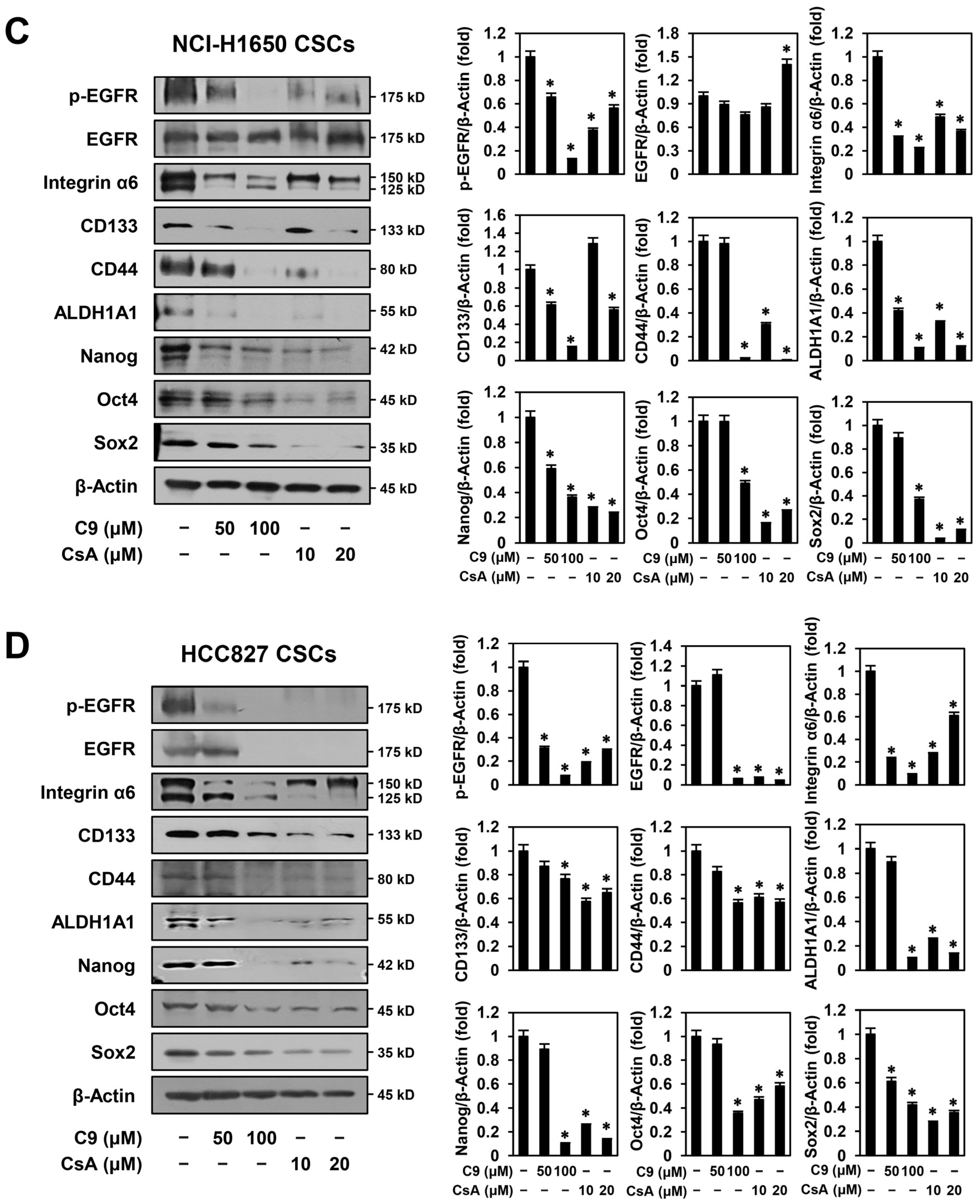
2.4. C9 and CsA Inhibit CSC Biomarker Expression through Dual Downregulation of CypA/CD147 and EGFR in NSCLC CSCs
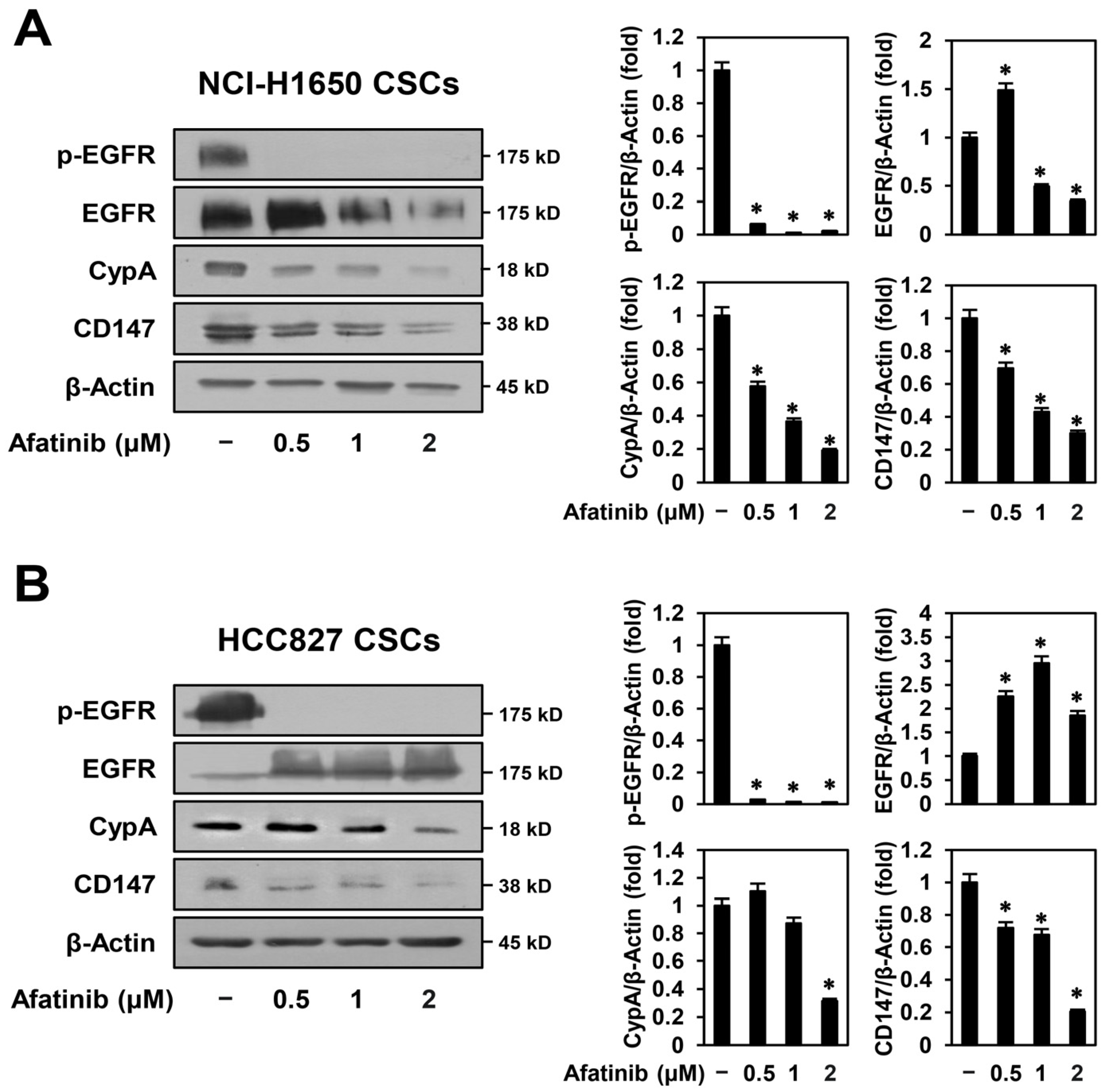
2.5. Afatinib Inhibits CypA/CD147 Expression in NSCLC CSCs
2.6. Combined Treatment with CypA Inhibitors and Afatinib More Potently Inhibits the Growth of EGFR-Mutated NSCLC CSCs than Single-Compound Treatments
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. NSCLC CSC Culture
4.3. Cell Proliferation Assay
4.4. Limiting Dilution Assay
4.5. Chick Embryo Chorioallantoic Membrane (CAM) Assay
4.6. Analysis of Apoptosis
4.7. Western Blot Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rabe, K.F.; Longo, D.L. Precision diagnosis and treatment for advanced non-small-cell lung cancer. N. Engl. J. Med. 2017, 377, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; Casaluce, F. Frontline immunotherapy for NSCLC: Alone or not alone? Nat. Rev. Clin. Oncol. 2018, 15, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.Y.; Xu, Y.P.; Jin, W.; Lou, L.G. Antitumor activity of lobaplatin alone or in combination with antitubulin agents in non-small-cell lung cancer. Anticancer Drugs 2012, 23, 698–705. [Google Scholar] [CrossRef]
- Jackman, D.; Pao, W.; Riely, G.J.; Engelman, J.A.; Kris, M.G.; Jänne, P.A.; Lynch, T.; Johnson, B.E.; Miller, V.A. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 357–360. [Google Scholar] [CrossRef]
- Clarke, M.F. Clinical and therapeutic implications of cancer stem cells. N. Engl. J. Med. 2019, 380, 2237–2245. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Leung, E.L.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.; Cheng, L.C.; Sihoe, A.D.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE 2010, 5, e14062. [Google Scholar] [CrossRef]
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A.; et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef]
- Lang, K.; Schmid, F.X.; Fischer, G. Catalysis of protein folding by prolyl isomerase. Nature 1987, 329, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Schiene, C.; Fischer, G. Enzymes that catalyse the restructuring of proteins. Curr. Opin. Struct. Biol. 2000, 10, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Jung, H.J. Cyclophilin A/CD147 interaction: A promising target for anticancer therapy. Int. J. Mol. Sci. 2022, 23, 9341. [Google Scholar] [CrossRef]
- Jin, Z.G.; Melaragno, M.G.; Liao, D.F.; Yan, C.; Haendeler, J.; Suh, Y.A.; Lambeth, J.D.; Berk, B.C. Cyclophilin A is a secreted growth factor induced by oxidative stress. Circ. Res. 2000, 87, 789–796. [Google Scholar] [CrossRef]
