
Mining the Australian Grains Gene Bank for Rust Resistance in Barley
 , , and
, , and
Abstract
:1. Introduction
2. Results
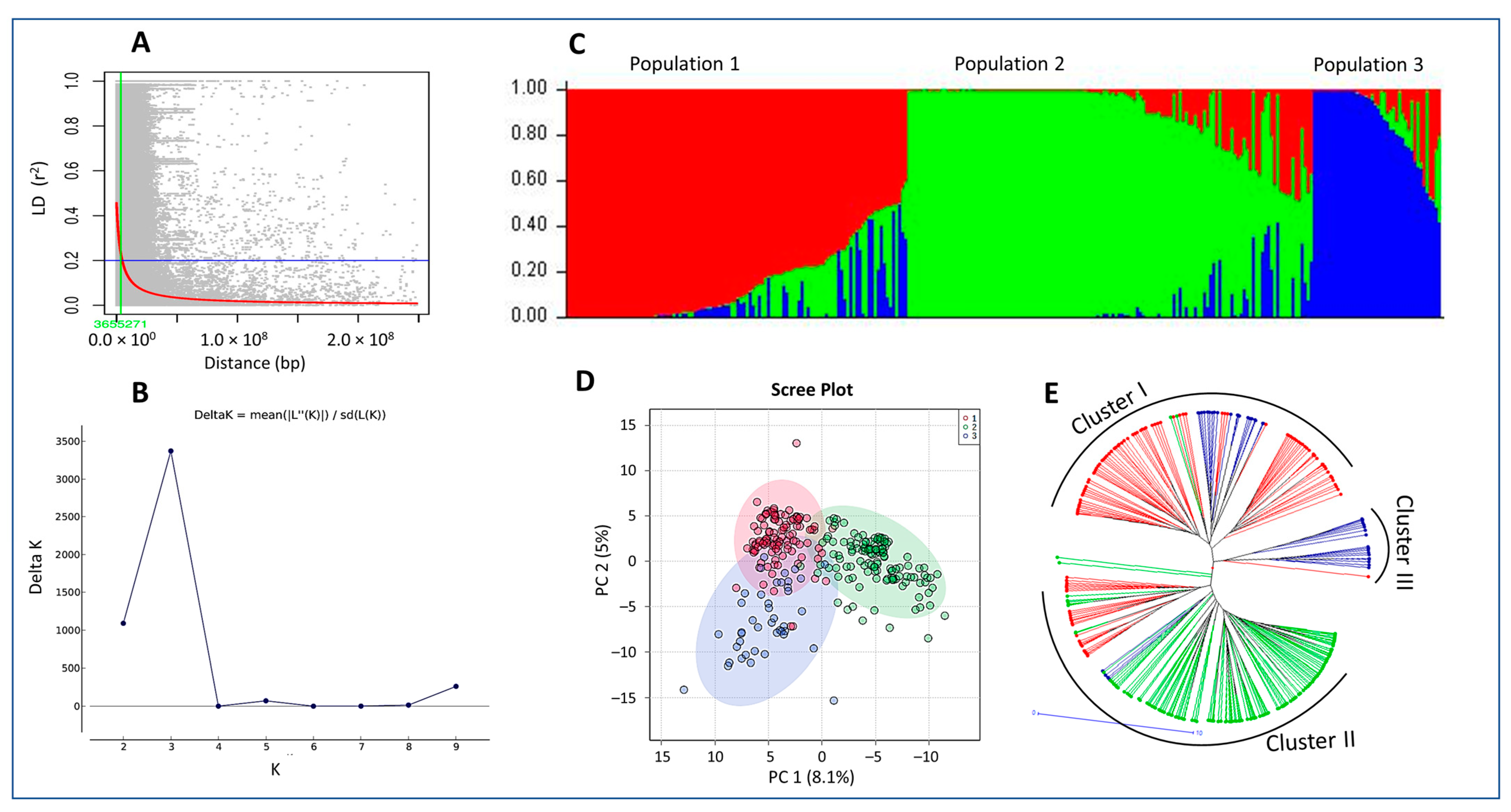
2.1. Population Structure
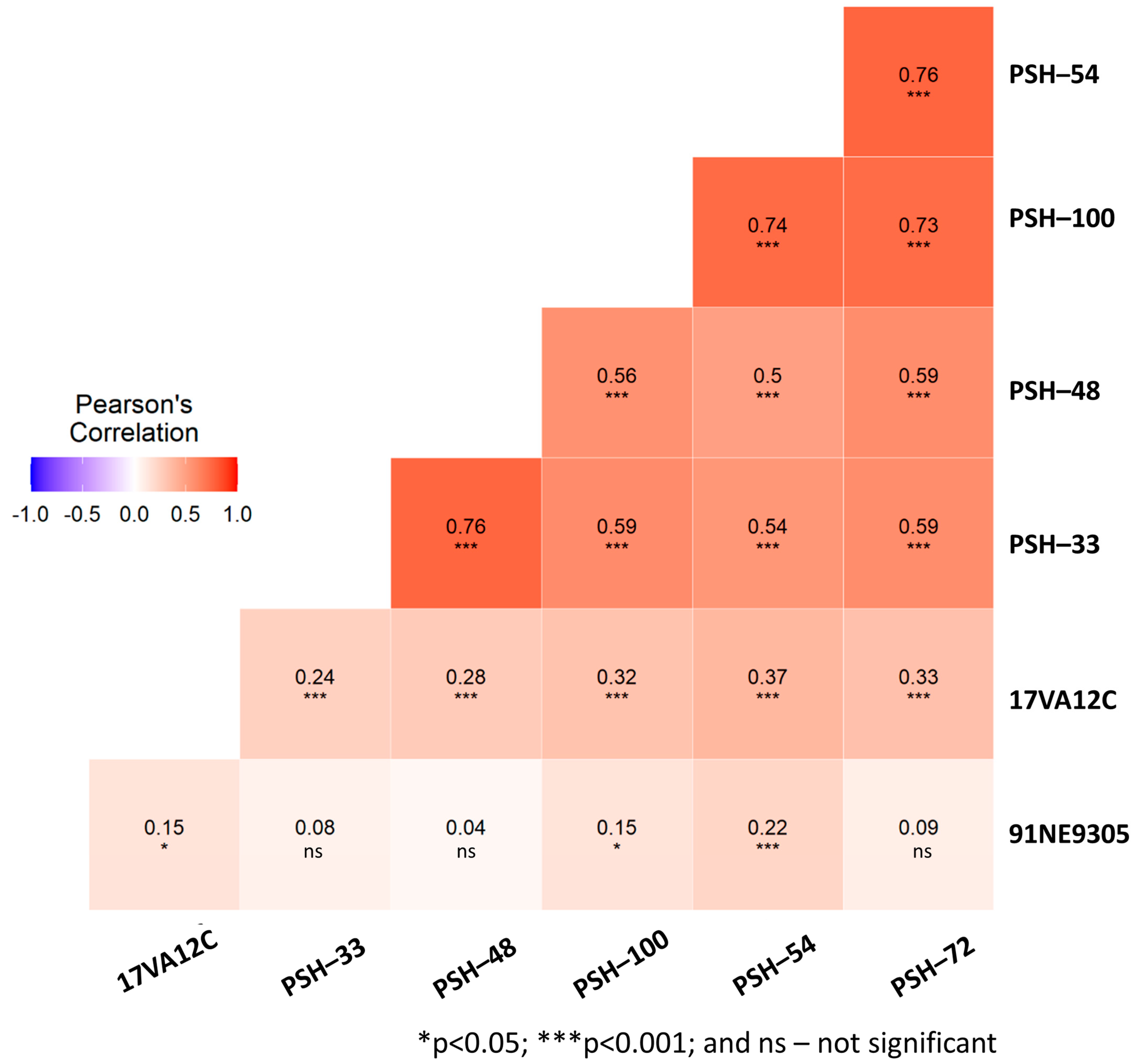
2.2. Phenotypic Rust Response under Controlled Greenhouse Conditions
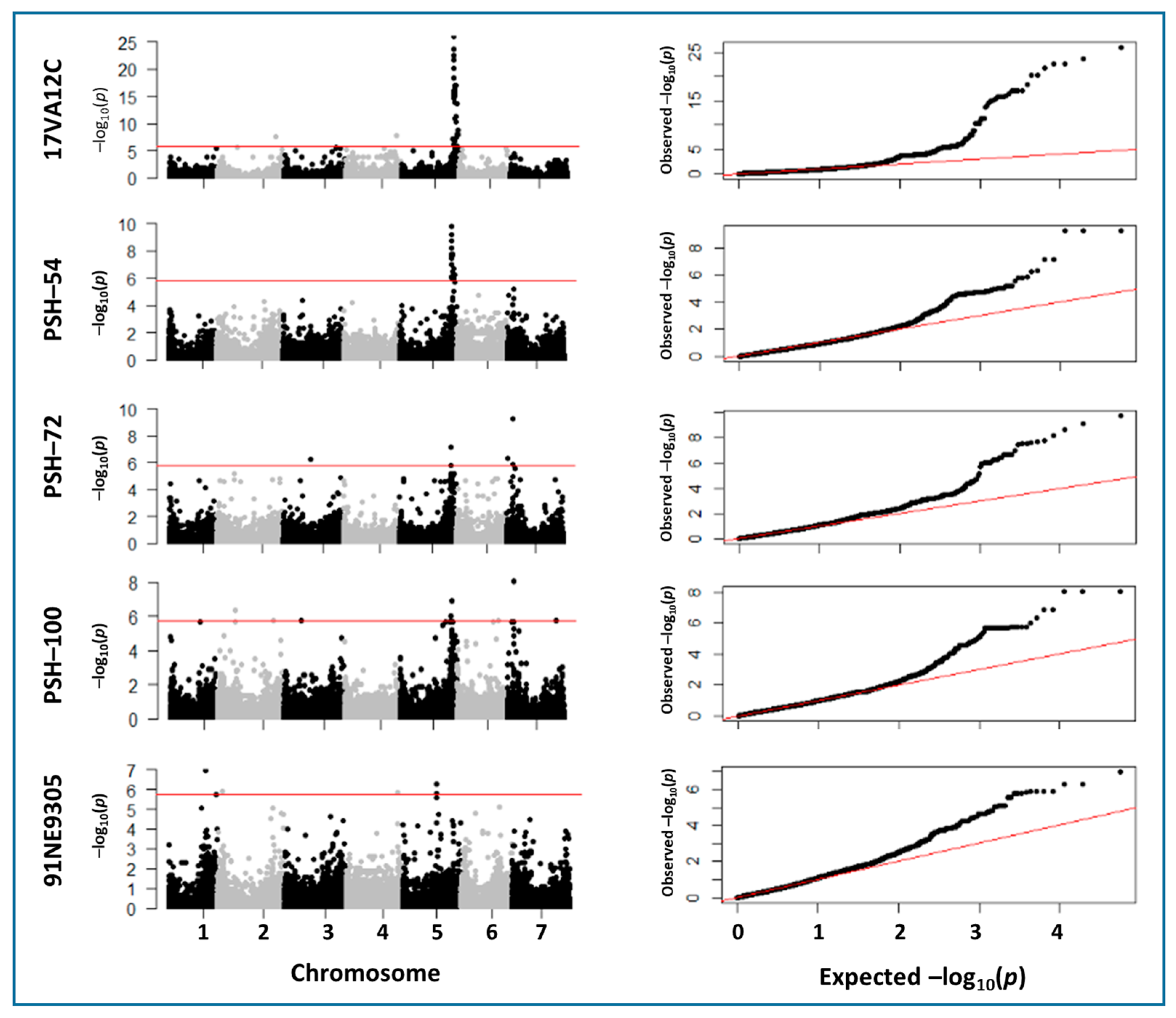
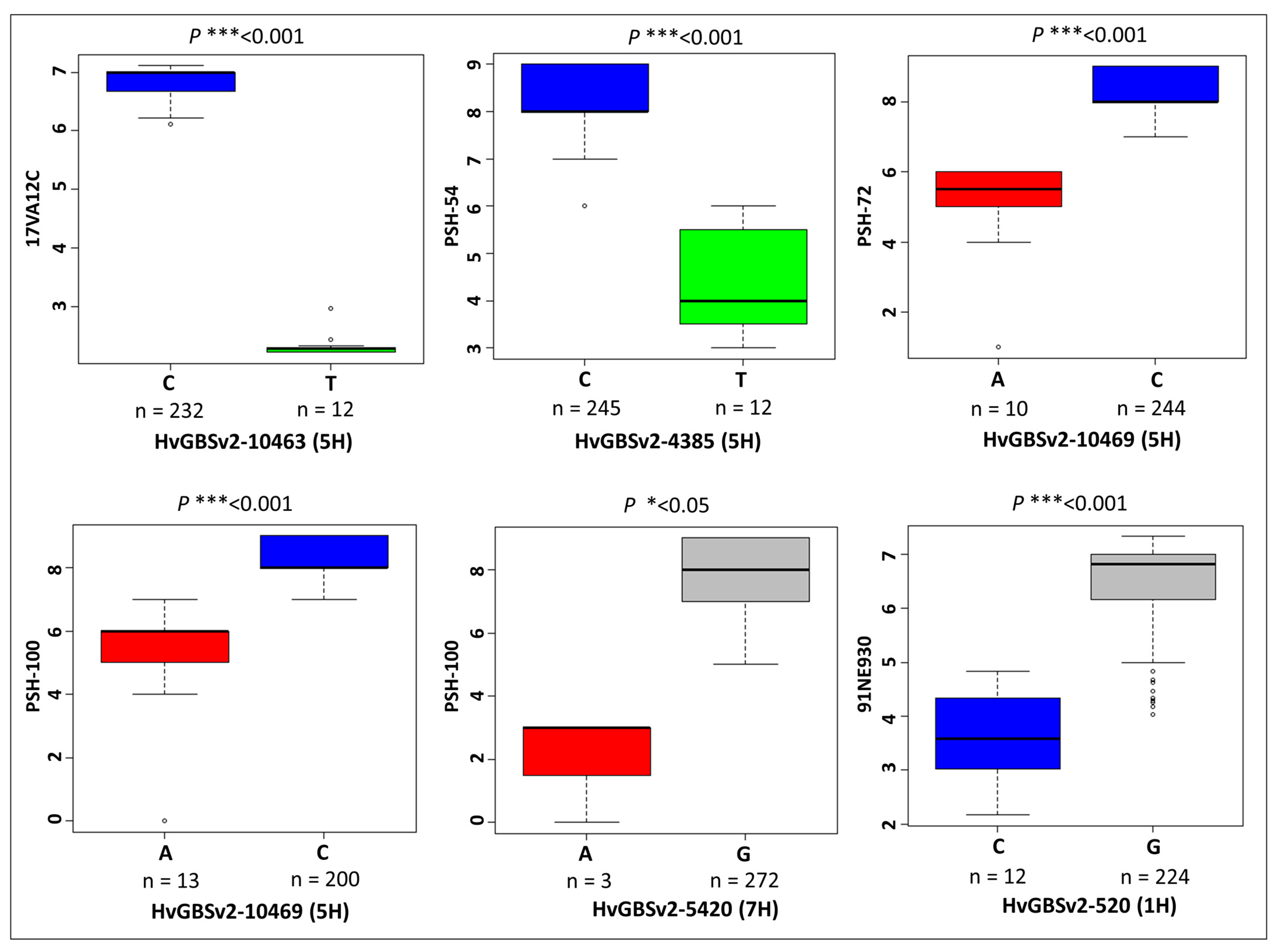
2.3. Marker–Trait Associations
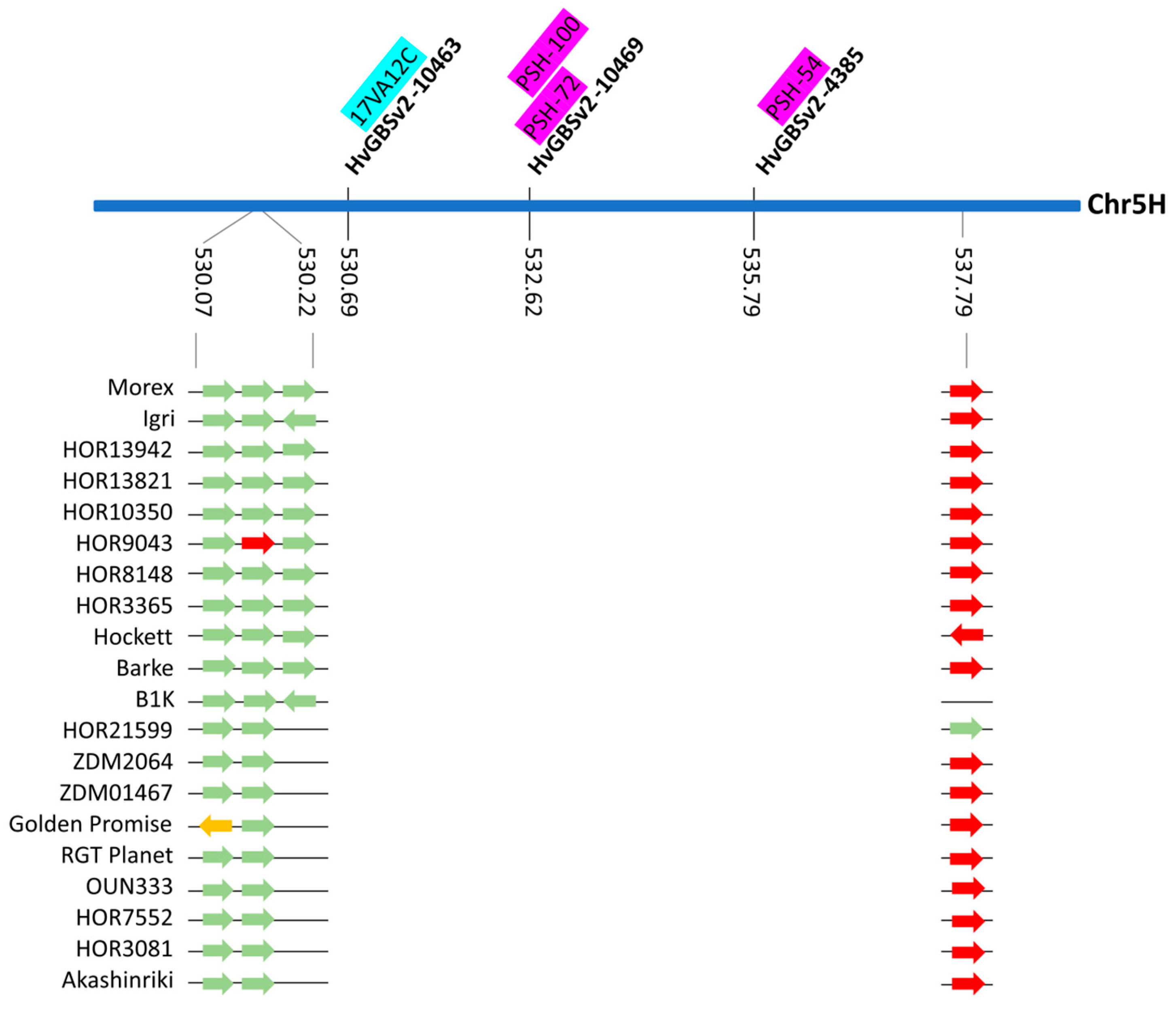
2.4. Investigating Genomic Regions Carrying Markers with Association to Multiple Pathogens
3. Materials and Methods
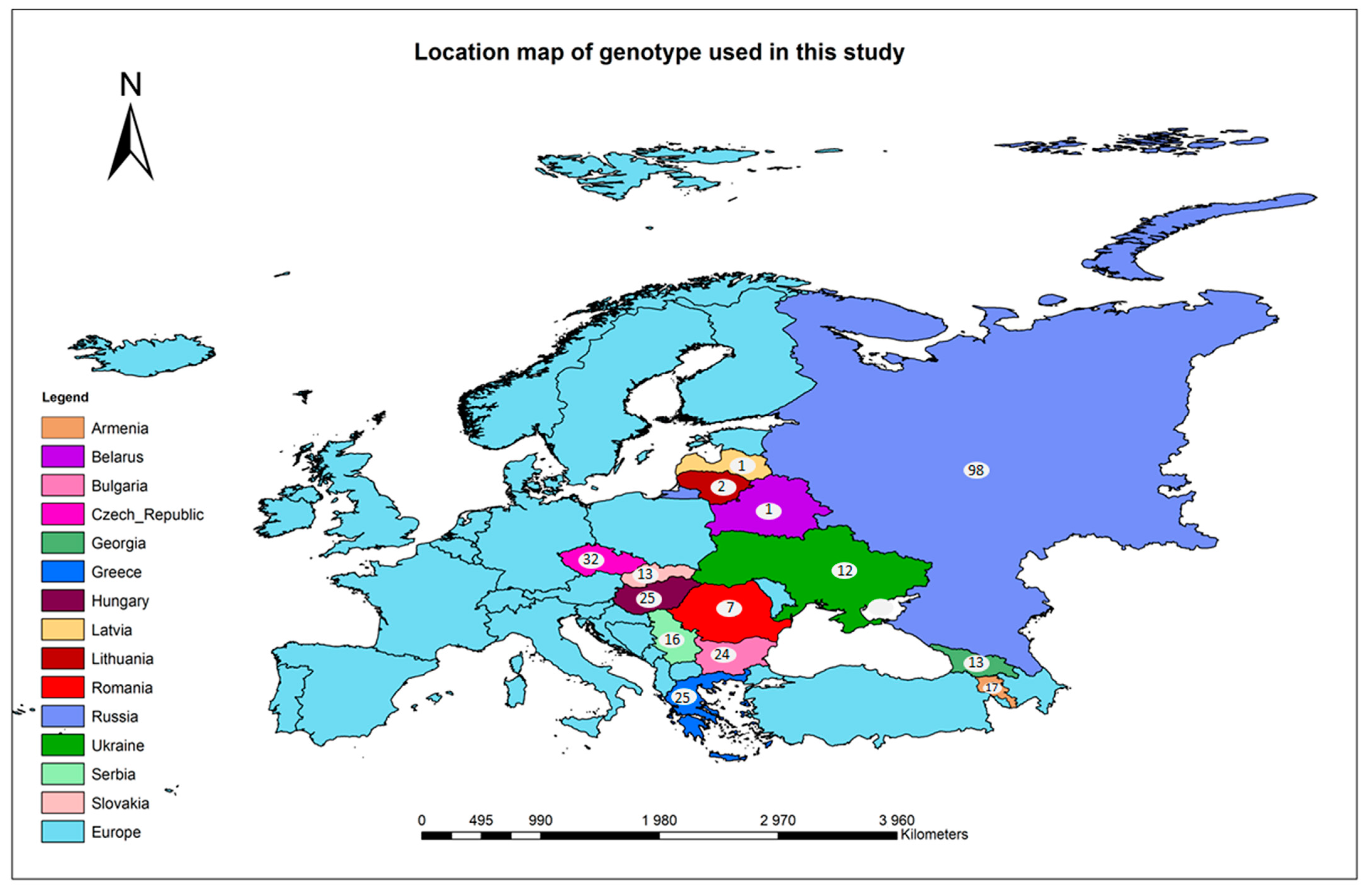
3.1. Plant Materials
3.2. Pathogen Isolates
3.3. Inoculation and Phenotypic Assessment of Rust Resistance
3.4. Genomic DNA Isolation and Quantification
3.5. Genotyping of Barley Genotypes
3.6. Identifying Genetically Redundant Accessions
3.7. Imputation
3.8. Population Structure Analysis
3.9. Linkage Disequilibrium
3.10. Detection of Significant SNP Markers Using GWAS
3.11. Pan-Genomic Evaluation of Candidate Loci
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aune, D.; Keum, N.; Giovannucci, E.; Fadnes, L.; Boffetta, P.; Greenwood, D.; Tonstad, S.; Vatten, L.J.; Riboli, E.; Norat, T. Whole grain consumption and risk of cardiovascular disease, cancer, and all cause and cause specific mortality: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2016, 353, i2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRoberts, N.; Nelson, N. