
LC-MS/MS-Based Serum Metabolomics and Transcriptome Analyses for the Mechanism of Augmented Renal Clearance

Abstract
:1. Introduction
2. Results
2.1. Results of Metabolomics
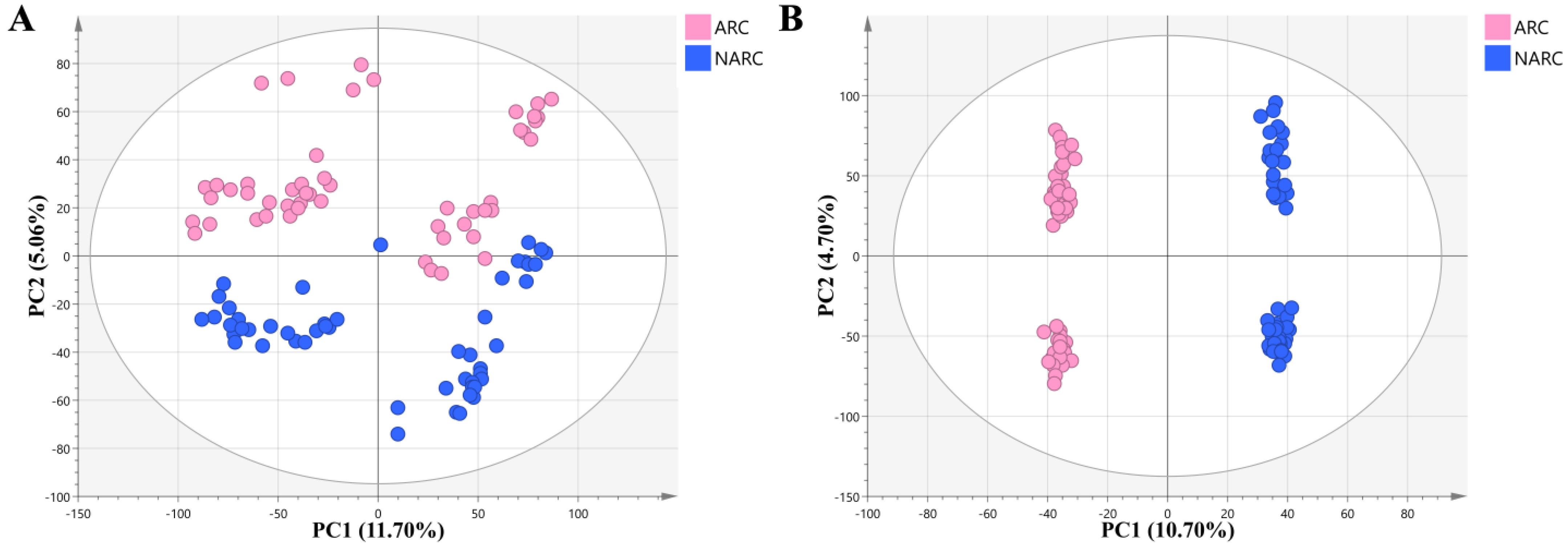
2.1.1. Clinical Evaluation of ARC
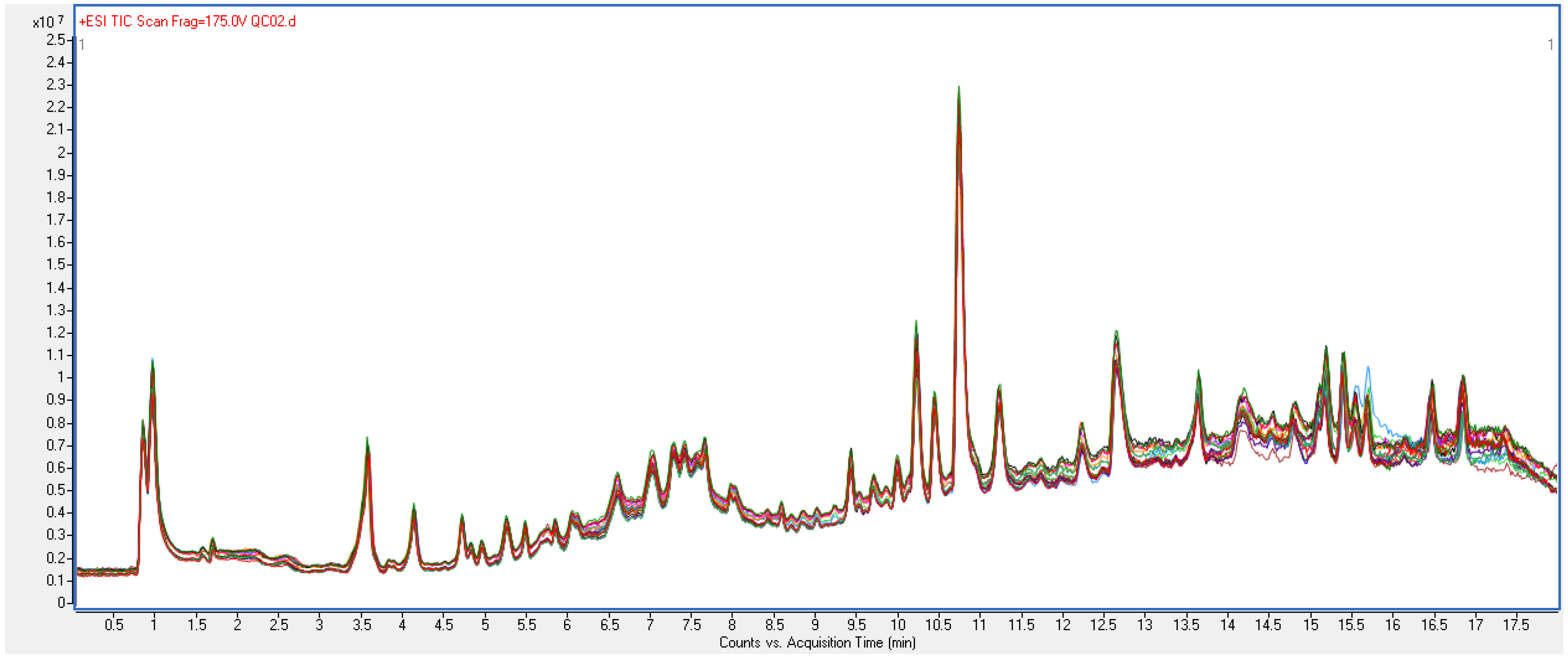
2.1.2. Quality Control of Untargeted Metabolic Profiling
2.1.3. Screening of Differential Metabolites
2.1.4. Annotation Results of Differential Metabolites
2.2. Results of Transcriptomics
2.2.1. Included Transcriptome Datasets and Their Basic Characteristics
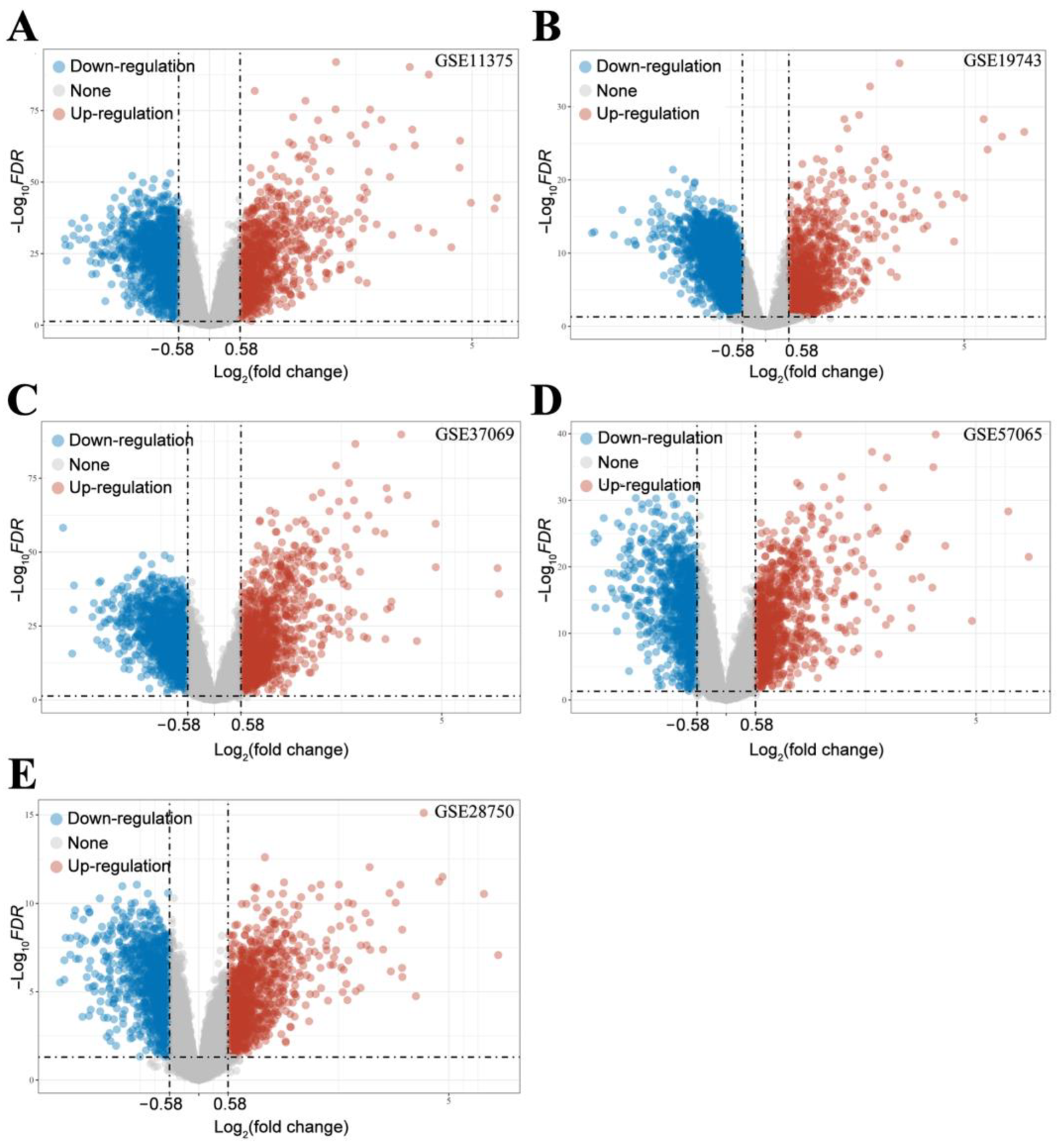
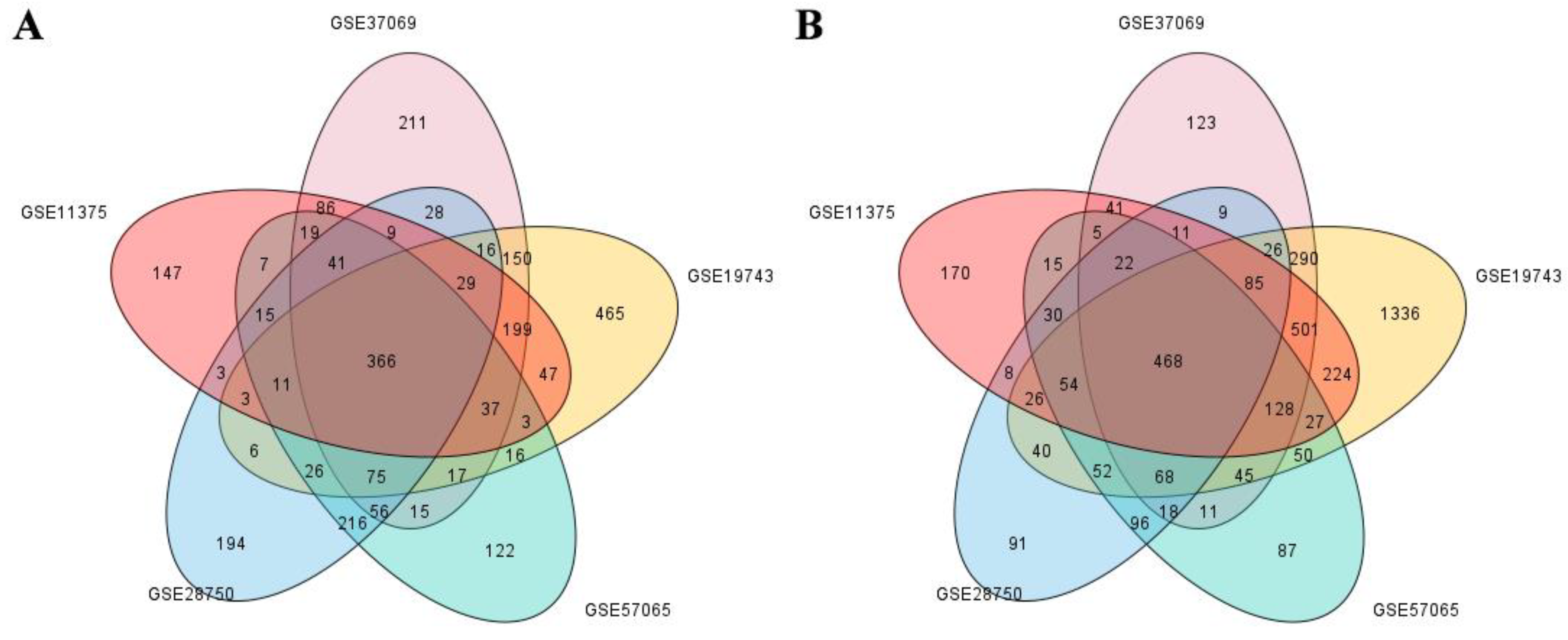
2.2.2. Screening of DEGs
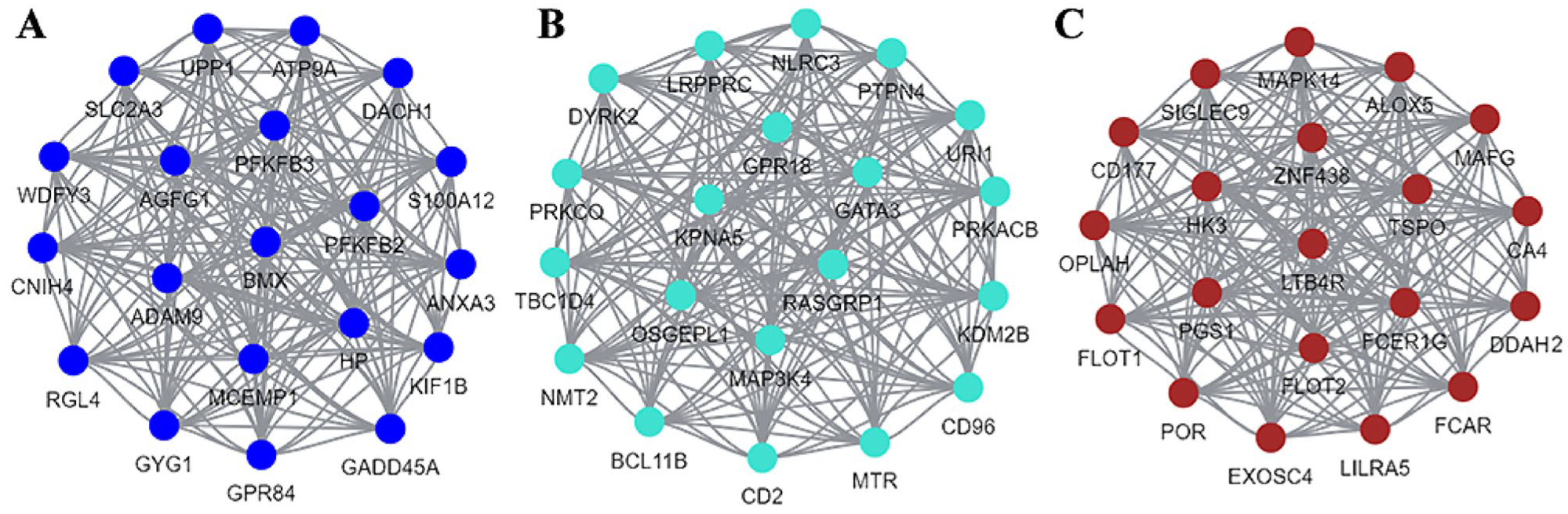
2.2.3. The Results of WGCNA
2.3. Results of Pathway Analysis
2.3.1. The Result of Network Analysis
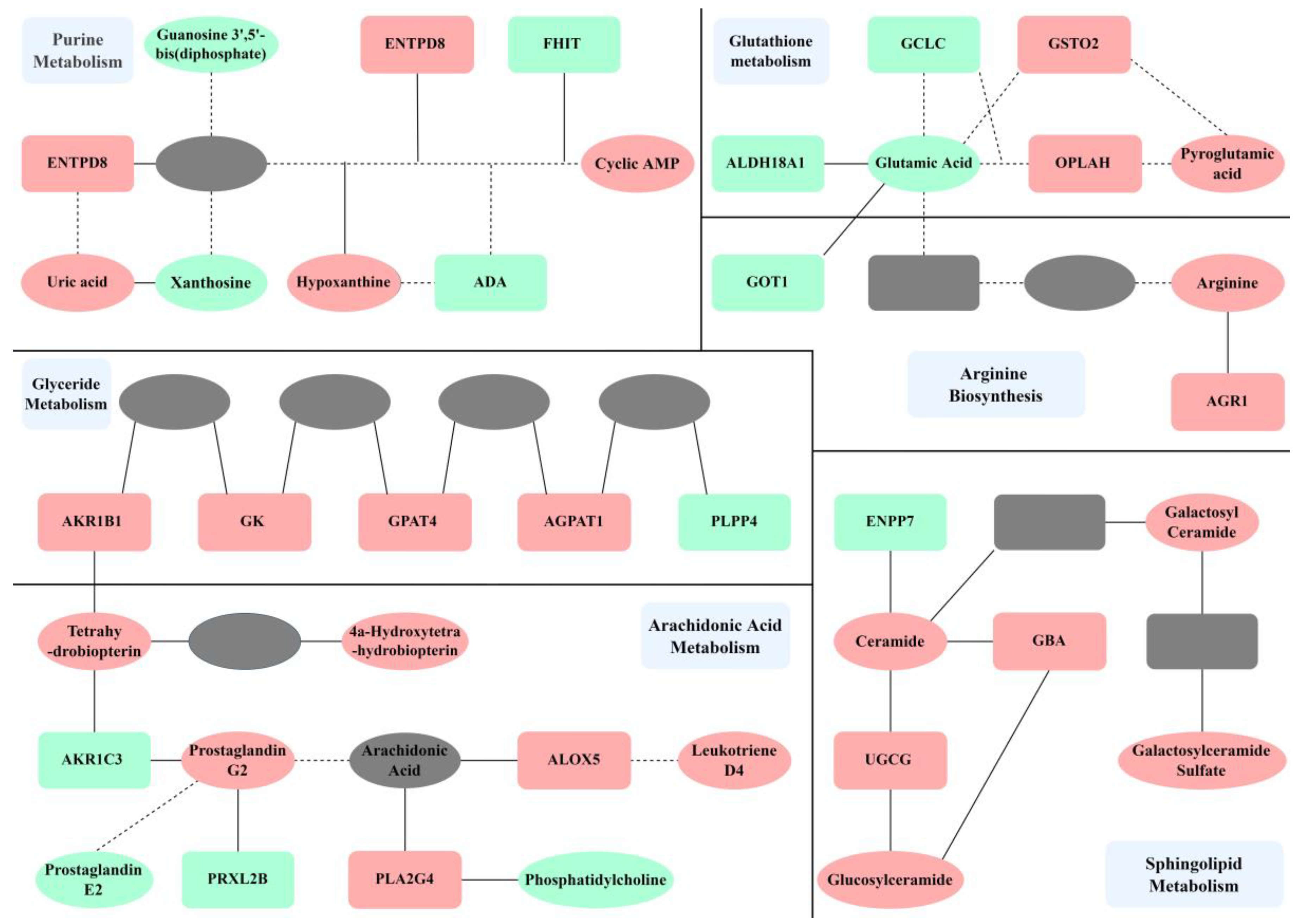
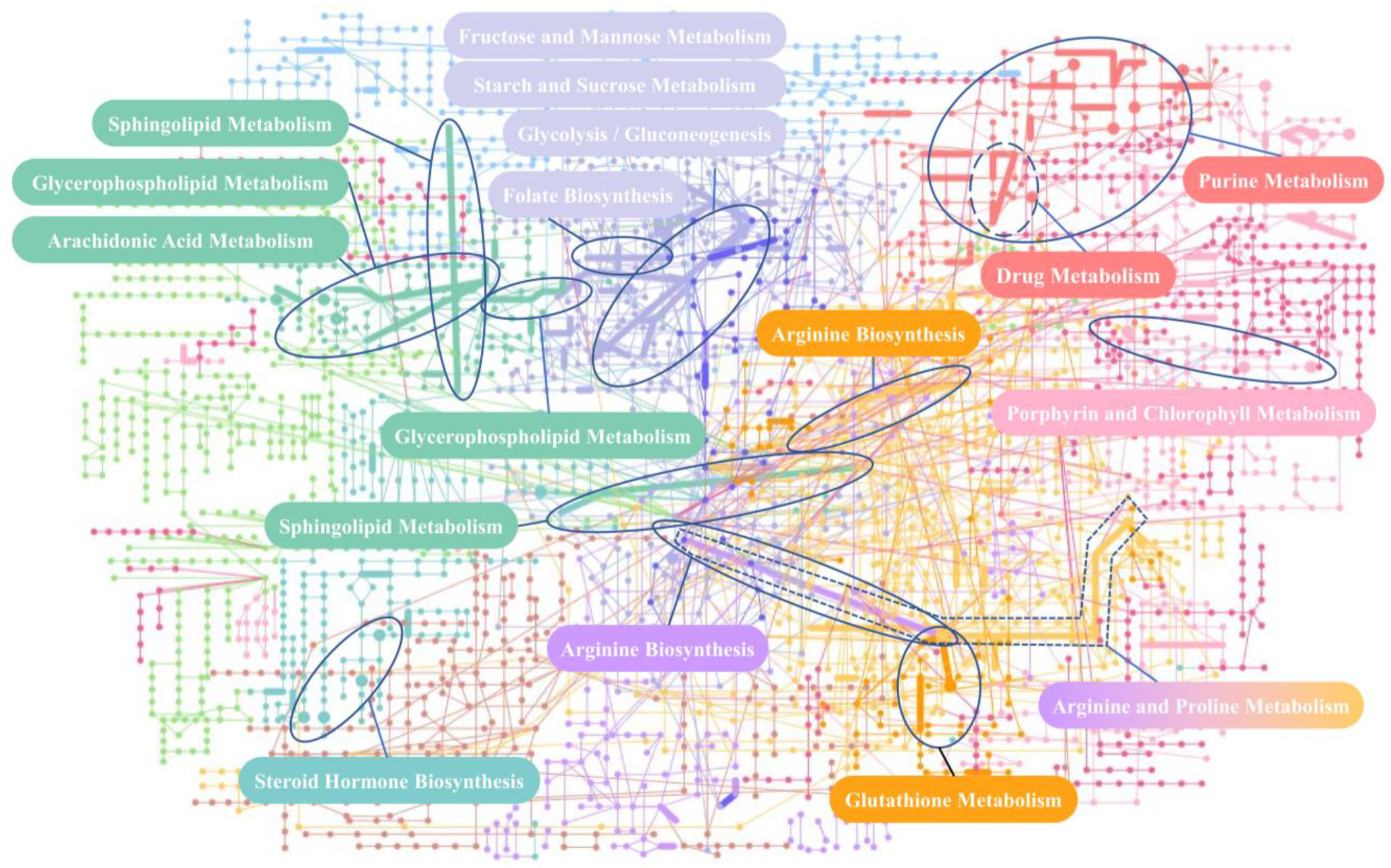
2.3.2. Pathway Analysis
2.3.3. Mechanism Analysis
3. Discussion
3.1. Improving Renal Perfusion
3.2. Improving Glomerular Capillary Wall Permeability
3.3. Limitations of This Study
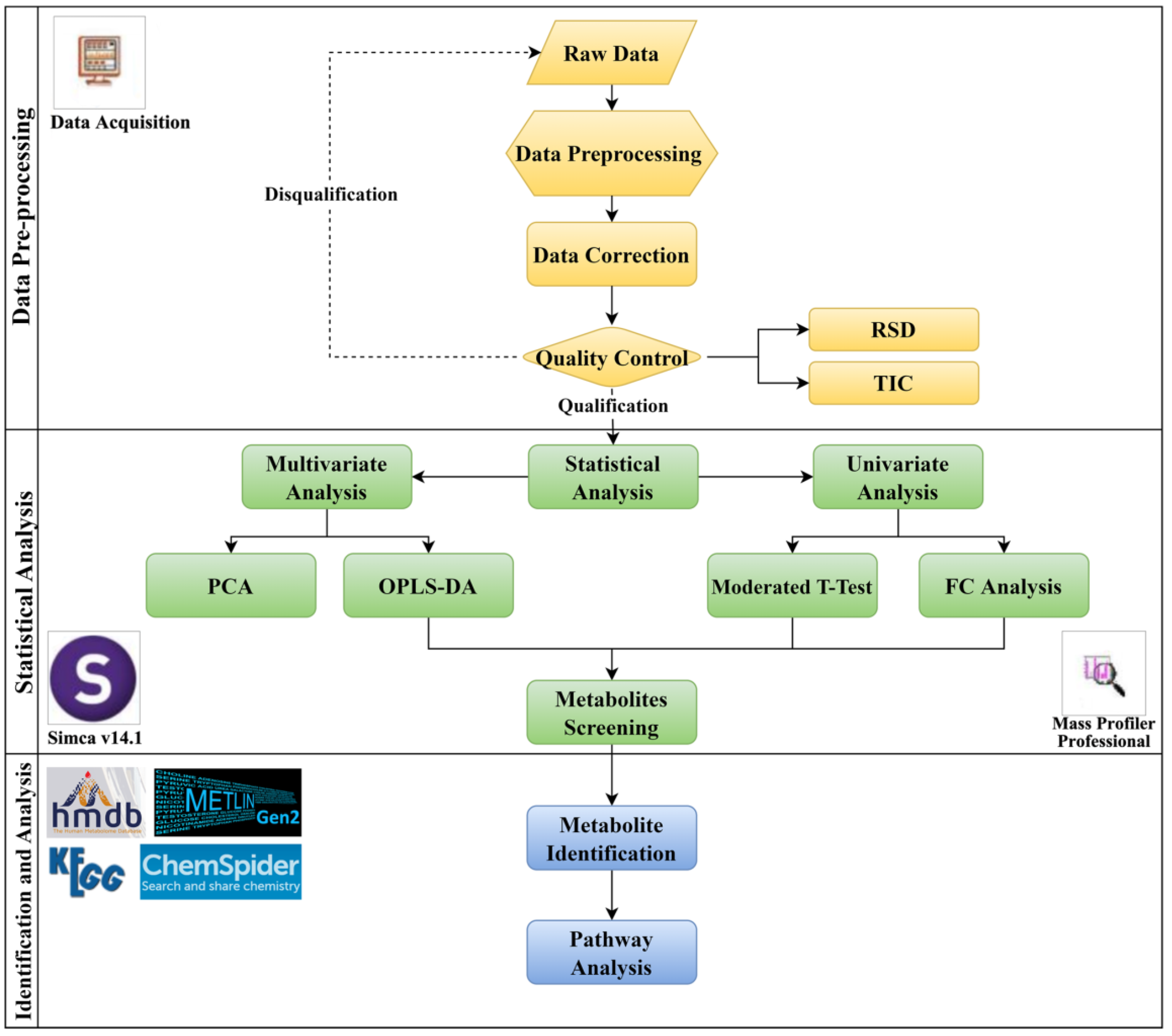
4. Materials and Methods
4.1. Methods for Metabolomics
4.1.1. Information Collection and Sample Screening
4.1.2. Sample Preparation
4.1.3. Quality Control
4.1.4. LC–MS Analysis
4.1.5. Data Processing
4.1.6. Metabolite Annotation
4.2. Transcriptomics Method
4.2.1. Dataset Filtering and Preparation
4.2.2. DEGs Screening and Annotation
4.2.3. WGCNA Analysis
4.2.4. Gene Function Enrichment Analysis
4.3. Method of Pathway Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Udy, A.A.; Roberts, J.A.; Boots, R.J.; Paterson, D.L.; Lipman, J. Augmented Renal Clearance. Clin. Pharm. 2010, 49, 1–16. [Google Scholar] [CrossRef]
- Claus, B.O.M.; Hoste, E.A.; Colpaert, K.; Robays, H.; Decruyenaere, J.; De Waele, J.J. Augmented Renal Clearance Is a Common Finding with Worse Clinical Outcome in Critically Ill Patients Receiving Antimicrobial Therapy. J. Crit. Care 2013, 28, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Cojutti, P.G.; Lazzarotto, D.; Candoni, A.; Dubbini, M.V.; Zannier, M.E.; Fanin, R.; Pea, F. Real-Time TDM-Based Optimization of Continuous-Infusion Meropenem for Improving Treatment Outcome of Febrile Neutropenia in Oncohaematological Patients: Results from a Prospective, Monocentric, Interventional Study. J. Antimicrob. Chemother. 2020, 75, 3029–3037. [Google Scholar] [CrossRef] [PubMed]
- De Waele, J.J.; Dumoulin, A.; Janssen, A.; Hoste, E.A. Epidemiology of Augmented Renal Clearance in Mixed ICU Patients. Minerva Anestesiol. 2015, 81, 1079–1085. [Google Scholar] [PubMed]
- Saito, K.; Kamio, S.; Ito, K.; Suzuki, N.; Abe, K.; Goto, T. A Simple Scoring Method to Predict Augmented Renal Clearance in Haematologic Malignancies. J. Clin. Pharm. Ther. 2020, 45, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Lannou, A.; Carrie, C.; Rubin, S.; Cane, G.; Cottenceau, V.; Petit, L.; Biais, M. Salt Wasting Syndrome in Brain Trauma Patients: A Pathophysiologic Approach Using Sodium Balance and Urinary Biochemical Analysis. BMC Neurol. 2020, 20, 190. [Google Scholar] [CrossRef] [PubMed]
- Mikami, R.; Hayakawa, M.; Imai, S.; Sugawara, M.; Takekuma, Y. Onset Timing and Duration of Augmented Renal Clearance in a Mixed Intensive Care Unit. J. Intensive Care 2023, 11, 13. [Google Scholar] [CrossRef]
- Tomasa-Irriguible, T.M.; Martínez-Vega, S.; Mor-Marco, E.; Herraiz-Ruiz, A.; Raguer-Pardo, L.; Cubells-Larrosa, C. Low Molecular Weight Heparins in COVID-19 Patients: Beware of Augmented Renal Clearance! Crit. Care 2020, 24, 325. [Google Scholar] [CrossRef]
- Deenen, S.; Fransen, L.F.C.; Jaspers, T.C.C.; Workum, J.D. Treatment Implications of Augmented Renal Clearance in a Critically Ill COVID-19 Patient: A Case Report. Fundam. Clin. Pharmacol. 2023, ahead of print. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Güiza, F.; Gijsen, M.; Spriet, I.; Dauwe, D.; Debaveye, Y.; Peetermans, M.; Wauters, J.; Van den Berghe, G.; Meyfroidt, G.; et al. External Validation of the Augmented Renal Clearance Predictor in Critically Ill COVID-19 Patients. Antibiotics 2023, 12, 698. [Google Scholar] [CrossRef]