- Pushkarsky, T.; Zybarth, G.; Dubrovsky, L.; Yurchenko, V.; Tang, H.; Guo, H.; Toole, B.; Sherry, B.; Bukrinsky, M. CD147 facilitates HIV-1 infection by interacting with virus-associated cyclophilin A. Proc. Natl. Acad. Sci. USA 2001, 98, 6360–6365. [Google Scholar] [CrossRef]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: A specific cytosolic binding protein for cyclosporin A. Science 1984, 226, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, V.; Zybarth, G.; O’Connor, M.; Dai, W.W.; Franchin, G.; Hao, T.; Guo, H.; Hung, H.C.; Toole, B.; Gallay, P.; et al. Active site residues of cyclophilin A are crucial for its signaling activity via CD147. J. Biol. Chem. 2002, 277, 22959–22965. [Google Scholar] [CrossRef]
- Shou, J.; You, L.; Yao, J.; Xie, J.; Jing, J.; Jing, Z.; Jiang, L.; Sui, X.; Pan, H.; Han, W. Cyclosporine A sensitizes human non-small cell lung cancer cells to gefitinib through inhibition of STAT3. Cancer Lett. 2016, 379, 124–133. [Google Scholar] [CrossRef]
- Dhakal, D.; Han, J.M.; Mishra, R.; Pandey, R.P.; Kim, T.S.; Rayamajhi, V.; Jung, H.J.; Yamaguchi, T.; Sohng, J.K. Characterization of tailoring steps of nargenicin A1 biosynthesis reveals a novel analogue with anticancer activities. ACS Chem. Biol. 2020, 15, 1370–1380. [Google Scholar] [CrossRef]
- Han, J.M.; Sohng, J.K.; Lee, W.H.; Oh, T.J.; Jung, H.J. Identification of cyclophilin A as a potential anticancer target of novel nargenicin A1 analog in AGS gastric cancer cells. Int. J. Mol. Sci. 2021, 22, 2473. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Choi, Y.S.; Dhakal, D.; Sohng, J.K.; Jung, H.J. Novel nargenicin A1 analog inhibits angiogenesis by downregulating the endothelial VEGF/VEGFR2 signaling and tumoral HIF-1α/VEGF pathway. Biomedicines 2020, 8, 252. [Google Scholar] [CrossRef]
- Cho, H.J.; Jung, H.J. Cyclophilin A inhibitors suppress proliferation and induce apoptosis of MKN45 gastric cancer stem-like cells by regulating CypA/CD147-mediated signaling pathway. Int. J. Mol. Sci. 2023, 24, 4734. [Google Scholar] [CrossRef] [PubMed]
- Yakisich, J.S.; Azad, N.; Venkatadri, R.; Kulkarni, Y.; Wright, C.; Kaushik, V.; Iyer, A.K. Formation of tumorspheres with increased stemness without external mitogens in a lung cancer model. Stem Cells Int. 2016, 2016, 5603135. [Google Scholar] [CrossRef]