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef]
- Park, R.; Golegaonkar, P.; Derevnina, L.; Sandhu, K.; Karaoglu, H.; Elmansour, H.; Dracatos, P.; Singh, D. Leaf rust of cultivated barley: Pathology and control. Annu. Rev. Phytopathol. 2015, 53, 565–589. [Google Scholar] [CrossRef] [PubMed]
- Steffenson, B.; Case, A.; Pretorius, Z.; Coetzee, V.; Kloppers, F.; Zhou, H.; Chai, Y.; Wanyera, R.; Macharia, G.; Bhavani, S.; et al. Vulnerability of Barley to African Pathotypes of Puccinia graminis f. sp. tritici and Sources of Resistance. Phytopathology 2017, 107, 950–962. [Google Scholar] [PubMed] [Green Version]
- Hundie, B.; Girma, B.; Tadesse, Z.; Edae, E.; Olivera, P.; Abera, E.H.; Bulbula, W.D.; Abeyo, B.; Badebo, A.; Cisar, G.; et al. Characterization of Ethiopian wheat germplasm for resistance to four Puccinia graminis f. sp. tritici races facilitated by single-race nurseries. Plant Dis. 2019, 103, 2359–2366. [Google Scholar] [PubMed] [Green Version]
- Dracatos, P.; Khatkar, M.; Singh, D.; Stefanato, F.; Park, R.; Boyd, L. Resistance in Australian barley (Hordeum vulgare) germplasm to the exotic pathogen Puccinia striiformis f. sp. hordei, causal agent of stripe rust. Plant Pathol. 2016, 65, 734–743. [Google Scholar]
