
Diseases from the Spectrum of Dermatitis and Eczema: Can “Omics” Sciences Help with Better Systematics and More Accurate Differential Diagnosis?


Abstract
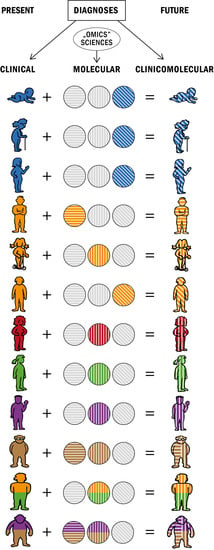
:
1. Introduction
2. Present Diagnostic Difficulties
3. How to Classify Diseases from the Spectrum of Dermatitis and Eczema (SoDE)
4. An Overview of Diseases from the Spectrum
4.1. Atopic Dermatitis (AD)
4.2. Irritant Contact Dermatitis (ICD)
4.3. Phototoxic Dermatitis
4.4. Radiodermatitis (RD)
4.5. Seborrheic Dermatitis (SD)
4.6. Allergic Contact Dermatitis (ACD)
4.7. Photoallergic Dermatitis
4.8. Protein Contact Dermatitis (PCD)
4.9. Autoimmune Dermatitis
4.10. Stasis Dermatitis
4.11. Deficiency Dermatitis
5. Discussion
6. Conclusions
7. Future Directions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balan, R.A.; Lozneanu, L.; Grigoras, A.; Caruntu, I.D.; Balan, T.A.; Giusca, S.E.; Amalinei, C. Spongiotic reaction patterns in autoimmune bullous dermatoses (Review). Exp. Ther. Med. 2021, 22, 1334. [Google Scholar] [CrossRef] [PubMed]
- Dizon, M.P.; Yu, A.M.; Singh, R.K.; Wan, J.; Chren, M.M.; Flohr, C.; Silverberg, J.I.; Margolis, D.J.; Langan, S.M.; Abuabara, K. Systematic review of atopic dermatitis disease definition in studies using routinely collected health data. Br. J. Dermatol. 2018, 178, 1280–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiewak, R. Pesticides and skin diseases in man. In Pesticides: Evaluation of Environmental Pollution; Rathore, H., Nolet, L.M., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp. 525–542. [Google Scholar]
- Dorynska, A.; Spiewak, R. Epidemiology of skin diseases from the spectrum of dermatitis and eczema. Malays. J. Dermatol. 2012, 29, 1–11. [Google Scholar]
- Fölster-Holst, R. Differential diagnoses of diaper dermatitis. Pediatr. Dermatol. 2018, 35 (Suppl. S1), s10–s18. [Google Scholar] [CrossRef] [Green Version]
- Wollenberg, A.; Barbarot, S.; Bieber, T.; Christen-Zaech, S.; Deleuran, M.; Fink-Wagner, A.; Gieler, U.; Girolomoni, G.; Lau, S.; Muraro, A.; et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: Part I. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 657–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollenberg, A.; Barbarot, S.; Bieber, T.; Christen-Zaech, S.; Deleuran, M.; Fink-Wagner, A.; Gieler, U.; Girolomoni, G.; Lau, S.; Muraro, A.; et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: Part II. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 850–878. [Google Scholar] [CrossRef] [Green Version]
- Johnston, G.A.; Exton, L.S.; Mohd Mustapa, M.F.; Slack, J.A.; Coulson, I.H.; English, J.S.; Bourke, J.F. British Association of Dermatologists’ guidelines for the management of contact dermatitis 2017. Br. J. Dermatol. 2017, 176, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Fonacier, L.; Bernstein, D.I.; Pacheco, K.; Holness, D.L.; Blessing-Moore, J.; Khan, D.; Lang, D.; Nicklas, R.; Oppenheimer, J.; Portnoy, J.; et al. Contact dermatitis: A practice parameter-update 2015. J. Allergy Clin. Immunol. Pract. 2015, 3 (Suppl. S3), S1–S39. [Google Scholar] [CrossRef]
- Naldi, L.; Diphoorn, J. Seborrhoeic dermatitis of the scalp. BMJ Clin. Evid. 2015, 2015, 1713. [Google Scholar]
- Victoire, A.; Magin, P.; Coughlan, J.; van Driel, M.L. Interventions for infantile seborrhoeic dermatitis (including cradle cap). Cochrane Database Syst. Rev. 2019, 3, CD011380. [Google Scholar] [CrossRef]
- Thyssen, J.P.; Schuttelaar, M.L.A.; Alfonso, J.H.; Andersen, K.E.; Angelova-Fischer, I.; Arents, B.W.M.; Bauer, A.; Brans, R.; Cannavo, A.; Christoffers, W.A.; et al. Guidelines for diagnosis, prevention, and treatment of hand eczema. Contact Dermat. 2022, 86, 357–378. [Google Scholar] [CrossRef] [PubMed]
- Halling-Overgaard, A.S.; Zachariae, C.; Thyssen, J.P. Management of Atopic Hand Dermatitis. Dermatol. Clin. 2017, 35, 365–372. [Google Scholar] [CrossRef]
- Mailhol, C.; Lauwers-Cances, V.; Rancé, F.; Paul, C.; Giordano-Labadie, F. Prevalence and risk factors for allergic contact dermatitis to topical treatment in atopic dermatitis: A study in 641 children. Allergy 2009, 64, 801–806. [Google Scholar] [CrossRef]
- Flohr, C.; Weiland, S.K.; Weinmayr, G.; Björkstén, B.; Bråbäck, L.; Brunekreef, B.; Büchele, G.; Clausen, M.; Cookson, W.O.; von Mutius, E.; et al. The role of atopic sensitization in flexural eczema: Findings from the International Study of Asthma and Allergies in Childhood Phase Two. J. Allergy Clin. Immunol. 2008, 121, 141–147.e4. [Google Scholar] [CrossRef] [PubMed]
- Czarnobilska, E.; Obtulowicz, K.; Dyga, W.; Spiewak, R. A half of schoolchildren with ‘ISAAC eczema’ are ill with allergic contact dermatitis. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.E.; Goldenberg, A.; Nedorost, S.; Thyssen, J.P.; Fonacier, L.; Spiewak, R. Flexural eczema versus atopic dermatitis. Dermatitis 2015, 26, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Frings, V.G.; Böer-Auer, A.; Breuer, K. Histomorphology and Immunophenotype of Eczematous Skin Lesions Revisited-Skin Biopsies Are Not Reliable in Differentiating Allergic Contact Dermatitis, Irritant Contact Dermatitis, and Atopic Dermatitis. Am. J. Dermatopathol. 2018, 40, 7–16. [Google Scholar] [CrossRef]