- Herreros-Pomares, A.; de-Maya-Girones, J.D.; Calabuig-Fariñas, S.; Lucas, R.; Martínez, A.; Pardo-Sánchez, J.M.; Alonso, S.; Blasco, A.; Guijarro, R.; Martorell, M.; et al. Lung tumorspheres reveal cancer stem cell-like properties and a score with prognostic impact in resected non-small-cell lung cancer. Cell Death Dis. 2019, 10, 660. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H. Cancer stem cells and self-renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Rota, L.M.; Lazzarino, D.A.; Ziegler, A.N.; LeRoith, D.; Wood, T.L. Determining mammosphere-forming potential: Application of the limiting dilution analysis. J. Mammary Gland Biol. Neoplasia 2012, 17, 119–123. [Google Scholar] [CrossRef]
- Safa, A.R. Resistance to cell death and its modulation in cancer stem cells. Crit. Rev. Oncog. 2016, 21, 203–219. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Tamm, I.; Wang, Y.; Sausville, E.; Scudiero, D.A.; Vigna, N.; Oltersdorf, T.; Reed, J.C. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998, 58, 5315–5320. [Google Scholar]
- Zheng, Y.; Wang, L.; Yin, L.; Yao, Z.; Tong, R.; Xue, J.; Lu, Y. Lung cancer stem cell markers as therapeutic targets: An update on signaling pathways and therapies. Front. Oncol. 2022, 12, 873994. [Google Scholar] [CrossRef]
- Codony-Servat, J.; Codony-Servat, C.; Cardona, A.F.; Giménez-Capitán, A.; Drozdowskyj, A.; Berenguer, J.; Bracht, J.W.P.; Ito, M.; Karachaliou, N.; Rosell, R. Cancer stem cell biomarkers in EGFR-mutation-positive non-small-cell lung cancer. Clin. Lung Cancer 2019, 20, 167–177. [Google Scholar] [CrossRef]
- Hu, F.; Li, C.; Zheng, X.; Zhang, H.; Shen, Y.; Zhou, L.; Yang, X.; Han, B.; Zhang, X. Lung adenocarcinoma resistance to therapy with EGFR-tyrosine kinase inhibitors is related to increased expression of cancer stem cell markers SOX2, OCT4 and NANOG. Oncol. Rep. 2020, 43, 727–735. [Google Scholar] [CrossRef]
- Gupta, R.; Dastane, A.M.; Forozan, F.; Riley-Portuguez, A.; Chung, F.; Lopategui, J.; Marchevsky, A.M. Evaluation of EGFR abnormalities in patients with pulmonary adenocarcinoma: The need to test neoplasms with more than one method. Mod. Pathol. 2009, 22, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Ouhtit, A.; Thouta, R.; Zayed, H.; Gaur, R.L.; Fernando, A.; Rahman, M.; Welsh, D.A. Cd44 mediates stem cell mobilization to damaged lung via its novel transcriptional targets, Cortactin and Survivin. Int. J. Med. Sci. 2020, 17, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.W.; Song, Y.; Kim, S.H.; Kim, J.; Seo, H.R. Potential mechanisms of Cd133 in cancer stem cells. Life Sci. 2017, 184, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and cancer: Regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef]