- Line, R.; Konzak, C.; Allan, R. Evaluating resistance to Puccinia striiformis in wheat. In Proceedings of the Research Co-ordination Meeting on Induced Mutations for Disease Resistance in Crop Plants, Novi Sad, Serbia, 4 June 1974. [Google Scholar]
- Chen, X.; Penman, L. Stripe rust epidemic and races of Puccinia striiformis in the United States in 2004. Phytopathology 2005, 95, S19. [Google Scholar]
- Agrama, H.; Dahleen, L.; Wentz, M.; Jin, Y.; Steffenson, B. Molecular mapping of the crown rust resistance gene rpc1 in barley. Phytopathology 2004, 94, 858–861. [Google Scholar] [CrossRef] [Green Version]
- Milner, S.; Jost, M.; Taketa, S.; Mazon, E.; Himmelbach, A.; Oppermann, M.; Weise, S.; Knupffer, H.; Basterrechea, M.; Konig, P.; et al. Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 2019, 51, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Mascher, M.; Wicker, T.; Jenkins, J.; Plott, C.; Lux, T.; Koh, C.; Ens, J.; Gundlach, H.; Boston, L.B.; Tulpová, Z.; et al. Long-read sequence assembly: A technical evaluation in barley. Plant Cell 2021, 33, 1888–1906. [Google Scholar] [CrossRef] [PubMed]