- Diepgen, T.L.; Ofenloch, R.F.; Bruze, M.; Bertuccio, P.; Cazzaniga, S.; Coenraads, P.J.; Elsner, P.; Gonçalo, M.; Svensson, A.; Naldi, L. Prevalence of contact allergy in the general population in different European regions. Br. J. Dermatol. 2016, 174, 319–329. [Google Scholar] [CrossRef]
- Winnicki, M.; Shear, N.H. A systematic approach to systemic contact dermatitis and symmetric drug-related intertriginous and flexural exanthema (SDRIFE): A closer look at these conditions and an approach to intertriginous eruptions. Am. J. Clin. Dermatol. 2011, 12, 171–180. [Google Scholar] [CrossRef]
- Zalewska-Janowska, A.; Spiewak, R.; Kowalski, M.L. Cutaneous Manifestation of Drug Allergy and Hypersensitivity. Immunol. Allergy Clin. N. Am. 2017, 37, 165–181. [Google Scholar] [CrossRef]
- Li, Y.; Li, L. Contact Dermatitis: Classifications and Management. Clin. Rev. Allergy Immunol. 2021, 61, 245–281. [Google Scholar] [CrossRef] [PubMed]
- Radi, G.; Campanti, A.; Diotallevi, F.; Martina, E.; Marani, A.; Offidani, A. A Systematic Review of Atopic Dermatitis: The Intriguing Journey Starting from Physiopathology to Treatment, from Laboratory Bench to Bedside. Biomedicines 2022, 25, 2700. [Google Scholar] [CrossRef] [PubMed]
- Schuler, C.F., 4th; Billi, A.C.; Maverakis, E.; Tsoi, L.C.; Gudjonsson, J.E. Novel insights into atopic dermatitis. J. Allergy Clin. Immunol. 2022, 151, 1145–1154. [Google Scholar] [CrossRef]
- Pinto, L.M.; Chiricozzi, A.; Calabrese, L.; Mannino, M.; Peris, K. Novel Therapeutic Strategies in the Topical Treatment of Atopic Dermatitis. Pharmaceutics 2022, 14, 2767. [Google Scholar] [CrossRef] [PubMed]
- Kondratuk, K.; Netravali, I.A.; Castelo-Soccio, L. Modern Interventions for Pediatric Atopic Dermatitis: An Updated Pharmacologic Approach. Dermatol. Ther. 2023, 13, 367–389. [Google Scholar] [CrossRef] [PubMed]
- David, E.; Ungar, B.; Renert-Yuval, Y.; Facheris, P.; Del Duca, E.; Guttman-Yassky, E. The evolving landscape of biologic therapies for atopic dermatitis: Present and future perspective. Clin. Exp. Allergy 2023, 53, 156–172. [Google Scholar] [CrossRef] [PubMed]
- De Simoni, E.; Rizzetto, G.; Molinelli, E.; Lucarini, G.; Mattioli-Belmonte, M.; Capodaglio, I.; Ferretti, G.; Bacchetti, T.; Offidani, A.; Simonetti, O. Metabolic Comorbidities in Pediatric Atopic Dermatitis: A Narrative Review. Life 2022, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Mesjasz, A.; Zawadzka, M.; Chałubiński, M.; Trzeciak, M. Is Atopic Dermatitis Only a Skin Disease? Int. J. Mol. Sci. 2023, 24, 837. [Google Scholar] [CrossRef]
- Szczepańska, M.; Blicharz, L.; Nowaczyk, J.; Makowska, K.; Goldust, M.; Waśkiel-Burnat, A.; Czuwara, J.; Samochocki, Z.; Rudnicka, L. The Role of the Cutaneous Mycobiome in Atopic Dermatitis. J. Fungi 2022, 8, 1153. [Google Scholar] [CrossRef]
- Hrestak, D.; Matijašić, M.; Čipčić Paljetak, H.; Ledić Drvar, D.; Ljubojević Hadžavdić, S.; Perić, M. Skin Microbiota in Atopic Dermatitis. Int. J. Mol. Sci. 2022, 23, 3503. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, L.; Gao, S.; Su, S. Guidelines for the Management of Atopic Dermatitis in Children: A Systematic Review. Int. Arch. Allergy Immunol. 2023, 184, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Hanifin, J.M.; Rajka, G. Diagnostic features of atopic dermatitis. Acta Derm. Venereol. Suppl. 1980, 92, 44–47. [Google Scholar] [CrossRef]
- Hanifin, J.M. Diagnostic criteria for atopic dermatitis: Consider the context. Arch. Dermatol. 1999, 135, 1551. [Google Scholar] [CrossRef] [PubMed]
- Rajka, G. Atopic Dermatitis; W. B. Saunders: London, UK, 1975; Volume 3, p. 2. [Google Scholar]
- Johansson, S.G.; Hourihane, J.O.; Bousquet, J.; Bruijnzeel-Koomen, C.; Dreborg, S.; Haahtela, T.; Kowalski, M.L.; Mygind, N.; Ring, J.; van Cauwenberge, P.; et al. EAACI (the European Academy of Allergology and Cinical Immunology) nomenclature task force: A revised nomenclature for allergy: An EAACI position statement from the EAACI nomenclature task force. Allergy 2001, 56, 813–824. [Google Scholar] [CrossRef]
- Bos, J.D.; Brenninkmeijer, E.E.; Schram, M.E.; Middelkamp-Hup, M.A.; Spuls, P.I.; Smitt, J.H. Atopic eczema or atopiform dermatitis. Exp. Dermatol. 2010, 19, 325–331. [Google Scholar] [CrossRef]
- Bax, M.C.; Flodmark, O.; Tydeman, C. From syndrome toward disease. Dev. Med. Child Neurol. Suppl. 2007, 109, 39–41. [Google Scholar] [CrossRef]
- Kaufman, B.P.; Guttman-Yassky, E.; Alexis, A.F. Atopic dermatitis in diverse racial and ethnic groups—Variations in epidemiology, genetics, clinical presentation and treatment. Exp. Dermatol. 2018, 27, 340–357. [Google Scholar] [CrossRef] [Green Version]
- Chiricozzi, A.; Maurelli, M.; Calabrese, L.; Peris, K.; Girolomoni, G. Overview of Atopic Dermatitis in Different Ethnic Groups. J. Clin. Med. 2023, 12, 2701. [Google Scholar] [CrossRef]
- Tokura, Y.; Hayano, S. Subtypes of atopic dermatitis: From phenotype to endotype. Allergol. Int. 2022, 71, 14–24. [Google Scholar] [CrossRef]
- Martin, P.E.; Eckert, J.K.; Koplin, J.J.; Lowe, A.J.; Gurrin, L.C.; Dharmage, S.C.; Vuillermin, P.; Tang, M.L.; Ponsonby, A.L.; Matheson, M.; et al. Which infants with eczema are at risk of food allergy? Results from a population-based cohort. Clin. Exp. Allergy 2015, 45, 255–264. [Google Scholar] [CrossRef]