- Naeem, H.E.M.A.E.; Abdelhamid, M.H.E.; Atteya, D.A.M. Impact of Augmented Renal Clearance on Enoxaparin Therapy in Critically Ill Patients. Egypt. J. Anaesth. 2017, 33, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Tai, C.-H.; Liao, W.-Y.; Wang, C.-C.; Kuo, C.-H.; Lin, S.-W.; Ku, S.-C. Augmented Renal Clearance Is Associated with Inadequate Antibiotic Pharmacokinetic/Pharmacodynamic Target in Asian ICU Population: A Prospective Observational Study. Infect. Drug Resist. 2019, 12, 2531–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttner, A.; Von Dach, E.; Renzoni, A.; Huttner, B.D.; Affaticati, M.; Pagani, L.; Daali, Y.; Pugin, J.; Karmime, A.; Fathi, M.; et al. Augmented Renal Clearance, Low β-Lactam Concentrations and Clinical Outcomes in the Critically Ill: An Observational Prospective Cohort Study. Int. J. Antimicrob. Agents 2015, 45, 385–392. [Google Scholar] [CrossRef]
- Barletta, J.F.; Mangram, A.J.; Byrne, M.; Sucher, J.F.; Hollingworth, A.K.; Ali-Osman, F.R.; Shirah, G.R.; Haley, M.; Dzandu, J.K. Identifying augmented renal clearance in trauma patients: Validation of the Augmented Renal Clearance in Trauma Intensive Care scoring system. J. Trauma Acute Care Surg. 2017, 82, 665. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Morimoto, S.; Izutani, Y.; Muranishi, K.; Kaneyama, H.; Hoshino, K.; Nishida, T.; Ishikura, H. Augmented Renal Clearance in Japanese Intensive Care Unit Patients: A Prospective Study. J. Intensive Care 2016, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Egea, A.; Dupuis, C.; de Montmollin, E.; Wicky, P.-H.; Patrier, J.; Jaquet, P.; Lefèvre, L.; Sinnah, F.; Marzouk, M.; Sonneville, R.; et al. Augmented Renal Clearance in the ICU: Estimation, Incidence, Risk Factors and Consequences-a Retrospective Observational Study. Ann. Intensive Care 2022, 12, 88. [Google Scholar] [CrossRef]
- Hirai, K.; Ihara, S.; Kinae, A.; Ikegaya, K.; Suzuki, M.; Hirano, K.; Itoh, K. Augmented Renal Clearance in Pediatric Patients with Febrile Neutropenia Associated with Vancomycin Clearance. Ther. Drug Monit. 2016, 38, 393–397. [Google Scholar] [CrossRef]
- Minkutė, R.; Briedis, V.; Steponavičiūtė, R.; Vitkauskienė, A.; Mačiulaitis, R. Augmented Renal Clearance—An Evolving Risk Factor to Consider during the Treatment with Vancomycin. J. Clin. Pharm. Ther. 2013, 38, 462–467. [Google Scholar] [CrossRef]
- Yu, Y.-X.; Lu, J.; Lu, H.; Li, L.; Li, J.-J.; Shi, L.; Duan, L.-F.; Zhuang, Z.-W.; Xue, S.-D.; Shen, Y.; et al. Predictive Performance of Reported Vancomycin Population Pharmacokinetic Model in Patients with Different Renal Function Status, Especially Those with Augmented Renal Clearance. Eur. J. Hosp. Pharm. 2021, 29, e6–e14. [Google Scholar] [CrossRef]
- Villanueva, R.D.; Talledo, O.; Neely, S.; White, B.; Celii, A.; Cross, A.; Kennedy, R. Vancomycin dosing in critically ill trauma patients: The VANCTIC Study. J. Trauma Acute Care Surg. 2019, 87, 1164. [Google Scholar] [CrossRef]
- Ishigo, T.; Ibe, Y.; Fujii, S.; Kazuma, S.; Aigami, T.; Kashiwagi, Y.; Takada, R.; Takahashi, S.; Fukudo, M.; Toda, T. Effect of Renal Clearance on Vancomycin Area under the Concentration-Time Curve Deviations in Critically Ill Patients. J. Infect. Chemother. 2023, 29, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Barrasa, H.; Soraluce, A.; Usón, E.; Sainz, J.; Martín, A.; Sánchez-Izquierdo, J.Á.; Maynar, J.; Rodríguez-Gascón, A.; Isla, A. Impact of Augmented Renal Clearance on the Pharmacokinetics of Linezolid: Advantages of Continuous Infusion from a Pharmacokinetic/Pharmacodynamic Perspective. Int. J. Infect. Dis. 2020, 93, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, J.; Carrié, C.; d’Houdain, N.; Djabarouti, S.; Petit, L.; Xuereb, F.; Legeron, R.; Biais, M.; Breilh, D. Are Standard Dosing Regimens of Ceftriaxone Adapted for Critically Ill Patients with Augmented Creatinine Clearance? Antimicrob. Agents Chemother. 2019, 63, e02134-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, G.; Briscoe, S.; McWhinney, B.; Ally, M.; Ungerer, J.; Lipman, J.; Roberts, J.A. Therapeutic Drug Monitoring of β-Lactam Antibiotics in the Critically Ill: Direct Measurement of Unbound Drug Concentrations to Achieve Appropriate Drug Exposures. J. Antimicrob. Chemother. 2018, 73, 3087–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulla, A.; Ewoldt, T.M.J.; Purmer, I.M.; Muller, A.E.; Gommers, D.; Endeman, H.; Koch, B.C.P. A Narrative Review of Predictors for β-Lactam Antibiotic Exposure during Empirical Treatment in Critically Ill Patients. Expert Opin. Drug Metab. Toxicol. 2021, 17, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Bakdach, D.; Elajez, R.; Bakdach, A.R.; Awaisu, A.; De Pascale, G.; Ait Hssain, A. Pharmacokinetics, Pharmacodynamics, and Dosing Considerations of Novel β-Lactams and β-Lactam/β-Lactamase Inhibitors in Critically Ill Adult Patients: Focus on Obesity, Augmented Renal Clearance, Renal Replacement Therapies, and Extracorporeal Membrane Oxygenation. J. Clin. Med. 2022, 11, 6898. [Google Scholar] [CrossRef]