- Jayakodi, M.; Padmarasu, S.; Haberer, G.; Bonthala, V.S.; Gundlach, H.; Monat, C.; Lux, T.; Kamal, N.; Lang, D.; Himmelbach, A.; et al. The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 2020, 588, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Steuernagel, B.; Witek, K.; Krattinger, S.; Ramirez-Gonzalez, R.H.; Schoonbeek, H.J.; Yu, G.; Baggs, E.; Witek, A.I.; Yadav, I.; Krasileva, K.V.; et al. The NLR-Annotator Tool Enables Annotation of the Intracellular Immune Receptor Repertoire. Plant Physiol. 2020, 183, 468–482. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Line, R.; Leung, H. Virulence and polymorphic DNA relationships of Puccinia striiformis f. sp. hordei to other rusts. Phytopathology 1995, 85, 1335–1342. [Google Scholar]
- Mehnaz, M.; Dracatos, P.; Dinh, H.; Forrest, K.; Rouse, M.; Park, R.; Singh, D. A novel locus conferring resistance to Puccinia hordei maps to the genomic region corresponding to Rph14 on barley chromosome 2HS. Front. Plant Sci. 2022, 13, 980870. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Turner, M.; Chao, S.; Kolmer, J.; Anderson, J. Genome wide association study of seedling and adult plant leaf rust resistance in elite spring wheat breeding lines. PLoS ONE 2016, 11, e0148671. [Google Scholar] [CrossRef] [Green Version]