- Holm, J.G.; Agner, T.; Sand, C.; Thomsen, S.F. Omalizumab for atopic dermatitis: Case series and a systematic review of the literature. Int. J. Dermatol. 2017, 56, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Holm, J.G.; Thomsen, S.F. Omalizumab for atopic dermatitis: Evidence for and against its use. G. Ital. Dermatol. Venereol. 2019, 154, 480–487. [Google Scholar] [CrossRef]
- Nečas, M.; Vašků, V.; Březinová, E. Lack of omalizumab efficacy in severe atopic dermatitis with extremely elevated IgE levels: Two case reports and a literature review. Acta Dermatovenerol. Alp. Pannonica Adriat. 2019, 28, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.; Cro, S.; Cornelius, V.; Jahan, R.; Radulovic, S.; Lack, G. Omalizumab for Severe Atopic Dermatitis in 4- to 19-Year-Olds: The ADAPT RCT; National Institute for Health and Care Research: Southampton, UK, 2022. [Google Scholar]
- Thijs, J.L.; Strickland, I.; Bruijnzeel-Koomen, C.A.; Nierkens, S.; Giovannone, B.; Csomor, E.; Sellman, B.R.; Mustelin, T.; Sleeman, M.A.; de Bruin-Weller, M.S.; et al. Moving toward endotypes in atopic dermatitis: Identification of patient clusters based on serum biomarker analysis. J. Allergy Clin. Immunol. 2017, 140, 730–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, D.S.; Nierkens, S.; Knol, E.F.; Giovannone, B.; Delemarre, E.M.; van der Schaft, J.; van Wijk, F.; de Bruin-Weller, M.S.; Drylewicz, J.; Thijs, J.L. Confirmation of multiple endotypes in atopic dermatitis based on serum biomarkers. J. Allergy Clin. Immunol. 2021, 147, 189–198. [Google Scholar] [CrossRef]
- Bakker, D.S.; de Graaf, M.; Nierkens, S.; Delemarre, E.M.; Knol, E.; van Wijk, F.; de Bruin-Weller, M.S.; Drylewicz, J.; Thijs, J.L. Unraveling heterogeneity in pediatric atopic dermatitis: Identification of serum biomarker based patient clusters. J. Allergy Clin. Immunol. 2022, 149, 125–134. [Google Scholar] [CrossRef]
- Afghani, J.; Traidl-Hoffmann, C.; Schmitt-Kopplin, P.; Reiger, M.; Mueller, C. An Overview of the Latest Metabolomics Studies on Atopic Eczema with New Directions for Study. Int. J. Mol. Sci. 2022, 23, 8791. [Google Scholar] [CrossRef]
- Brunner, P.M.; Israel, A.; Leonard, A.; Pavel, A.B.; Kim, H.J.; Zhang, N.; Czarnowicki, T.; Patel, K.; Murphrey, M.; Ramsey, K.; et al. Distinct transcriptomic profiles of early-onset atopic dermatitis in blood and skin of pediatric patients. Ann. Allergy Asthma Immunol. 2019, 122, 318–330.e3. [Google Scholar] [CrossRef]
- Pavel, A.B.; Zhou, L.; Diaz, A.; Ungar, B.; Dan, J.; He, H.; Estrada, Y.D.; Xu, H.; Fernandes, M.; Renert-Yuval, Y.; et al. The proteomic skin profile of moderate-to-severe atopic dermatitis patients shows an inflammatory signature. J. Am. Acad. Dermatol. 2020, 82, 690–699. [Google Scholar] [CrossRef]
- Goleva, E.; Calatroni, A.; LeBeau, P.; Berdyshev, E.; Taylor, P.; Kreimer, S.; Cole, R.N.; Leung, D.Y.M. Skin tape proteomics identifies pathways associated with transepidermal water loss and allergen polysensitization in atopic dermatitis. J. Allergy Clin. Immunol. 2020, 146, 1367–1378. [Google Scholar] [CrossRef]
- Rojahn, T.B.; Vorstandlechner, V.; Krausgruber, T.; Bauer, W.M.; Alkon, N.; Bangert, C.; Thaler, F.M.; Sadeghyar, F.; Fortelny, N.; Gernedl, V.; et al. Single-cell transcriptomics combined with interstitial fluid proteomics defines cell type-specific immune regulation in atopic dermatitis. J. Allergy Clin. Immunol. 2020, 146, 1056–1069. [Google Scholar] [CrossRef]
- Park, J.; Lutz, S.M.; Choi, S.; Lee, S.; Park, S.C.; Kim, K.; Choi, H.; Park, H.; Lee, S.Y.; Weiss, S.T.; et al. Multi-omics analyses implicate EARS2 in the pathogenesis of atopic dermatitis. Allergy 2021, 76, 2602–2604. [Google Scholar] [CrossRef] [PubMed]
- Wongvibulsin, S.; Sutaria, N.; Kannan, S.; Alphonse, M.P.; Belzberg, M.; Williams, K.A.; Brown, I.D.; Choi, J.; Roh, Y.S.; Pritchard, T.; et al. Transcriptomic analysis of atopic dermatitis in African Americans is characterized by Th2/Th17-centered cutaneous immune activation. Sci. Rep. 2021, 27, 11175. [Google Scholar] [CrossRef]
- Morelli, P.; Gaspari, M.; Gabriele, C.; Dastoli, S.; Bennardo, L.; Pavel, A.B.; Patruno, C.; Del Duca, E.; Nistico, S.P. Proteomic analysis from skin swabs reveals a new set of proteins identifying skin impairment in atopic dermatitis. Exp. Dermatol. 2021, 30, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Shima, K.; Inoue, T.; Uehara, Y.; Iwamura, M.; Fukagawa, S.; Kuwano, T.; Tanida, K.; Takada, N.; Saito-Abe, M.; Yamamoto-Hanada, K.; et al. Non-invasive transcriptomic analysis using mRNAs in skin surface lipids obtained from children with mild-to-moderate atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1477–1485. [Google Scholar] [CrossRef]
- Zhou, X.; Xiao, B.; Zeng, J.; Zhou, L.; Wang, X.; Zhao, S.; Li, X.; Zhang, H.; Su, Y.; Zhao, Z.; et al. Identification of Cofilin-1 as a novel biomarker of atopic dermatitis using iTRAQ quantitative proteomics. J. Clin. Lab. Anal. 2022, 36, e24751. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, R.; Halstead, S.; McKeone, D.; Hicks, S.D. Understanding immunological origins of atopic dermatitis through multi-omic analysis. Pediatr. Allergy Immunol. 2022, 33, e13817. [Google Scholar] [CrossRef]
- Koch, M.; Kockmann, T.; Rodriguez, E.; Wehkamp, U.; Hiebert, P.; Ben-Yehuda Greenwald, M.; Stölzl, D.; Beer, H.D.; Tschachler, E.; Weidinger, S.; et al. Quantitative Proteomics Identifies Reduced NRF2 Activity and Mitochondrial Dysfunction in Atopic Dermatitis. J. Investig. Dermatol. 2023, 143, 220–231.e7. [Google Scholar] [CrossRef]