- Baptista, J.P.; Sousa, E.; Martins, P.J.; Pimentel, J.M. Augmented Renal Clearance in Septic Patients and Implications for Vancomycin Optimisation. Int. J. Antimicrob. Agents 2012, 39, 420–423. [Google Scholar] [CrossRef]
- Udy, A.A.; Varghese, J.M.; Altukroni, M.; Briscoe, S.; McWhinney, B.C.; Ungerer, J.P.; Lipman, J.; Roberts, J.A. Subtherapeutic Initial β-Lactam Concentrations in Select Critically Ill Patients: Association between Augmented Renal Clearance and Low Trough Drug Concentrations. Chest 2012, 142, 30–39. [Google Scholar] [CrossRef]
- Conil, J.-M.; Georges, B.; Mimoz, O.; Dieye, E.; Ruiz, S.; Cougot, P.; Samii, K.; Houin, G.; Saivin, S. Influence of Renal Function on Trough Serum Concentrations of Piperacillin in Intensive Care Unit Patients. Intensive Care Med. 2006, 32, 2063–2066. [Google Scholar] [CrossRef]
- De Waele, J.J.; Lipman, J.; Akova, M.; Bassetti, M.; Dimopoulos, G.; Kaukonen, M.; Koulenti, D.; Martin, C.; Montravers, P.; Rello, J.; et al. Risk Factors for Target Non-Attainment during Empirical Treatment with β-Lactam Antibiotics in Critically Ill Patients. Intensive Care Med. 2014, 40, 1340–1351. [Google Scholar] [CrossRef]
- Vincent, J.-L.; Bassetti, M.; François, B.; Karam, G.; Chastre, J.; Torres, A.; Roberts, J.A.; Taccone, F.S.; Rello, J.; Calandra, T.; et al. Advances in Antibiotic Therapy in the Critically Ill. Crit. Care 2016, 20, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torian, S.C.; Wiktor, A.J.; Roper, S.E.; Laramie, K.E.; Miller, M.A.; Mueller, S.W. Burn Injury and Augmented Renal Clearance: A Case for Optimized Piperacillin-Tazobactam Dosing. J. Burn. Care Res. 2023, 44, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Hefny, F.; Sambhi, S.; Morris, C.; Kung, J.Y.; Stuart, A.; Mahmoud, S.H. Drug Dosing in Critically Ill Adult Patients with Augmented Renal Clearance. Eur. J. Drug Metab. Pharm. 2022, 47, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, Y.; Ma, Y.; Wang, P.; Zhong, J.; Chu, Y. Augmented Renal Clearance: What Have We Known and What Will We Do? Front. Pharmacol. 2021, 12, 723731. [Google Scholar] [CrossRef] [PubMed]
- Beunders, R.; Schütz, M.J.; van Groenendael, R.; Leijte, G.P.; Kox, M.; van Eijk, L.T.; Pickkers, P. Endotoxemia-Induced Release of Pro-Inflammatory Mediators Are Associated with Increased Glomerular Filtration Rate in Humans In Vivo. Front. Med. 2020, 7, 559671. [Google Scholar] [CrossRef]
- Chakraborty, R.K.; Burns, B. Systemic Inflammatory Response Syndrome; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Margraf, A.; Ludwig, N.; Zarbock, A.; Rossaint, J. Systemic Inflammatory Response Syndrome after Surgery: Mechanisms and Protection. Anesth. Analg. 2020, 131, 1693–1707. [Google Scholar] [CrossRef]
- Sharma, A.; Mucino, M.J.; Ronco, C. Renal Functional Reserve and Renal Recovery after Acute Kidney Injury. Nephron Clin. Pract. 2014, 127, 94–100. [Google Scholar] [CrossRef]
- Fliser, D.; Zeier, M.; Nowack, R.; Ritz, E. Renal Functional Reserve in Healthy Elderly Subjects. J. Am. Soc. Nephrol. 1993, 3, 1371–1377. [Google Scholar] [CrossRef]
- Fuhrman, D.Y. The Role of Renal Functional Reserve in Predicting Acute Kidney Injury. Crit. Care Clin. 2021, 37, 399–407. [Google Scholar] [CrossRef]
- Dias, C.; Gaio, A.R.; Monteiro, E.; Barbosa, S.; Cerejo, A.; Donnelly, J.; Felgueiras, Ó.; Smielewski, P.; Paiva, J.-A.; Czosnyka, M. Kidney-Brain Link in Traumatic Brain Injury Patients? A Preliminary Report. Neurocrit. Care 2015, 22, 192–201. [Google Scholar] [CrossRef]
- Pesonen, A.; Ben-Hamouda, N.; Schneider, A. Acute Kidney Injury after Brain Injury: Does It Exist? Minerva Anestesiol. 2021, 87, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Afsar, B.; Sag, A.A.; Yalcin, C.E.; Kaya, E.; Siriopol, D.; Goldsmith, D.; Covic, A.; Kanbay, M. Brain-Kidney Cross-Talk: Definition and Emerging Evidence. Eur. J. Intern. Med. 2016, 36, 7–12. [Google Scholar] [CrossRef]