- Fulton, T.; Chunwongse, J.; Tanksley, S. Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol. Biol. Rep. 1995, 13, 207–209. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBNet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.; Daly, M.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 1 November 2022).
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Int. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Money, D.; Gardner, K.; Migicovsky, Z.; Schwaninger, H.; Zhong, G.; Myles, S. LinkImpute: Fast and Accurate Genotype Imputation for Nonmodel Organisms. G3 Genes Genomes Genet. 2015, 5, 2383–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, J.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.; Von Holdt, B. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Richards, J.; Friesen, T.; Brueggeman, R. Association mapping utilizing diverse barley lines reveals net form net blotch seedling resistance/susceptibility loci. Theor. Appl. Genet. 2017, 130, 915–927. [Google Scholar] [CrossRef]
- Bradbury, P.; Zhang, Z.; Kroon, D.; Casstevens, T.; Ramdoss, Y.; Buckler, E. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [Green Version]
- Weir, B. Inferences about linkage disequilibrium. Biometrics 1979, 35, 235–254. [Google Scholar] [CrossRef]
- Remington, D.; Thornsberry, J.; Matsuoka, Y.; Wilson, L.; Whitt, S.; Doebley, J.; Kresovich, S.; Goodman, M.; Buckler, E. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 2001, 98, 11479–11484. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Steffenson, B. Sources and genetics of crown rust resistance in barley. Phytopathology 2002, 92, 1064–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcher, A.; Cuesta-Marcos, A.; Smith, K.; Mundt, C.; Chen, X.; Hayes, P. TCAP FAC-WIN6 elite barley GWAS panel QTL. I. Barley stripe rust resistance QTL in facultative and winter six-rowed malt barley breeding programs identified via GWAS. Crop Sci. 2018, 58, 103–119. [Google Scholar] [CrossRef]
- Jin, Y.; Statler, G.; Franckowiak, J.; Steffenson, B. Linkage between leaf rust resistance genes and morphological markers in barley. Phytopathology 1993, 83, 230–233. [Google Scholar] [CrossRef]
- Wang, Y.; Subedi, S.; de Vries, H.; Doornenbal, P.; Vels, A.; Hensel, G.; Kumlehn, J.; Johnston, P.A.; Qi, X.; Blilou, I.; et al. Orthologous receptor kinases quantitatively affect the host status of barley to leaf rust fungi. Nat. Plants 2019, 5, 1129–1135. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Races | Topper | Heils Franken | Emir | Astrix | Hiproly | Varunda | Adeb Binder | Trumpf | Mazurka | Bigo | I5 | Bancroft |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PSH-33 | V | A | A | A | A | A | V | A | A | A | A | A |