- Ilves, L.; Ottas, A.; Kaldvee, B.; Abram, K.; Soomets, U.; Zilmer, M.; Jaks, V.; Kingo, K. Metabolomic Differences between the Skin and Blood Sera of Atopic Dermatitis and Psoriasis. Int. J. Mol. Sci. 2022, 23, 13001. [Google Scholar] [CrossRef]
- Sutaria, N.; Alphonse, M.P.; Roh, Y.S.; Choi, J.; Parthasarathy, V.; Deng, J.; Bordeaux, Z.A.; Taylor, M.T.; Pritchard, T.; Kim, N.; et al. Cutaneous Transcriptomics Identifies Fibroproliferative and Neurovascular Gene Dysregulation in Prurigo Nodularis Compared with Psoriasis and Atopic Dermatitis. J. Investig. Dermatol. 2022, 142, 2537–2540. [Google Scholar] [CrossRef]
- Wong, L.S.; Yen, Y.T. Chronic Nodular Prurigo: An Update on the Pathogenesis and Treatment. Int. J. Mol. Sci. 2022, 23, 12390. [Google Scholar] [CrossRef] [PubMed]
- Agelopoulos, K.; Renkhold, L.; Wiegmann, H.; Dugas, M.; Süer, A.; Zeidler, C.; Schmelz, M.; Pereira, M.P.; Ständer, S. Transcriptomic, Epigenomic, and Neuroanatomic Signatures Differ in Chronic Prurigo, Atopic Dermatitis, and Brachioradial Pruritus. J. Investig. Dermatol. 2023, 143, 264–272.e3. [Google Scholar] [CrossRef] [PubMed]
- Bains, S.N.; Nash, P.; Fonacier, L. Irritant Contact Dermatitis. Clin. Rev. Allergy Immunol. 2019, 56, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Nixon, R. Irritant Contact Dermatitis—A Review. Curr. Dermatol. Rep. 2022, 11, 41–51. [Google Scholar] [CrossRef]
- Kendall, A.C.; Pilkington, S.M.; Sassano, G.; Rhodes, L.E.; Nicolaou, A. N-Acyl ethanolamide and eicosanoid involvement in irritant dermatitis. Br. J. Dermatol. 2016, 175, 163–171. [Google Scholar] [CrossRef]
- Fartasch, M.; Schnetz, E.; Diepgen, T.L. Characterization of detergent-induced barrier alterations—Effect of barrier cream on irritation. J. Investig. Dermatol. Symp. Proc. 1998, 3, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutlubay, Z.; Sevim, A.; Engin, B.; Tüzün, Y. Photodermatoses, including phototoxic and photoallergic reactions (internal and external). Clin. Dermatol. 2014, 32, 73–79. [Google Scholar] [CrossRef]
- Spiewak, R. The substantial differences between photoallergic and phototoxic reactions. Ann. Agric. Environ. Med. 2012, 19, 888–889. [Google Scholar]
- Hofmann, G.A.; Weber, B. Drug-induced photosensitivity: Culprit drugs, potential mechanisms and clinical consequences. J. Dtsch. Dermatol. Ges. 2021, 19, 19–29. [Google Scholar] [CrossRef]
- Hegedus, F.; Mathew, L.M.; Schwartz, R.A. Radiation dermatitis: An overview. Int. J. Dermatol. 2017, 56, 909–914. [Google Scholar] [CrossRef]
- Xie, G.; Ao, X.; Lin, T.; Zhou, G.; Wang, M.; Wang, H.; Chen, Y.; Li, X.; Xu, B.; He, W.; et al. E-Cadherin-Mediated Cell Contact Controls the Epidermal Damage Response in Radiation Dermatitis. J. Investig. Dermatol. 2017, 137, 1731–1739. [Google Scholar] [CrossRef] [PubMed]
- Rognoni, E.; Walko, G. The Roles of YAP/TAZ and the Hippo Pathway in Healthy and Diseased Skin. Cells 2019, 8, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, Y.M.; Egeberg, A. Seborrhoeic dermatitis—Understood or understudied? Br. J. Dermatol. 2019, 181, 659. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.L.; Pye, R.J. Seborrhoea is not a feature of seborrhoeic dermatitis. Br. Med. J. 1983, 286, 1169–1170. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, C.G.; Burkhart, C.N. Qualitative, not quantitative, alterations of sebum important in seborrhoeic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 441. [Google Scholar] [CrossRef]
- Wikramanayake, T.C.; Borda, L.J.; Miteva, M.; Paus, R. Seborrheic dermatitis—Looking beyond Malassezia. Exp. Dermatol. 2019, 28, 991–1001. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Feng, Y.; Liu, C.; Yang, Z.; de Hoog, S.; Qu, Y.; Chen, B.; Li, D.; Xiong, H.; Shi, D. Presence of Malassezia Hyphae Is Correlated with Pathogenesis of Seborrheic Dermatitis. Microbiol. Spectr. 2022, 10, e0116921. [Google Scholar] [CrossRef]
- Goularte-Silva, V.; Paulino, L.C. Ketoconazole beyond antifungal activity: Bioinformatics-based hypothesis on lipid metabolism in dandruff and seborrheic dermatitis. Exp. Dermatol. 2022, 31, 821–822. [Google Scholar] [CrossRef]
- Foley, P.; Zuo, Y.; Plunkett, A.; Merlin, K.; Marks, R. The frequency of common skin conditions in preschool-aged children in Australia: Seborrheic dermatitis and pityriasis capitis (cradle cap). Arch. Dermatol. 2003, 139, 318–322. [Google Scholar] [CrossRef] [Green Version]
- Yates, V.M.; Kerr, R.E.; MacKie, R.M. Early diagnosis of infantile seborrhoeic dermatitis and atopic dermatitis—Clinical features. Br. J. Dermatol. 1983, 108, 633–638. [Google Scholar] [CrossRef]
- Mills, K.J.; Hu, P.; Henry, J.; Tamura, M.; Tiesman, J.P.; Xu, J. Dandruff/seborrhoeic dermatitis is characterized by an inflammatory genomic signature and possible immune dysfunction: Transcriptional analysis of the condition and treatment effects of zinc pyrithione. Br. J. Dermatol. 2012, 166 (Suppl. S2), 33–40. [Google Scholar] [CrossRef] [PubMed]
- Mraz, V.; Geisler, C.; Bonefeld, C.M. Dendritic Epidermal T Cells in Allergic Contact Dermatitis. Front. Immunol. 2020, 11, 874. [Google Scholar] [CrossRef]
