
Iridium Complexes with BIAN-Type Ligands: Synthesis, Structure and Redox Chemistry
 , , , ,
, , , ,  and
and
Abstract
:
1. Introduction
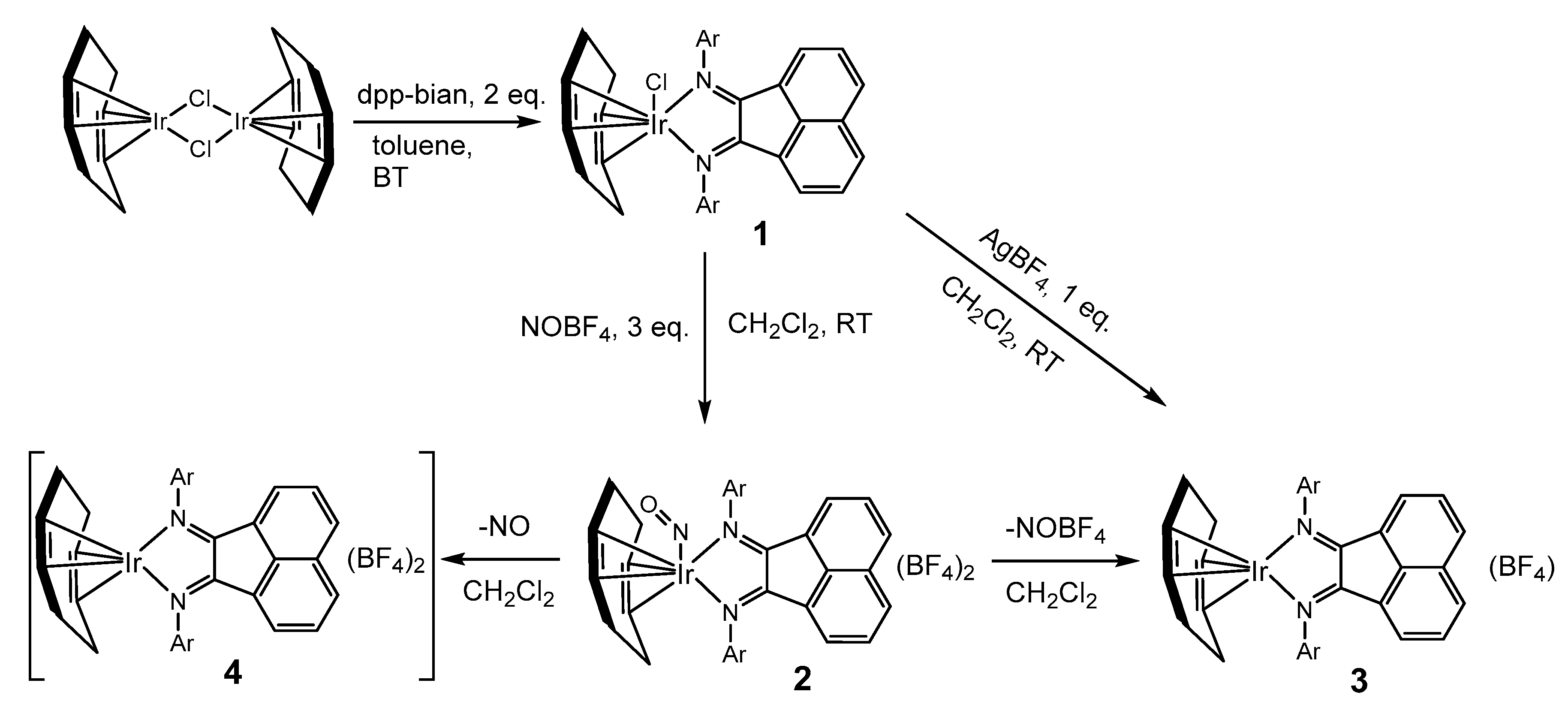
2. Results and Discussion
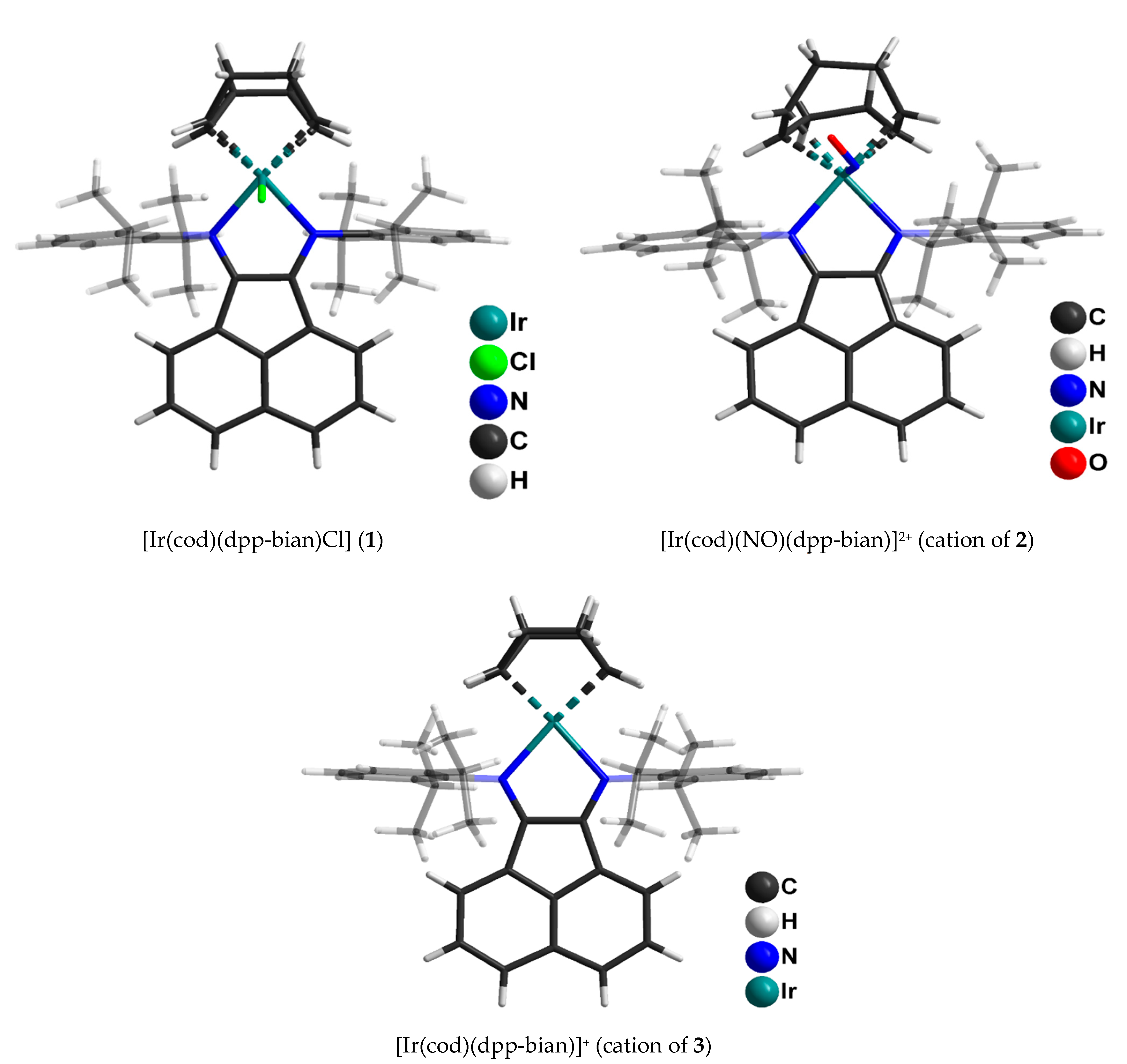