- Lei, H.M.; Zhang, K.R.; Wang, C.H.; Wang, Y.; Zhuang, G.L.; Lu, L.M.; Zhang, J.; Shen, Y.; Chen, H.Z.; Zhu, L. Aldehyde dehydrogenase 1a1 confers erlotinib resistance via facilitating the reactive oxygen species-reactive carbonyl species metabolic pathway in lung adenocarcinomas. Theranostics 2019, 9, 7122–7139. [Google Scholar] [CrossRef] [PubMed]
- Ferone, G.; Song, J.Y.; Sutherland, K.D.; Bhaskaran, R.; Monkhorst, K.; Lambooij, J.P.; Proost, N.; Gargiulo, G.; Berns, A. Sox2 is the determining oncogenic switch in promoting lung squamous cell carcinoma from different cells of origin. Cancer Cell 2016, 30, 519–532. [Google Scholar] [CrossRef]
- Mohiuddin, I.S.; Wei, S.J.; Kang, M.H. Role of Oct4 in cancer stem-like cells and chemotherapy resistance. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165432. [Google Scholar] [CrossRef]
- Chang, B.; Park, M.J.; Choi, S.I.; In, K.H.; Kim, C.H.; Lee, S.H. Nanog as an adverse predictive marker in advanced non-small cell lung cancer treated with platinum-based chemotherapy. Onco. Targets Ther. 2017, 10, 4625–4633. [Google Scholar] [CrossRef] [PubMed]
- Atikcan, S.; Unsal, E.; Demirag, F.; Köksal, D.; Yilmaz, A. Correlation between survivin expression and prognosis in non-small cell lung cancer. Respir. Med. 2006, 100, 2220–2226. [Google Scholar] [CrossRef]
- Chen, X.; Duan, N.; Zhang, C.; Zhang, W. Survivin and tumorigenesis: Molecular mechanisms and therapeutic strategies. J. Cancer 2016, 7, 314–323. [Google Scholar] [CrossRef]
- Jung, J.H.; Choi, J.W.; Lee, M.K.; Choi, Y.H.; Nam, T.J. Effect of cyclophilin from Pyropia Yezoensis on the proliferation of intestinal epithelial cells by epidermal growth factor receptor/Ras signaling pathway. Mar. Drugs 2019, 17, 297. [Google Scholar] [CrossRef]
- Grass, G.D.; Tolliver, L.B.; Bratoeva, M.; Toole, B.P. CD147, CD44, and the epidermal growth factor receptor (EGFR) signaling pathway cooperate to regulate breast epithelial cell invasiveness. J. Biol. Chem. 2013, 288, 26089–26104. [Google Scholar] [CrossRef]
- Xu, B.Q.; Fu, Z.G.; Meng, Y.; Wu, X.Q.; Wu, B.; Xu, L.; Jiang, J.L.; Li, L.; Chen, Z.N. Gemcitabine enhances cell invasion via activating HAb18G/CD147-EGFR-pSTAT3 signaling. Oncotarget 2016, 7, 62177–62193. [Google Scholar] [CrossRef]
- Dai, L.; Guinea, M.C.; Slomiany, M.G.; Bratoeva, M.; Grass, G.D.; Tolliver, L.B.; Maria, B.L.; Toole, B.P. CD147-dependent heterogeneity in malignant and chemoresistant properties of cancer cells. Am. J. Pathol. 2013, 182, 577–585. [Google Scholar] [CrossRef]