- Massion, P.B.; Feron, O.; Dessy, C.; Balligand, J.-L. Nitric Oxide and Cardiac Function: Ten Years after, and Continuing. Circ. Res. 2003, 93, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurhaluk, N. The Effectiveness of L-Arginine in Clinical Conditions Associated with Hypoxia. Int. J. Mol. Sci. 2023, 24, 8205. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.S.; Wolin, M.S. Nitric Oxide: Pathophysiological Mechanisms. Annu. Rev. Physiol. 1995, 57, 737–769. [Google Scholar] [CrossRef]
- Zhu, L.-J.; Li, F.; Zhu, D.-Y. NNOS and Neurological, Neuropsychiatric Disorders: A 20-Year Story. Neurosci. Bull. 2023, 2023, 1–15. [Google Scholar] [CrossRef]
- Bagang, N.; Gupta, K.; Singh, G.; Kanuri, S.H.; Mehan, S. Protease-Activated Receptors in Kidney Diseases: A Comprehensive Review of Pathological Roles, Therapeutic Outcomes and Challenges. Chem. Biol. Interact. 2023, 377, 110470. [Google Scholar] [CrossRef]
- Xia, Y.; Tsai, A.L.; Berka, V.; Zweier, J.L. Superoxide Generation from Endothelial Nitric-Oxide Synthase. A Ca2+/Calmodulin-Dependent and Tetrahydrobiopterin Regulatory Process. J. Biol. Chem. 1998, 273, 25804–25808. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.S.; Koppenol, W.H. Nitric Oxide, Superoxide, and Peroxynitrite: The Good, the Bad, and Ugly. Am. J. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandy, P.; Danial, H.; Ambrus, I.; Rothery, R.A.; Schulz, R. Peroxynitrite Is a Major Contributor to Cytokine-Induced Myocardial Contractile Failure. Circ. Res. 2000, 87, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Dawson, V.L.; Dawson, T.M.; Snyder, S.H.; Zweier, J.L. Nitric Oxide Synthase Generates Superoxide and Nitric Oxide in Arginine-Depleted Cells Leading to Peroxynitrite-Mediated Cellular Injury. Proc. Natl. Acad. Sci. USA 1996, 93, 6770–6774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lortie, M.J.; Satriano, J.; Gabbai, F.B.; Thareau, S.; Khang, S.; Deng, A.; Pizzo, D.P.; Thomson, S.C.; Blantz, R.C.; Munger, K.A. Production of Arginine by the Kidney Is Impaired in a Model of Sepsis: Early Events Following LPS. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R1434–R1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuczeriszka, M.; Walkowska, A.; Olszynski, K.H.; Rafalowska, J.; Sadowski, J.; Kompanowska-Jezierska, E. Arginine and Tetrahydrobiopterin Supplementation in Rats with Salt-Induced Blood Pressure Increase: Minor Hypotensive Effect but Improvement of Renal Haemodynamics. J. Physiol. Pharmacol. 2019, 70, 219–227. [Google Scholar] [CrossRef]
- Valdivielso, J.M.; Eritja, À.; Caus, M.; Bozic, M. Glutamate-Gated NMDA Receptors: Insights into the Function and Signaling in the Kidney. Biomolecules 2020, 10, 1051. [Google Scholar] [CrossRef] [PubMed]
- Van de Poll, M.C.G.; Soeters, P.B.; Deutz, N.E.P.; Fearon, K.C.H.; Dejong, C.H.C. Renal Metabolism of Amino Acids: Its Role in Interorgan Amino Acid Exchange. Am. J. Clin. Nutr. 2004, 79, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Haddad, J.J. N-Methyl-D-Aspartate (NMDA) and the Regulation of Mitogen-Activated Protein Kinase (MAPK) Signaling Pathways: A Revolving Neurochemical Axis for Therapeutic Intervention? Prog. Neurobiol. 2005, 77, 252–282. [Google Scholar] [CrossRef]
- Bozic, M.; Valdivielso, J.M. The Potential of Targeting NMDA Receptors Outside the CNS. Expert. Opin. Ther. Targets 2015, 19, 399–413. [Google Scholar] [CrossRef]
- Bozic, M.; Valdivielso, J.M. Functional Distribution and Regulation of the NMDAR in the Kidney, Heart and Parathyroid Gland. In The NMDA Receptors; Hashimoto, K., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 51–68. ISBN 978-3-319-49795-2. [Google Scholar]
- Yang, C.-C.; Chien, C.-T.; Wu, M.-H.; Ma, M.-C.; Chen, C.-F. NMDA Receptor Blocker Ameliorates Ischemia-Reperfusion-Induced Renal Dysfunction in Rat Kidneys. Am. J. Physiol. Renal. Physiol. 2008, 294, F1433–F1440. [Google Scholar] [CrossRef] [Green Version]
- Deng, A.; Thomson, S.C. Renal NMDA Receptors Independently Stimulate Proximal Reabsorption and Glomerular Filtration. Am. J. Physiol. Renal. Physiol. 2009, 296, F976–F982. [Google Scholar] [CrossRef] [Green Version]
- Diaz, M.F.; Li, N.; Lee, H.J.; Adamo, L.; Evans, S.M.; Willey, H.E.; Arora, N.; Torisawa, Y.; Vickers, D.A.; Morris, S.A.; et al. Biomechanical Forces Promote Blood Development through Prostaglandin E2 and the CAMP–PKA Signaling Axis. J. Exp. Med. 2015, 212, 665–680. [Google Scholar] [CrossRef]