2.1. X-ray Structure Description
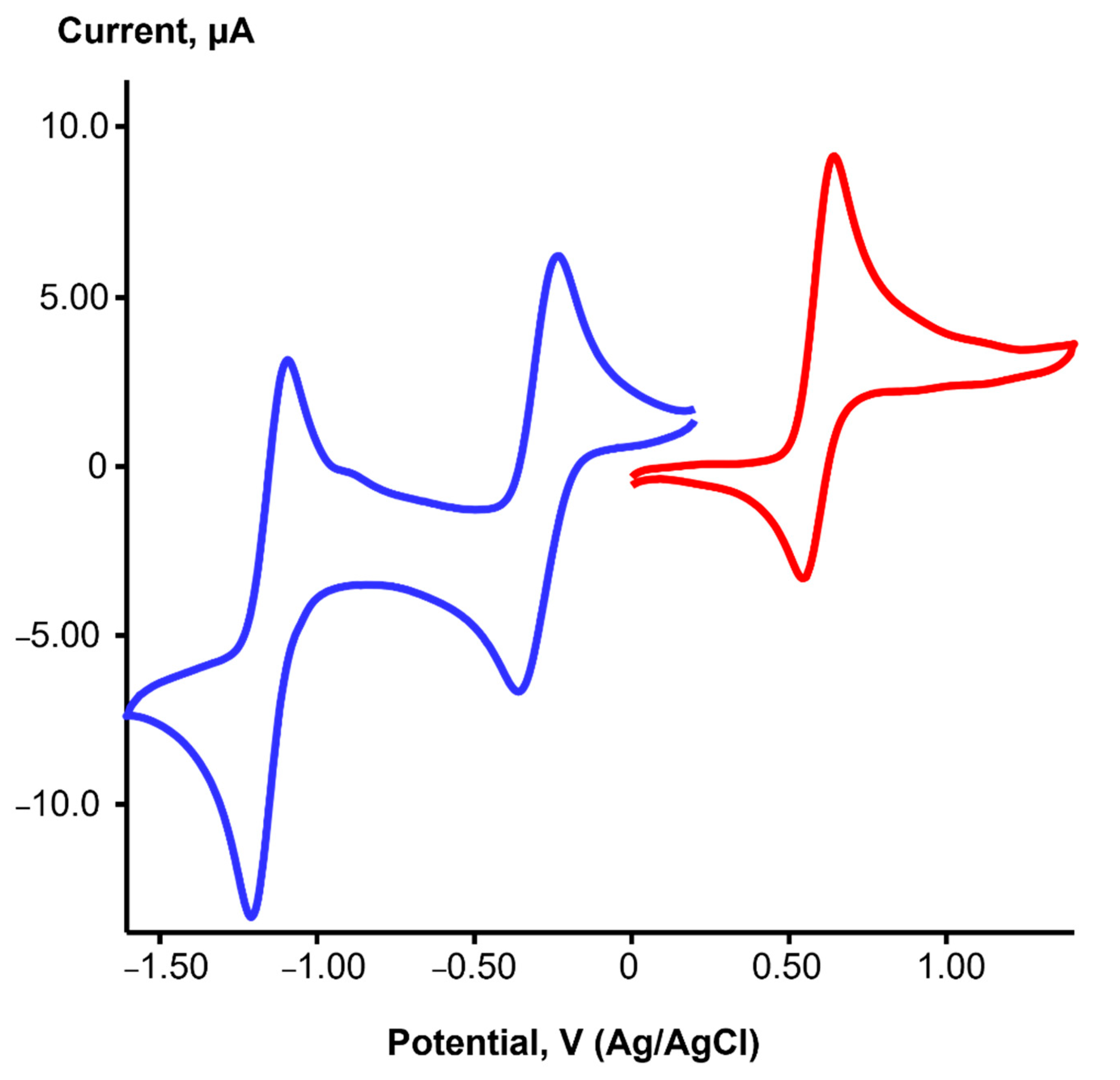
2.2. Redox Properties