- Hata, A.; Katakami, N.; Kaji, R.; Yokoyama, T.; Kaneda, T.; Tamiya, M.; Inoue, T.; Kimura, H.; Yano, Y.; Tamura, D.; et al. Afatinib plus bevacizumab combination after acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: Multicenter, single-arm, phase 2 trial (ABC Study). Cancer 2018, 124, 3830–3838. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Weiss, J.; Sos, M.L.; Koker, M.; Heynck, S.; Netzer, C.; Fischer, S.; Rode, H.; Rauh, D.; Rahnenführer, J.; et al. Analysis of compound synergy in high-throughput cellular screens by population-based lifetime modeling. PLoS ONE 2010, 5, e8919. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Hobor, S.; Bertotti, A.; Zecchin, D.; Huang, S.; Galimi, F.; Cottino, F.; Prahallad, A.; Grernrum, W.; Tzani, A.; et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014, 7, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.C.; Jablons, D.M.; Yang, C.T.; You, L. Epidermal growth factor receptor (EGFR) pathway, yes-associated protein (YAP) and the regulation of programmed death-ligand 1 (PD-L1) in non-small cell lung cancer (NSCLC). Int. J. Mol. Sci. 2019, 20, 3821. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Kim, Y.J.; Jung, H.J. Discovery of a new CaMKII-targeted synthetic lethal therapy against glioblastoma stem-like cells. Cancers 2022, 14, 1315. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 Values (µM) | |||
|---|---|---|---|---|
| A549 CSCs | NCI-H1299 CSCs | NCI-H1650 CSCs | HCC827 CSCs | |
| C9 | 49.03 ± 0.015 | 57.56 ± 0.036 | 29.18 ± 0.027 | 25.57 ± 0.014 |
| CsA | 9.34 ± 0.051 | 9.72 ± 0.059 | 2.47 ± 0.035 | 8.90 ± 0.009 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.M.; Kim, S.M.; Kim, H.L.; Cho, H.J.; Jung, H.J. Natural Cyclophilin A Inhibitors Suppress the Growth of Cancer Stem Cells in Non-Small Cell Lung Cancer by Disrupting Crosstalk between CypA/CD147 and EGFR. Int. J. Mol. Sci. 2023, 24, 9437. https://doi.org/10.3390/ijms24119437
Han JM, Kim SM, Kim HL, Cho HJ, Jung HJ. Natural Cyclophilin A Inhibitors Suppress the Growth of Cancer Stem Cells in Non-Small Cell Lung Cancer by Disrupting Crosstalk between CypA/CD147 and EGFR. International Journal of Molecular Sciences. 2023; 24(11):9437. https://doi.org/10.3390/ijms24119437
Chicago/Turabian StyleHan, Jang Mi, Sung Min Kim, Hong Lae Kim, Hee Jeong Cho, and Hye Jin Jung. 2023. "Natural Cyclophilin A Inhibitors Suppress the Growth of Cancer Stem Cells in Non-Small Cell Lung Cancer by Disrupting Crosstalk between CypA/CD147 and EGFR" International Journal of Molecular Sciences 24, no. 11: 9437. https://doi.org/10.3390/ijms24119437