- Li, Y.; Wei, Y.; Zheng, F.; Guan, Y.; Zhang, X. Prostaglandin E2 in the Regulation of Water Transport in Renal Collecting Ducts. Int. J. Mol. Sci. 2017, 18, 2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, S.K.; Butler, K.L.; Hayden, D.; Tompkins, R.G.; Serhan, C.N.; Irimia, D. Gene Expression of Proresolving Lipid Mediator Pathways Is Associated with Clinical Outcomes in Trauma Patients. Crit. Care Med. 2015, 43, 2642–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Cui, Y.; Ding, X.; Liu, S.; Han, B.; Duan, X.; Zhang, H.; Sun, T. Analysis of MRNA-lncRNA and MRNA-lncRNA-Pathway Co-expression Networks Based on WGCNA in Developing Pediatric Sepsis. Bioengineered 2021, 12, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Chen, Y.; Cai, Q. The Role of the LTB4-BLT1 Axis in Health and Disease. Pharmacol. Res. 2020, 158, 104857. [Google Scholar] [CrossRef]
- Chen, X.; Xie, H.; Liu, Y.; Ou, Q.; Deng, S. Interference of ALOX5 Alleviates Inflammation and Fibrosis in High Glucose-induced Renal Mesangial Cells. Exp. Ther. Med. 2022, 25, 34. [Google Scholar] [CrossRef]
- Satchell, S. The Role of the Glomerular Endothelium in Albumin Handling. Nat. Rev. Nephrol. 2013, 9, 717–725. [Google Scholar] [CrossRef]
- Szrejder, M.; Rachubik, P.; Rogacka, D.; Audzeyenka, I.; Rychłowski, M.; Angielski, S.; Piwkowska, A. Extracellular ATP Modulates Podocyte Function through P2Y Purinergic Receptors and Pleiotropic Effects on AMPK and CAMP/PKA Signaling Pathways. Arch. Biochem. Biophys. 2020, 695, 108649. [Google Scholar] [CrossRef]
- Ding, H.; Bai, F.; Cao, H.; Xu, J.; Fang, L.; Wu, J.; Yuan, Q.; Zhou, Y.; Sun, Q.; He, W.; et al. PDE/CAMP/Epac/C/EBP-β Signaling Cascade Regulates Mitochondria Biogenesis of Tubular Epithelial Cells in Renal Fibrosis. Antioxid. Redox Signal. 2018, 29, 637–652. [Google Scholar] [CrossRef]
- Bankir, L.; Ahloulay, M.; Devreotes, P.N.; Parent, C.A. Extracellular CAMP Inhibits Proximal Reabsorption: Are Plasma Membrane CAMP Receptors Involved? Am. J. Physiol. Ren. Physiol. 2002, 282, F376–F392. [Google Scholar] [CrossRef] [Green Version]
- Cockcroft, D.W.; Gault, M.H. Prediction of Creatinine Clearance from Serum Creatinine. Nephron 1976, 16, 31–41. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Janková, J.; van de Geer, S. Confidence Intervals for High-Dimensional Inverse Covariance Estimation. Electron. J. Stat. 2015, 9, 1205–1229. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Duren, W.; Evans, C.R.; Burant, C.F.; Michailidis, G.; Karnovsky, A. Sparse Network Modeling and Metscape-Based Visualization Methods for the Analysis of Large-Scale Metabolomics Data. Bioinformatics 2017, 33, 1545–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | ARC (N = 76) | NARC (N = 81) | p-Value |
|---|---|---|---|
| Gender (Male/Female) | 42/34 | 53/28 | 0.193 |
| Age (years) | 62 (55.25–66.75) | 50 (38–59) | 0.000 * |
| Vancomycin concentration (mg/L) | 14.1 (12.15–16.68) | 7.1 (5.25–8.2) | 0.000 * |
| BMI (kg/m2) | 23.68 (21.48–26.28) | 25.66 (23.13–27.89) | 0.001 * |
| Creatinine clearance (mL/min) | 101.55 (92.21–115.59) | 171.45 (148.38–208.7) | 0.000 * |
| White blood cells (109/L) | 9.05 (5.93–13.23) | 8.98 (5.51–12.68) | 0.562 |
| Lymphocyte count (109/L) | 1.13 (0.84–1.49) | 1.25 (0.78–1.73) | 0.497 |
| Neutrophil count (109/L) | 6.55 (3.96–11) | 6.66 (3.39–9.61) | 0.256 |
| Monocytes count (109/L) | 0.54 (0.4–0.76) | 0.57 (0.35–0.81) | 0.868 |
| Proportion of neutrophils (%) | 76.5 (69–86.7) | 73.4 (63.7–81.35) | 0.074 |
| Red blood cells (1012/L) | 3.26 ± 0.71 | 3.34 ± 0.82 | 0.517 |
| Hemoglobin (g/L) | 97 (82–110) | 98 (79.5–118) | 0.685 |
| Platelet count (109/L) | 204 (116–300) | 252 (133–346.5) | 0.198 |
| Aspartate aminotransferase (U/L) | 20 (15.5–35) | 23 (14–49.5) | 0.299 |
| Alpine aminotransferase (U/L) | 27 (17–49) | 33 (20.5–62.5) | 0.156 |
| Alkaline phosphatase (U/L) | 90 (69–125) | 87 (61.5–132) | 0.566 |
| Glutamyl transpeptidase (U/L) | 47 (24–100) | 65 (31–102) | 0.150 |
| Total protein (g/L) | 55.8 (49.6–63) | 55.8 (51.95–59.25) | 0.744 |
| Albumin (g/L) | 29.94 ± 6.13 | 29.30 ± 5.57 | 0.493 |
| Globulin (mg/L) | 15.05 (8.9–21.65) | 15.2 (7.88–21.45) | 0.893 |
| Urea (mmol/L) | 5.38 (4.09–8.83) | 4.2 (2.99–5.92) | 0.000 * |
| Creatinine (μmol/L) | 57 (47–67) | 42 (33–54) | 0.000 * |
| Serum cystatin C (mg/L) | 1.07 (0.89–1.26) | 0.79 (0.66–0.96) | 0.000 * |
| Antithrombin III (mg/L) | 61.90 ± 24.40 | 75.80 ± 19.93 | 0.053 |
| Prothrombin time (s) | 14.7 (13.7–16.5) | 14.5 (13.8–15.7) | 0.753 |