- Lefevre, M.A.; Vocanson, M.; Nosbaum, A. Role of tissue-resident memory T cells in the pathophysiology of allergic contact dermatitis. Curr. Opin. Allergy Clin. Immunol. 2021, 21, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Kalicińska, J.; Wiśniowska, B.; Polak, S.; Spiewak, R. Artificial Intelligence That Predicts Sensitizing Potential of Cosmetic Ingredients with Accuracy Comparable to Animal and In Vitro Tests-How Does the Infotechnomics Compare to Other “Omics” in the Cosmetics Safety Assessment? Int. J. Mol. Sci. 2023, 24, 6801. [Google Scholar] [CrossRef]
- Lepoittevin, J.P. Metabolism versus chemical transformation or pro- versus prehaptens? Contact Dermat. 2006, 54, 73–74. [Google Scholar] [CrossRef]
- de Groot, A. Linalool Hydroperoxides. Dermatitis 2019, 30, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.; Hotchkiss, S.A.; Pease, C.K. Cinnamic compound metabolism in human skin and the role metabolism may play in determining relative sensitisation potency. J. Dermatol. Sci. 2003, 31, 9–19. [Google Scholar] [CrossRef]
- Martin, S.F.; Rustemeyer, T.; Thyssen, J.P. Recent advances in understanding and managing contact dermatitis. F1000Res 2018, 7, 810. [Google Scholar] [CrossRef] [Green Version]
- Gru, A.A.; Salavaggione, A.L. Common spongiotic dermatoses. Semin. Diagn. Pathol. 2017, 34, 226–236. [Google Scholar] [CrossRef]
- Silvestre, M.C.; Sato, M.N.; Reis, V.M.S.D. Innate immunity and effector and regulatory mechanisms involved in allergic contact dermatitis. An. Bras. Dermatol. 2018, 93, 242–250. [Google Scholar] [CrossRef] [Green Version]
- Brys, A.K.; Rodriguez-Homs, L.G.; Suwanpradid, J.; Atwater, A.R.; MacLeod, A.S. Shifting Paradigms in Allergic Contact Dermatitis: The Role of Innate Immunity. J. Investig. Dermatol. 2020, 140, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Azeem, M.; Kader, H.; Kerstan, A.; Hetta, H.F.; Serfling, E.; Goebeler, M.; Muhammad, K. Intricate Relationship Between Adaptive and Innate Immune System in Allergic Contact Dermatitis. Yale J. Biol. Med. 2020, 93, 699–709. [Google Scholar]
- Andersen, K.E.; Hjorth, N.; Menné, T. The baboon syndrome: Systemically-induced allergic contact dermatitis. Contact Dermat. 1984, 10, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, A.; Kawashima, H.; Okubo, Y.; Hoshika, A. A new proposal for a clinical-oriented subclassification of baboon syndrome and a review of baboon syndrome. Asian Pac. J. Allergy Immunol. 2011, 29, 150–160. [Google Scholar] [PubMed]
- Nowowiejska, J.; Baran, A.; Flisiak, I. Baboon syndrome. Postep. Dermatol. Alergol. 2022, 39, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, N.; Kimber, I.; Williams, J.; Maxwell, G. Skin sensitization: Uncertainties, challenges, and opportunities for improved risk assessment. Contact Dermat. 2019, 80, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Chilton, M.L.; Api, A.M.; Foster, R.S.; Gerberick, G.F.; Lavelle, M.; Macmillan, D.S.; Na, M.; O’Brien, D.; O’Leary-Steele, C.; Patel, M.; et al. Updating the Dermal Sensitisation Thresholds using an expanded dataset and an in silico expert system. Regul. Toxicol. Pharmacol. 2022, 133, 105200. [Google Scholar] [CrossRef]
- Sebastião, A.I.; Ferreira, I.; Brites, G.; Silva, A.; Neves, B.M.; Teresa Cruz, M. NLRP3 Inflammasome and Allergic Contact Dermatitis: A Connection to Demystify. Pharmaceutics 2020, 12, 867. [Google Scholar] [CrossRef]
- Kiecka, A.; Macura, B.; Szczepanik, M. Modulation of allergic contact dermatitis via gut microbiota modified by diet, vitamins, probiotics, prebiotics, and antibiotics. Pharmacol. Rep. 2023, 75, 236–248. [Google Scholar] [CrossRef]
- Esser, P.R.; Huber, M.; Martin, S.F. Endoplasmic reticulum stress and the inflammatory response in allergic contact dermatitis. Eur. J. Immunol. 2023; e2249984, online ahead of print. [Google Scholar] [CrossRef]
- Höper, T.; Mussotter, F.; Haase, A.; Luch, A.; Tralau, T. Application of proteomics in the elucidation of chemical-mediated allergic contact dermatitis. Toxicol. Res. 2017, 6, 595–610. [Google Scholar] [CrossRef] [Green Version]
- Honari, G. Photoallergy. Rev Environ Health 2014, 29, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.F.; Rato, M.; Martins, C. Drug-induced photosensitivity: Photoallergic and phototoxic reactions. Clin. Dermatol. 2016, 34, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Ludriksone, L.; Elsner, P. Adverse Reactions to Sunscreens. Curr. Probl. Dermatol. 2021, 55, 223–235. [Google Scholar] [CrossRef] [PubMed]
- European Multicentre Photopatch Test Study (EMCPPTS) Taskforce. A European multicentre photopatch test study. Br. J. Dermatol. 2012, 166, 1002–1009. [Google Scholar] [CrossRef]
- Barbaud, A. Mechanism and diagnosis of protein contact dermatitis. Curr. Opin. Allergy Clin. Immunol. 2020, 20, 117–121. [Google Scholar] [CrossRef]
- Alfonso, J.H.; Afanou, A.K.; Holm, J.O.; Stylianou, E. Skin bioengineering in the diagnosis of occupational protein contact dermatitis. Occup. Med. 2020, 70, 282–285. [Google Scholar] [CrossRef]
- Pesonen, M.; Koskela, K.; Aalto-Korte, K. Contact urticaria and protein contact dermatitis in the Finnish Register of Occupational Diseases in a period of 12 years. Contact Dermat. 2020, 83, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, A. The question of sensitiveness to non-bacterial toxins and proteins. Br. J. Dermatol. 1922, 34, 331–338. [Google Scholar] [CrossRef]