| PSH-48 | V | A | A | A | A | A | A | A | A | A | A | A |
| PSH-54 | V | A | A | A | A | A | V | V | A | A | A | V |
| PSH-72 | V | V | V | V | V | V | V | V | V | V | V | V |
| PSH-100 | V | A | A | A | V | V | V | V | A | V | A | V |
| Trait | Marker | Chr | Pos (bp) * | Allele | QTL Interval | Flanking Markers | −log10(p-Value) | Effect on Trait |
|---|---|---|---|---|---|---|---|---|
| 17VA12C | HvGBSv2-7726 | 2H | 77796488 | A | 585594328 | HvGBSv2-7726 | 7.63 | −4.41 |
| HvGBSv2-9587 | 4H | 558532757 | C | 558532757 | HvGBSv2-9587 | 7.8 | −2.64 | |
| HvGBSv2-10463 | 5H | 530696479 | C | 530058811–571321539 | HvGBSv2-4369–HvGBSv2-4479 | 25.95 | 4.62 | |
| PSH-54 | HvGBSv2-4385 | 5H | 535795731 | C | 530058811–566858977 | HvGBSv2-4369–HvGBSv2-10559 | 9.79 | 4.51 |
| PSH-72 | HvGBSv2-8315 | 3H | 291597999 | C | 291597999 | HvGBSv2-8315 | 6.26 | 1.93 |
| HvGBSv2-10469 | 5H | 532621998 | A | 530696479–532621998 | HvGBSv2-10463–HvGBSv2-10469 | 7.16 | −2.24 | |
| HvGBSv2-5420 | 7H | 74751061 | A | 74751061 | HvGBSv2-5420 | 9.25 | −4.36 | |
| PSH-100 | HvGBSv2-1099 | 2H | 169669002 | A | 169669002 | HvGBSv2-1099 | 6.38 | 7.33 |
| HvGBSv2-10469 | 5H | 532621998 | A | 532621998 | HvGBSv2-10469 | 6.88 | −2.45 | |
| HvGBSv2-5420 | 7H | 74751061 | A | 74751061 | HvGBSv2-5420 | 8.05 | −4.36 | |