| Prothrombin activity (%) | 80 (63.5–92.5) | 79 (70.5–88) | 0.706 |
| International normalized ratio | 1.15 (1.05–1.34) | 1.15 (1.08–1.25) | 0.733 |
| Activated partial thromboplastin time (s) | 42.4 (36.7–47.7) | 42.6 (38.3–49.75) | 0.488 |
| Plasma fibrin (g/L) | 4.56 ± 1.76 | 4.96 ± 1.64 | 0.214 |
| Fibrinogen degradation products (μg/mL) | 13.03 (7.49–24.98) | 17.08 (9.79–21.77) | 0.383 |
| D-dimer (μg/mL) | 2.79 (1.71–6.12) | 3.08 (1.45–5.2) | 0.987 |
| C-reactive protein (mg/L) | 44.95 (14.93–105.75) | 52.25 (13.9–133.48) | 0.444 |
| Procalcitonin (ng/mL) | 0.2 (0.08–0.71) | 0.2 (0.07–0.58) | 0.453 |
| B-type natriuretic peptide (pg/mL) | 141 (54.25–307.25) | 61 (36–219.5) | 0.136 |
| Items | ARC (N = 76) | NARC (N = 81) | p-Value |
|---|---|---|---|
| Gender (Male/Female) | 29/22 | 33/18 | 0.190 |
| Age (years) | 59 (53–65) | 56 (46–61) | 0.063 |
| Vancomycin concentration (mg/L) | 14.1 (11.9–17.2) | 7.3 (5.6–8.5) | 0.000 * |
| BMI (kg/m2) | 24.22 (22.15–27.1) | 24.46 (22.86–26.3) | 0.817 |
| Creatinine clearance (ml/min) | 9.27 (6.55–13.89) | 8.62 (4.61–12.8) | 0.238 |
| White blood cells (109/L) | 1.22 (0.82–1.73) | 1.2 (0.66–1.85) | 0.608 |
| Lymphocyte count (109/L) | 6.68 (3.89–10.33) | 5.41 (1.65–8.52) | 0.064 |
| Neutrophil count (109/L) | 0.55 (0.4–0.84) | 0.56 (0.21–0.72) | 0.469 |
| Monocytes count (109/L) | 75.6 (64.3–87.95) | 69.6 (56–82.5) | 0.086 |
| Proportion of neutrophils (%) | 3.16 (2.8–3.89) | 3.42 (2.69–4.03) | 0.820 |
| Red blood cells (1012/L) | 98.12 ± 20.56 | 96.37 ± 29.21 | 0.731 |
| Hemoglobin (g/L) | 203 (96.5–308.75) | 227 (113–332.5) | 0.869 |
| Platelet count (109/L) | 21.5 (16.25–47.5) | 26 (14–61) | 0.471 |
| Aspartate aminotransferase (U/L) | 32 (18.75–57) | 31.5 (20–56) | 0.869 |
| Alpine aminotransferase (U/L) | 88.5 (69.5–128.75) | 90.5 (61.75–129.75) | 0.687 |
| Alkaline phosphatase (U/L) | 47 (26.5–97.75) | 62.5 (34–98.25) | 0.343 |
| Glutamyl transpeptidase (U/L) | 57.05 (49.98–63.8) | 56.1 (53.9–60.53) | 0.677 |
| Total protein (g/L) | 30.10 ± 6.19 | 29.658 ± 4.75 | 0.689 |
| Albumin (g/L) | 14.65 (8.6–21.75) | 15.4 (8.2–21.2) | 0.971 |
| Globulin (mg/L) | 6.5 (4.55–10.02) | 3.97 (2.81–5.47) | 0.000 * |
| Urea (mmol/L) | 61 (50–70) | 42 (34–50) | 0.000 * |
| Creatinine (μmol/L) | 1.09 (0.91–1.45) | 0.82 (0.64–0.98) | 0.000 * |
| Serum cystatin C (mg/L) | 62.18 ± 15.39 | 42.06 ± 11.24 | 0.085 |
| Antithrombin III (mg/L) | 14.65 (13.85–17.48) | 14.5 (13.7–15.5) | 0.547 |
| Prothrombin time(s) | 79.5 (57.5–89.25) | 80 (71–89) | 0.558 |
| Prothrombin activity (%) | 1.16 (1.07–1.43) | 1.14 (1.07–1.24) | 0.566 |
| International normalized ratio | 40.85 (36.83–48.03) | 42.1 (38.3–50) | 0.347 |
| Activated partial thromboplastin time (s) | 4.57 ± 1.86 | 5.20 ± 1.48 | 0.119 |
| Plasma fibrin (g/L) | 13.03 (8.4–24.4) | 17.22 (10.48–20.93) | 0.423 |
| Fibrinogen degradation products (μg/mL) | 2.79 (1.7–6.19) | 2.49 (1.38–4.58) | 0.326 |
| D-dimer (μg/mL) | 53.5 (16.8–116.8) | 39.4 (10.13–127.23) | 0.911 |
| C-reactive protein (mg/L) | 0.2 (0.09–0.64) | 0.17 (0.07–0.39) | 0.194 |
| Procalcitonin (ng/mL) | 144 (58–332) | 129.5 (42.25–237.25) | 0.429 |
| B-type natriuretic peptide (pg/mL) | 29/22 | 33/18 | 0.190 |
| RSD (%) | 837.8305 @0.85 | 165.0791 @3.63 | 563.3502 @5.24 | 431.3025 @7.68 | 1485.992 @10.74 | 328.1513 @13.64 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Area | RT | Area | RT | Area | RT | Area | RT | Area | RT | Area | RT | |
| System repeatability | 2.62 | 0.12 | 0.90 | 0.03 | 2.44 | 0.02 | 1.65 | 0.09 | 2.56 | 0.06 | 2.08 | 0.06 |
| Method replicability | 2.55 | 0.70 | 1.90 | 0.30 | 3.37 | 0.14 | 1.56 | 0.07 | 4.50 | 0.03 | 1.71 | 0.02 |
| 4 h Stability | 3.46 | 0.23 | 0.69 | 0.05 | 1.43 | 0.00 | 1.60 | 0.03 | 2.61 | 0.05 | 1.50 | 0.04 |
| 8 h Stability | 4.49 | 0.30 | 4.52 | 0.53 | 1.24 | 0.04 | 2.67 | 0.08 | 1.74 | 0.09 | 1.42 | 0.07 |
| 12 h Stability | 3.54 | 0.19 | 0.71 | 0.21 | 1.34 | 0.03 | 2.75 | 0.10 | 2.20 | 0.02 | 1.94 | 0.01 |
| No. | m/z@RT | Compound Name | Molecular Formula | HMDB ID | KEGG ID | Up/Down | p-Value |
|---|---|---|---|---|---|---|---|
| 1 | 199.0335@0.90 | O-Phosphohomoserine | C4H10NO6P | HMDB0003484 | C01102 | Up | 0.000 |
| 2 | 725.6688@0.93 | Galactosylceramide (d18:1/18:1(9Z)) | C42H79NO8 | HMDB0010714 | C02686 | Down | 0.000 |
| 3 | 284.8999@0.94 | Xanthosine | C10H12N4O6 | HMDB0000299 | C01762 | Down | 0.000 |