- Young, E.; Bruijnzeel-Koomen, C.; Berrens, L. Delayed type hypersensitivity in atopic dermatitis. Acta Derm. Venereol. Suppl. 1985, 114, 77–81. [Google Scholar]
- Yu, B.; Sawai, T.; Uehara, M.; Ishida, T.; Horiike, K.; Nozaki, M. Immediate hypersensitivity skin reactions to human dander in atopic dermatitis. Partial purification and characterization of human dander allergens. Arch. Dermatol. 1988, 124, 1530–1533. [Google Scholar] [CrossRef]
- Ivert, L.U.; Wahlgren, C.F.; Lindelöf, B.; Dal, H.; Bradley, M.; Johansson, E.K. Association between atopic dermatitis and autoimmune diseases: A population-based case-control study. Br. J. Dermatol. 2021, 185, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Chester, J.; Kaleci, S.; Liberati, S.; Alicandro, T.; Rivi, M.; Bonzano, L.; Guanti, M.; Andreone, P.; Pellacani, G. Atopic dermatitis associated with autoimmune, cardiovascular and mental health comorbidities: A systematic review and meta-analysis. Eur. J. Dermatol. 2022, 32, 34–48. [Google Scholar] [CrossRef]
- Floca, E.; Gaga, R.; Sur, G.; Lupan, I.; Armat, I.; Samasca, G.; Sur, L.M. A new autoimmune disease: Atopic dermatitis in children. Allergol. Immunopathol. 2022, 50, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Lipman, Z.M.; Labib, A.; Vander Does, A.; Yosipovitch, G. Autoimmune Progesterone Dermatitis: A Systematic Review. Dermatitis 2022, 33, 249–256. [Google Scholar] [CrossRef] [PubMed]
- James, T.; Ghaferi, J.; LaFond, A. The histopathologic features of autoimmune progesterone dermatitis. J. Cutan. Pathol. 2017, 44, 70–74. [Google Scholar] [CrossRef] [Green Version]
- Halevy, S.; Cohen, A.D.; Lunenfeld, E.; Grossman, N. Autoimmune progesterone dermatitis manifested as erythema annulare centrifugum: Confirmation of progesterone sensitivity by in vitro interferon-gamma release. J. Am. Acad. Dermatol. 2002, 47, 311–313. [Google Scholar] [CrossRef]
- Lee, M.K.; Lee, W.Y.; Yong, S.J.; Shin, K.C.; Lee, S.N.; Lee, S.J.; Lee, J.H.; Kim, S.H. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis. Allergy Asthma Immunol. Res. 2011, 3, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Banwaith, T.; Acharya, J. Autoimmune Progesterone Dermatitis Imitating Eczematous Dermatitis. J. Clin. Dermatol. Ther. 2002, 8, 0100. [Google Scholar] [CrossRef]
- Vasconcelos, C.; Xavier, P.; Vieira, A.P.; Martinho, M.; Rodrigues, J.; Bodas, A.; Barros, M.A.; Mesquita-Guimaraes, J. Autoimmune progesterone urticaria. Gynecol. Endocrinol. 2000, 14, 245–247. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Jindal, R.; Chauhan, P. Autoimmune progesterone dermatitis presenting as fixed drug eruption. BMJ Case Rep. 2019, 12, e231873. [Google Scholar] [CrossRef]
- Aghazadeh, N.; Berry, N.A.; Torgerson, R.R.; Park, M.A.; Davis, D.M.R. Autoimmune progesterone dermatitis: A retrospective case series. Int. J. Womens Dermatol. 2022, 8, e009. [Google Scholar] [CrossRef] [PubMed]
- Yotsumoto, S.; Shimomai, K.; Hashiguchi, T.; Uchimiya, H.; Usuki, K.; Nishi, M.; Kanekura, T.; Kanzaki, T. Estrogen dermatitis: A dendritic-cell-mediated allergic condition. Dermatology 2003, 207, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Zachary, C.; Fackler, N.; Juhasz, M.; Pham, C.; Mesinkovska, N.A. Catamenial dermatoses associated with autoimmune, inflammatory, and systemic diseases: A systematic review. Int. J. Womens Dermatol. 2019, 5, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Sippel, K.; Mayer, D.; Ballmer, B.; Dragieva, G.; Läuchli, S.; French, L.E.; Hafner, J. Evidence that venous hypertension causes stasis dermatitis. Phlebology 2011, 26, 361–365. [Google Scholar] [CrossRef]
- Shaikh, S.; Patel, P.M.; Armbrecht, E.S.; Hurley, M.Y. Patient survey reports association between compression stocking use adherence and stasis dermatitis flare frequency. J. Am. Acad. Dermatol. 2021, 84, 1485–1487. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Kursewicz, C.D.; Fayne, R.A.; Nanda, S.; Shah, S.M.; Nattkemper, L.; Yokozeki, H.; Yosipovitch, G. Mechanisms of Itch in Stasis Dermatitis: Significant Role of IL-31 from Macrophages. J. Investig. Dermatol. 2020, 140, 850–859.e3. [Google Scholar] [CrossRef]
- Yosipovitch, G.; Nedorost, S.T.; Silverberg, J.I.; Friedman, A.J.; Canosa, J.M.; Cha, A. Stasis Dermatitis: An Overview of Its Clinical Presentation, Pathogenesis, and Management. Am. J. Clin. Dermatol. 2023, 24, 275–286. [Google Scholar] [CrossRef]
- Hashimoto, T.; Yokozeki, H.; Karasuyama, H.; Satoh, T. IL-31-generating network in atopic dermatitis comprising macrophages, basophils, thymic stromal lymphopoietin, and periostin. J. Allergy Clin. Immunol. 2023, 151, 737–746.e6. [Google Scholar] [CrossRef]
- Silverberg, J.I.; Hou, A.; Warshaw, E.M.; Maibach, H.I.; Belsito, D.V.; DeKoven, J.G.; Zug, K.A.; Taylor, J.S.; Sasseville, D.; Fransway, A.F.; et al. Prevalence and trend of allergen sensitization in patients with a diagnosis of stasis dermatitis referred for patch testing, North American contact dermatitis group data, 2001–2016. Arch. Dermatol. Res. 2022, 314, 857–867. [Google Scholar] [CrossRef]
- Ogawa, Y.; Kinoshita, M.; Shimada, S.; Kawamura, T. Zinc and Skin Disorders. Nutrients 2018, 11, 199. [Google Scholar] [CrossRef] [Green Version]