| 91NE9305 | HvGBSv2-520 | 1H | 400297507 | C | 400297507 | HvGBSv2-520 | 6.96 | −2.02 |
| HvGBSv2-926 | 2H | 47089419 | C | 47089419-55575144 | HvGBSv2-926–HvGBSv2-928 | 5.9 | 2.35 | |
| HvGBSv2-3492 | 4H | 556987599 | A | 556987599 | HvGBSv2-3492 | 5.84 | −2.86 | |
| HvGBSv2-10146 | 5H | 348514954 | C | 387529130-393006859 | HvGBSv2-10143–HvGBSv2-4058 | 6.28 | 2.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arifuzzaman, M.; Jost, M.; Wang, M.; Chen, X.; Perovic, D.; Park, R.F.; Rouse, M.; Forrest, K.; Hayden, M.; Khan, G.A.; et al. Mining the Australian Grains Gene Bank for Rust Resistance in Barley. Int. J. Mol. Sci. 2023, 24, 10860. https://doi.org/10.3390/ijms241310860
Arifuzzaman M, Jost M, Wang M, Chen X, Perovic D, Park RF, Rouse M, Forrest K, Hayden M, Khan GA, et al. Mining the Australian Grains Gene Bank for Rust Resistance in Barley. International Journal of Molecular Sciences. 2023; 24(13):10860. https://doi.org/10.3390/ijms241310860
Chicago/Turabian StyleArifuzzaman, Md, Matthias Jost, Meinan Wang, Xianming Chen, Dragan Perovic, Robert F. Park, Matthew Rouse, Kerrie Forrest, Matthew Hayden, Ghazanfar Abbas Khan, and et al. 2023. "Mining the Australian Grains Gene Bank for Rust Resistance in Barley" International Journal of Molecular Sciences 24, no. 13: 10860. https://doi.org/10.3390/ijms241310860