| 4 | 727.664@0.96 | GlcCer(d18:1/18:0) | C42H81NO8 | HMDB0004972 | C01190 | Down | 0.000 |
| 5 | 329.7503@0.97 | Inosine 2′,3′-cyclic phosphate | C10H11N4O7P | HMDB0011680 | C05768 | Up | 0.000 |
| 6 | 168.0808@0.98 | Uric acid | C5H4N4O3 | HMDB0000289 | C00366 | Up | 0.000 |
| 7 | 136.0409@0.99 | Hypoxanthine | C5H4N4O | HMDB0000157 | C00262 | Up | 0.000 |
| 8 | 561.7431@1.00 | Protoporphyrin IX | C5H4N4O | HMDB0000241 | C02191 | Down | 0.000 |
| 9 | 181.0735@1.00 | L-Tyrosine | C9H11NO3 | HMDB0000158 | C00082 | Up | 0.000 |
| 10 | 779.4283@1.01 | PS(14:0/22:6(4Z, 7Z, 10Z, 13Z, 16Z, 19Z)) | C34H34N4O4 | HMDB0012340 | - | Down | 0.000 |
| 11 | 983.2857@1.09 | Adrenorphin | C44H69N15O9S | HMDB0059791 | C16108 | Down | 0.001 |
| 12 | 173.985@1.72 | L-Arginine | C42H70NO10P | HMDB0000517 | C00062 | Up | 0.001 |
| 13 | 129.0428@1.80 | Pyroglutamic acid | C5H7NO3 | HMDB0000267 | C01879 | Up | 0.000 |
| 14 | 148.052@3.60 | L-Glutamic acid | C44H69N15O9S | HMDB0000148 | C00025 | Down | 0.000 |
| 15 | 242.1244@4.82 | Tetrahydrobiopterin | C6H14N4O2 | HMDB0000027 | C00272 | Down | 0.000 |
| 16 | 258.1433@5.13 | 4a-Hydroxytetrahydrobiopterin | C9H15N5O4 | HMDB0002281 | C15522 | Up | 0.001 |
| 17 | 352.2329@6.41 | Prostaglandin E2 | C25H40N2O6S | HMDB0001220 | C00584 | Down | 0.000 |
| 18 | 332.1375@6.48 | 17a-Hydroxypregnenolone | C5H9NO4 | HMDB0000363 | C05138 | Up | 0.000 |
| 19 | 779.5233@7.25 | 3-O-Sulfogalactosylceramide (d18:1/16:0) | C9H15N5O3 | HMDB0012313 | C06125 | Down | 0.000 |
| 20 | 398.2013@9.07 | PC(22:5(7Z, 10Z, 13Z, 16Z, 19Z)/14:1(9Z)) | C9H15N5O4 | HMDB0008690 | C00157 | Down | 0.000 |
| 21 | 474.282@9.36 | 10-Formyltetrahydrofolate | C20H32O5 | HMDB0000972 | C00234 | Down | 0.000 |
| 22 | 496.3461@9.64 | Leukotriene D4 | C21H32O3 | HMDB0003080 | C05951 | Up | 0.000 |
| 23 | 852.6166@9.68 | Heme A | C49H56FeN4O6 | HMDB0006901 | C15670 | Up | 0.000 |
| 24 | 330.2389@9.88 | Deoxycorticosterone | C40H77NO11S | HMDB0000016 | C03205 | Up | 0.000 |
| 25 | 565.299@10.01 | CDP-glucose | C44H76NO8P | HMDB0003369 | C00501 | Up | 0.000 |
| 26 | 457.8033@10.14 | FMNH2 | C20H23N7O7 | HMDB0001142 | C01847 | Down | 0.000 |
| 27 | 893.6207@10.15 | Octanoyl-CoA | C25H40N2O6S | HMDB0001070 | C01944 | Down | 0.000 |
| 28 | 541.3042@10.19 | Formamidopyrimidine nucleoside triphosphate | C10H18N5O15P3 | HMDB0006822 | C05922 | Down | 0.000 |
| 29 | 1338.8467@10.21 | Ganglioside GA1 (d18:1/24:0) | C25H40N2O6S | HMDB0004909 | C06136 | Up | 0.000 |
| 30 | 1142.8156@12.78 | Chenodeoxycholoyl-CoA | C45H74N7O19P3S | HMDB0006292 | C05337 | Down | 0.000 |
| 30 | 414.2053@10.45 | 4,4-Dimethyl-5a-cholesta-8-en-3b-ol | C21H30O3 | HMDB0006840 | C03541 | Up | 0.000 |
| 31 | 656.4657@10.83 | PA(P-16:0e/18:2(9Z, 12Z)) | C15H25N3O16P2 | HMDB0011155 | C15647 | Up | 0.000 |
| 32 | 907.6382@10.83 | 3-Oxooctanoyl-CoA | C29H48N7O18P3S | HMDB0003941 | C05267 | Down | 0.000 |
| 33 | 464.8111@10.90 | Testosterone glucuronide | C17H23N4O9P | HMDB0003193 | C11134 | Down | 0.000 |
| 34 | 442.7967@11.02 | Guanosine diphosphate | C29H50N7O17P3S | HMDB0001201 | C00035 | Down | 0.001 |
| 35 | 526.8658@11.20 | 3-Carboxy-1-hydroxypropylthiamine diphosphate | C10H18N5O15P3 | HMDB0006744 | C05381 | Down | 0.000 |
| 36 | 643.9412@11.24 | Phosphatidylinositol-3,4,5-trisphosphate | C68H126N2O23 | HMDB0004249 | C05981 | Up | 0.000 |
| 37 | 725.5357@11.51 | PE(20:3(8Z, 11Z, 14Z)/P-16:0) | C45H74N7O19P3S | HMDB0009378 | C00350 | Down | 0.000 |
| 38 | 1156.8068@11.62 | Choloyl-CoA | C29H50O | HMDB0001374 | C01794 | Up | 0.000 |
| 39 | 367.307@11.82 | Prostaglandin G2 | C37H69O7P | HMDB0003235 | C05956 | Up | 0.000 |
| 41 | 740.0203@13.15 | 1-Diphosinositol pentakisphosphate | C29H48N7O18P3S | HMDB0012494 | C11174 | Down | 0.000 |
| 42 | 718.0028@13.36 | 5-Methyltetrahydropteroyltri-L-glutamic acid | C30H39N9O12 | HMDB0012177 | C04489 | Up | 0.000 |
| 43 | 328.2594@13.52 | Cyclic AMP | C25H36O8 | HMDB0000058 | C00575 | Up | 0.000 |
| 44 | 602.9041@13.56 | Guanosine 3’,5’-bis(diphosphate) | C10H15N5O11P2 | HMDB0059638 | C01228 | Up | 0.000 |
| 45 | 647.4537@14.21 | Cer(d18:1/24:1(15Z)) | C16H25N4O10P2S | HMDB0004953 | C00195 | Down | 0.002 |
| Dataset | Disease | Platform | Experimental Group | Healthy Control Group |
|---|---|---|---|---|
| GSE11375 | Trauma | GPL570 | 158 | 26 |
| GSE37069 | Burns | GPL570 | 132 | 37 |
| GSE57065 | Burns | GPL570 | 71 | 25 |
| GSE19743 | Sepsis | GPL570 | 27 | 28 |