- Glutsch, V.; Hamm, H.; Goebeler, M. Zinc and skin: An update. J. Dtsch. Dermatol. Ges. 2019, 17, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, M.; Philippidou, M.; Duncan, M.; Goolamali, S.; Basu, T.; Walsh, S. Dietary deprivation during the COVID-19 pandemic producing acquired vulval zinc-deficiency dermatitis. Clin. Exp. Dermatol. 2021, 46, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Hattangdi-Haridas, S.; Lanham-New, S.A.; Wong, W.H.S.; Ho, M.H.K.; Darling, A.L. Vitamin D Deficiency and Effects of Vitamin D Supplementation on Disease Severity in Patients with Atopic Dermatitis: A Systematic Review and Meta-Analysis in Adults and Children. Nutrients 2019, 11, 1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Liu, J.; Li, W.; Zheng, H.; Dai, H.; Qiu, G.; Yu, D.; Yao, D.; Yin, X. Causal Associations between Vitamin D Levels and Psoriasis, Atopic Dermatitis, and Vitiligo: A Bidirectional Two-Sample Mendelian Randomization Analysis. Nutrients 2022, 11, 5284. [Google Scholar] [CrossRef]
- Brożyna, A.A.; Slominski, R.M.; Nedoszytko, B.; Zmijewski, M.A.; Slominski, A.T. Vitamin D Signaling in Psoriasis: Pathogenesis and Therapy. Int. J. Mol. Sci. 2022, 23, 8575. [Google Scholar] [CrossRef]
- Carlberg, C. A Pleiotropic Nuclear Hormone Labelled Hundred Years Ago Vitamin D. Nutrients 2022, 15, 171. [Google Scholar] [CrossRef] [PubMed]
- Gallo, D.; Baci, D.; Kustrimovic, N.; Lanzo, N.; Patera, B.; Tanda, M.L.; Piantanida, E.; Mortara, L. How Does Vitamin D Affect Immune Cells Crosstalk in Autoimmune Diseases? Int. J. Mol. Sci. 2023, 24, 4689. [Google Scholar] [CrossRef]
- Sirbe, C.; Rednic, S.; Grama, A.; Pop, T.L. An Update on the Effects of Vitamin D on the Immune System and Autoimmune Diseases. Int. J. Mol. Sci. 2022, 23, 9784. [Google Scholar] [CrossRef]
- Ellison, D.L.; Moran, H.R. Vitamin D: Vitamin or Hormone? Nurs. Clin. N. Am. 2021, 56, 47–57. [Google Scholar] [CrossRef]
- Ono, E.; Murota, H.; Mori, Y.; Yoshioka, Y.; Nomura, Y.; Munetsugu, T.; Yokozeki, H.; Katayama, I. Sweat Glucose and GLUT2 Expression in Atopic Dermatitis: Implication for Clinical Manifestation and Treatment. PLoS ONE 2018, 13, e0195960. [Google Scholar] [CrossRef] [Green Version]
- Sokołowska-Wojdyło, M.; Kłudkowska, J.; Olszewska, B.; Seredyńska, J.; Biernat, W.; Błażewicz, I.; Rustowska-Rogowska, A.; Nowicki, R.J. The first case of drug-induced pseudoscleroderma and eczema craquelé related to nab-paclitaxel pancreatic adenocarcinoma treatment. Postepy Dermatol. Alergol. 2018, 35, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.; Gu, X.; Luo, X.; Al-Mousa, H.; Arnaout, R.; Al-Saud, B.L.; Lopata, A.; Li, L.; Dasouki, M.; Rahman, A.M.A. Metabolomics Distinguishes DOCK8 Deficiency from Atopic Dermatitis: Towards a Biomarker Discovery. Metabolites 2019, 9, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Shaikhly, T.; Ochs, H.D. Hyper IgE syndromes: Clinical and molecular characteristics. Immunol. Cell Biol. 2019, 97, 368–379. [Google Scholar] [CrossRef]
- Pan, C.; Zhao, A.; Li, M. Atopic Dermatitis-like Genodermatosis: Disease Diagnosis and Management. Diagnostics 2022, 12, 2177. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Saeidian, A.H.; Youssefian, L.; Hsu, S.; Vahidnezhad, H.; Uitto, J. Acquired ichthyosis, asteatotic dermatitis or xerosis? An update on pathoetiology and drug-induced associations. J. Eur. Acad. Dermatol. Venereol. 2023, 37, 47–56. [Google Scholar] [CrossRef]
- Munera-Campos, M.; Chicharro, P.; Gonzalez Quesada, A.; Flórez Menéndez, Á.; de la Cueva Dobao, P.; Gimenez Arnau, A.M.; Gilaberte Calzada, Y.; Rodríguez Serna, M.; Montero-Vilchez, T.; Silvestre, J.F.; et al. BIOBADATOP Spanish Atopic Dermatitis Registry: Description and Early Findings. Actas Dermosifiliogr. 2023, 114, 479–487. [Google Scholar] [CrossRef]
- Simpson, E.L.; De Benedetto, A.; Boguniewicz, M.; Ong, P.Y.; Lussier, S.; Villarreal, M.; Schneider, L.C.; Paller, A.S.; Guttman-Yassky, E.; Hanifin, J.M.; et al. Phenotypic and Endotypic Determinants of Atopic Dermatitis Severity from the Atopic Dermatitis Research Network (ADRN) Registry. J. Allergy Clin. Immunol. Pract. 2023; online ahead of print. [Google Scholar] [CrossRef]

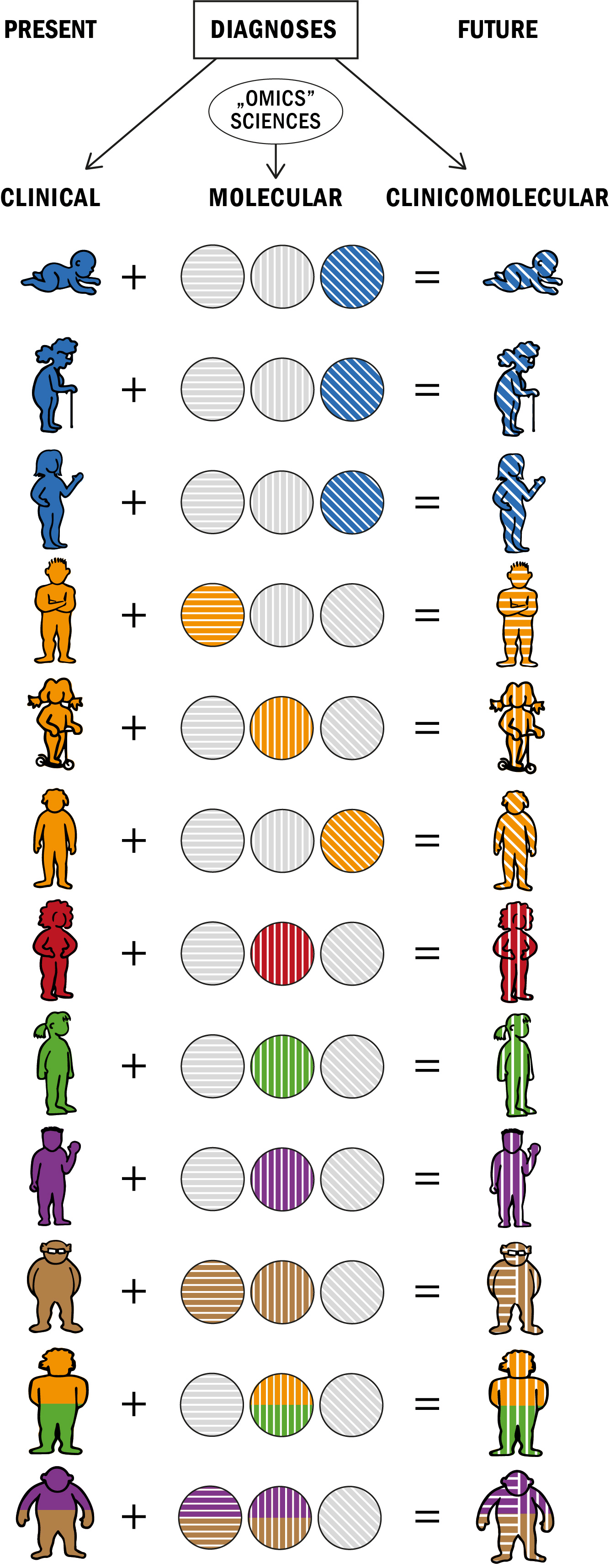{kind=link}
| Etiopathology Classes | Examples of Clinical Diagnoses 1 |
|---|---|
| No known/detectable triggers | Intrinsic atopic dermatitis (endogenous eczema) |
| Triggered by exogenous factors without the involvement of specific hypersensitivity | Irritant contact dermatitis Winter dermatitis (hand, foot, and generalized) Microtrauma dermatitis Friction dermatitis Phototoxic dermatitis Radiodermatitis (radiation dermatitis) Seborrheic dermatitis |
| Triggered by exogenous factors with the involvement of specific immunological hypersensitivity | Extrinsic atopic dermatitis (exogenous eczema) Allergic contact dermatitis SDRIFE/SRACD 2 Photoallergic dermatitis Protein contact dermatitis |
| Autoimmune reactions | Autoimmune dermatitis, including: Autoimmune progesterone dermatitis Autoimmune estrogen dermatitis |
| Homeostatic imbalance | Stasis dermatitis Deficiency dermatitis |
| Customary Term | Reason for Use |
|---|---|
| Airborne dermatitis | Specific route of exposure. |
| Asteatotic eczema (eczema craquelé; asteatotic dermatitis) | Characteristic clinical picture: skin fissures occurring in irregular, polygonal or curvilinear patterns, referred to as “crazy paving”. |