| GSE28750 | Sepsis | GPL570 | 10 | 20 |
| ID | Name | Degree | Betweenness | Expr. |
|---|---|---|---|---|
| C00025 | L-Glutamic Acid | 20 | 115.4780104 | −4.7288723 |
| C00062 | L-Arginine | 17 | 99.79187188 | 4.249939 |
| C00002 | Adenosine Triphosphate | 16 | 110.4030875 | NA |
| C00006 | NADP | 14 | 62.6121986 | NA |
| C00035 | Guanosine diphosphate | 13 | 43.00154164 | −4.1962013 |
| C00082 | L-tyrosine | 12 | 30.70040727 | 2.531392 |
| C00004 | NADH | 12 | 51.46153831 | NA |
| C00366 | Uric acid | 10 | 30.76239229 | 12.050512 |
| C00272 | Tetrahydrobiopterin | 10 | 22.48090304 | −9.704965 |
| C00262 | Hypoxanthine | 10 | 20.86003946 | 7.3225327 |
| C00584 | Prostaglandin E2 | 9 | 46.96623783 | −5.0245094 |
| C00575 | cAMP | 9 | 34.11316313 | 6.905841 |
| Pathway Name | Expected | Hits | p-Value | Impact |
|---|---|---|---|---|
| Nitrogen metabolism | 0.1967 | 2 | 0.0154 | 0.4444 |
| Porphyrin and chlorophyll metabolism | 1.0426 | 4 | 0.0191 | 0.0962 |
| Arachidonic acid metabolism | 1.5934 | 5 | 0.0199 | 0.1500 |
| Sphingolipid metabolism | 1.1410 | 4 | 0.0257 | 0.4912 |
| Folate biosynthesis | 1.2000 | 4 | 0.0303 | 0.3333 |
| Fructose and mannose metabolism | 0.7869 | 3 | 0.0424 | 0.1539 |
| Aminoacyl-tRNA biosynthesis | 1.4557 | 4 | 0.0555 | 0.0548 |
| Neomycin, kanamycin and gentamicin biosynthesis | 0.0787 | 1 | 0.0764 | 0.6667 |
| Glutathione metabolism | 1.1016 | 3 | 0.0959 | 0.1818 |
| Arginine biosynthesis | 0.5312 | 2 | 0.0975 | 0.3462 |
| Purine metabolism | 3.2656 | 6 | 0.1036 | 0.1879 |
| One carbon pool by folate | 0.6098 | 2 | 0.1230 | 0.4333 |
| D-Glutamine and D-glutamate metabolism | 0.1967 | 1 | 0.1805 | 0.3333 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | 0.2164 | 1 | 0.1967 | 0.6000 |
| Starch and sucrose metabolism | 0.8459 | 2 | 0.2066 | 0.1429 |
| Glycerophospholipid metabolism | 1.6918 | 3 | 0.2382 | 0.2118 |
| Ubiquinone and other terpenoid-quinone biosynthesis | 0.3344 | 1 | 0.2874 | 0.0625 |
| Linoleic acid metabolism | 0.3344 | 1 | 0.2874 | 0.0625 |
| Phenylalanine metabolism | 0.4131 | 1 | 0.3423 | 0.0500 |
| alpha-Linolenic acid metabolism | 0.4328 | 1 | 0.3553 | 0.0952 |
| Phosphatidylinositol signaling system | 1.4557 | 2 | 0.4311 | 0.0822 |
| Fatty acid elongation | 1.4754 | 2 | 0.4379 | 0.0946 |
| Butanoate metabolism | 0.5705 | 1 | 0.4399 | 0.0357 |
| Arginine and proline metabolism | 1.5344 | 2 | 0.4582 | 0.1169 |
| Glycosylphosphatidylinositol (GPI)-anchor biosynthesis | 0.6098 | 1 | 0.4619 | 0.1333 |
| Histidine metabolism | 0.6295 | 1 | 0.4727 | 0.0645 |
| Selenocompound metabolism | 0.6885 | 1 | 0.5036 | 0.0588 |
| Ether lipid metabolism | 0.7672 | 1 | 0.5421 | 0.0263 |
| Primary bile acid biosynthesis | 1.8098 | 2 | 0.5470 | 0.1099 |
| Citrate cycle (TCA cycle) | 0.8262 | 1 | 0.5690 | 0.0488 |
| Fatty acid degradation | 2.0066 | 2 | 0.6042 | 0.1089 |
| Galactose metabolism | 1.0033 | 1 | 0.6408 | 0.0800 |
| Glyoxylate and dicarboxylate metabolism | 1.1016 | 1 | 0.6755 | 0.0182 |
| Glycolysis or gluconeogenesis | 1.2000 | 1 | 0.7069 | 0.0667 |
| Alanine, aspartate and glutamate metabolism | 1.2000 | 1 | 0.7069 | 0.1167 |
| Inositol phosphate metabolism | 1.3574 | 1 | 0.7511 | 0.0441 |
| Drug metabolism—other enzymes | 1.3770 | 1 | 0.7561 | 0.0290 |
| Cysteine and methionine metabolism | 1.3967 | 1 | 0.7611 | 0.0286 |
| Steroid hormone biosynthesis | 3.9148 | 3 | 0.7644 | 0.0707 |
| Amino sugar and nucleotide sugar metabolism | 1.5541 | 1 | 0.7972 | 0.1282 |
| Tyrosine metabolism | 1.7311 | 1 | 0.8315 | 0.1264 |
| Pyrimidine metabolism | 1.9475 | 1 | 0.8657 | 0.0612 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Luo, Y.; Yang, S.; Jiang, M.; Chu, Y. LC-MS/MS-Based Serum Metabolomics and Transcriptome Analyses for the Mechanism of Augmented Renal Clearance. Int. J. Mol. Sci. 2023, 24, 10459. https://doi.org/10.3390/ijms241310459
Wang Y, Luo Y, Yang S, Jiang M, Chu Y. LC-MS/MS-Based Serum Metabolomics and Transcriptome Analyses for the Mechanism of Augmented Renal Clearance. International Journal of Molecular Sciences. 2023; 24(13):10459. https://doi.org/10.3390/ijms241310459
Chicago/Turabian StyleWang, Yidan, Yifan Luo, Shu Yang, Mingyan Jiang, and Yang Chu. 2023. "LC-MS/MS-Based Serum Metabolomics and Transcriptome Analyses for the Mechanism of Augmented Renal Clearance" International Journal of Molecular Sciences 24, no. 13: 10459. https://doi.org/10.3390/ijms241310459