| Connubial dermatitis (consort dermatitis; dermatitis by proxy) | Specific route of exposure (allergic symptoms in a seemingly non-exposed person due to transmission of haptens from an exposed person in close surroundings, typically a member of the same household). |
| Dyshidrotic eczema (dyshidrotic dermatitis, pompholyx) | Characteristic clinical picture (vesicles or bullae) and localization (palms and soles); persistence and recurrence. |
| Foot dermatitis (foot eczema) | Characteristic localization, similarity of clinical picture regardless of etiopathogenesis, frequent polyetiology, negative impact on daily routines and work and resistance to treatment. |
| Hand eczema (hand dermatitis) | Characteristic localization, similarity of clinical picture regardless of etiopathogenesis, frequent polyetiology, negative impact on daily routines and work and resistance to treatment. |
| Nummular eczema (nummular dermatitis) | Characteristic clinical picture, persistence and recurrence. |
| Occupational dermatitis (occupational eczema) | Specific exposure, legal status and economic impact. |
| Study Description | Selected Results of Possible use in Differential Diagnosis | Comment | Ref. |
|---|---|---|---|
| Transcriptomic analysis of blood and skin samples |
|
| [51] |
| Transcriptomic and proteomic analysis of skin and blood samples |
|
| [52] |
| Proteomic analysis of skin tape strips |
|
| [53] |
| Proteomic analysis of suction blister fluid |
|
| [54] |
| Multi-omics analysis of feces |
|
| [55] |
| Transcriptomic analysis of skin samples |
|
| [56] |
| Proteomic analysis of skin swabs | Swabs from non-lesional skin revealed significantly lower levels of 18 proteins, serpin B6, carboxypeptidase M, 20S core proteasome and its adaptors (PSMA2, PSMA5, PSMB2, PSMB4 and KCTD5), tissue alpha-L-fucosidase (FUCA1), zinc-alpha-2-glycoprotein (AZGP), N-acetylgalactosamine-6-sulfatase (GALNS), 7-dehydrocholesterol reductase (DHCR7), ester hydrolase (C11orf54), phospholipase B (PLB1; PLBD2), phospholipase D3 (PLD3), phospholipid-binding annexin A7 (ANXA7), acid phosphatase (ACPP) and nucleophosmin (NPM1), and an increased expression only of the protein TDRD15 in AD patients when compared with healthy controls. |
| [57] |
| Transcriptomic analysis of skin surface lipids |
|
| [58] |
| Proteomic analysis of serum samples confirmed via ELISA and immunohistochemistry | Significantly elevated expression of serum Cofilin-1 in AD patients when compared with healthy controls. |
| [59] |
| Proteomic and transcriptomic analyses of saliva samples |
|
| [60] |
| Proteomic analysis of skin samples |
|
| [61] |
| Metabolomic analysis of skin and blood samples |
|
| [62] |
| Transcriptomic study of skin biopsies |
|
| [63] |
| Transcriptomic and epigenomic study of skin biopsies |
|
| [65] |
| Clinical Form | Route of the Hapten Exposure |
|---|---|
| ACD limited to the site of contact | Direct skin contact with the sensitizing hapten. |
| ACD with local spreading | After entering into the skin, the hapten disperses from the primary focus via lymph vessels, resulting in the emergence of secondary foci (satellites) in close vicinity. |
| Hematogenous ACD | After passing through the skin in the exposed site (primary patch), the hapten enters systemic circulation via lymph vessels or veins and is redistributed to the skin (and other organs) via arteries. A primary patch can be found with subsequent secondary eczematous foci dispersed over the body, with possible predominance in flexures and skin folds. Involved areas may also be limited to previous localizations of ACD. As the route includes the portal vein and the liver, actual haptens may be metabolites of the substance entering the body; in such cases the primary patch may be absent. |
| Systemic reactivation of ACD (SRACD) | The (pro)hapten enters systemic circulation directly (via inhalation, oral absorption, injection or implant) or indirectly via the gut and liver passage and is further redistributed to the skin and other organs via the arteries. Actual haptens may be metabolites of the substances entering the body. The eczematous foci may be dispersed over entire skin areas, show a predominance in flexures and skin folds or be limited to previous locations of ACD. The primary patch is not present in SRACD. |
| Protein Group | Examples (Documented Cases) |
|---|---|
| Plant proteins, food |
|
| Plant proteins, non-food |
|
| Animal proteins, food |
|
| Animal proteins, non-food |
|
| Fungal proteins, food |
|
| Fungal proteins, non-food |
|
| Protein hydrolysates |
|
| Bioengineered proteins |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiewak, R. Diseases from the Spectrum of Dermatitis and Eczema: Can “Omics” Sciences Help with Better Systematics and More Accurate Differential Diagnosis? Int. J. Mol. Sci. 2023, 24, 10468. https://doi.org/10.3390/ijms241310468
Spiewak R. Diseases from the Spectrum of Dermatitis and Eczema: Can “Omics” Sciences Help with Better Systematics and More Accurate Differential Diagnosis? International Journal of Molecular Sciences. 2023; 24(13):10468. https://doi.org/10.3390/ijms241310468
Chicago/Turabian StyleSpiewak, Radoslaw. 2023. "Diseases from the Spectrum of Dermatitis and Eczema: Can “Omics” Sciences Help with Better Systematics and More Accurate Differential Diagnosis?" International Journal of Molecular Sciences 24, no. 13: 10468. https://doi.org/10.3390/ijms241310468