2.3. Non-Innocent Properties of Dpp-Bian and NO Ligands in 1 and 2
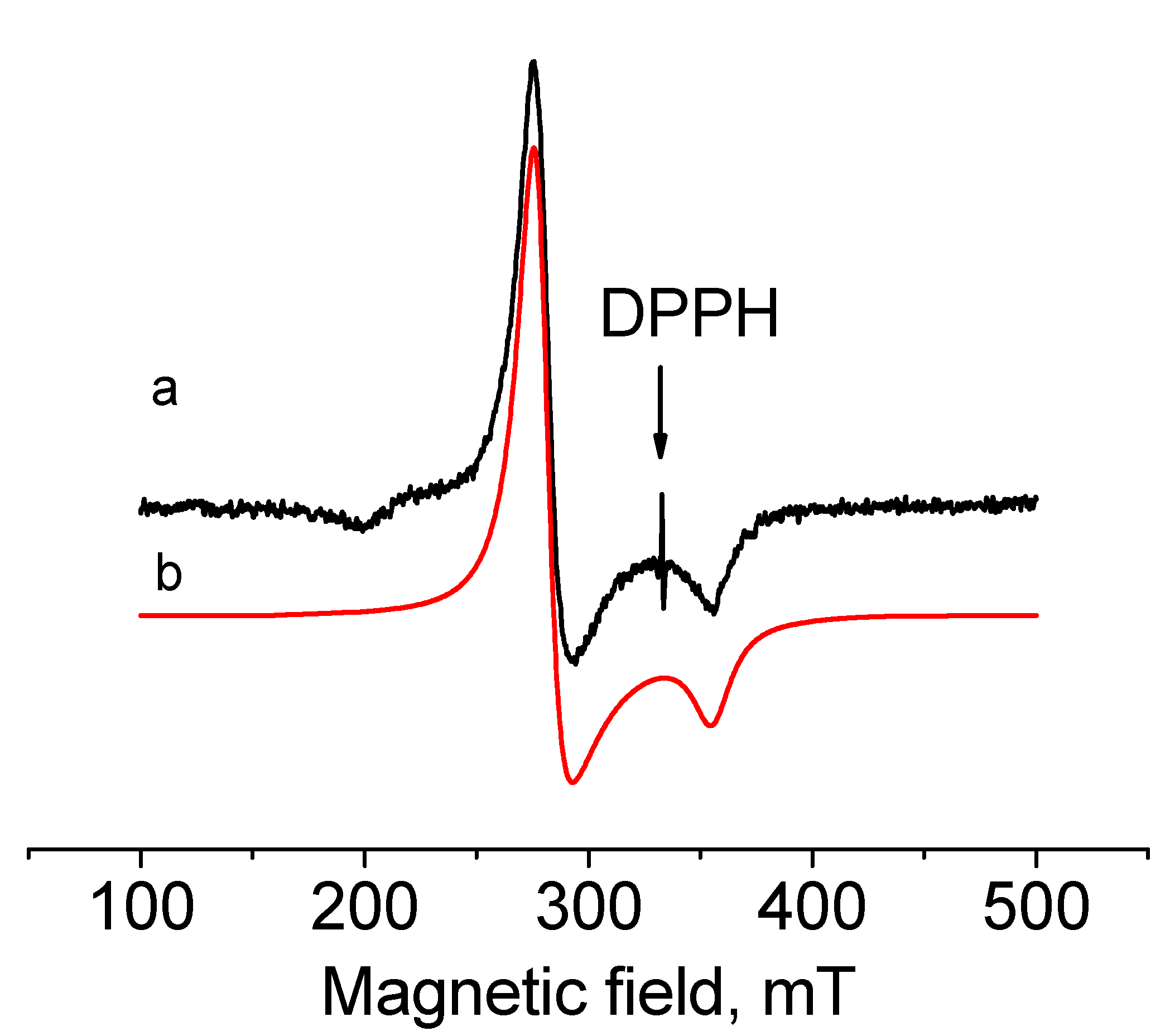
2.4. EPR Spectroscopy
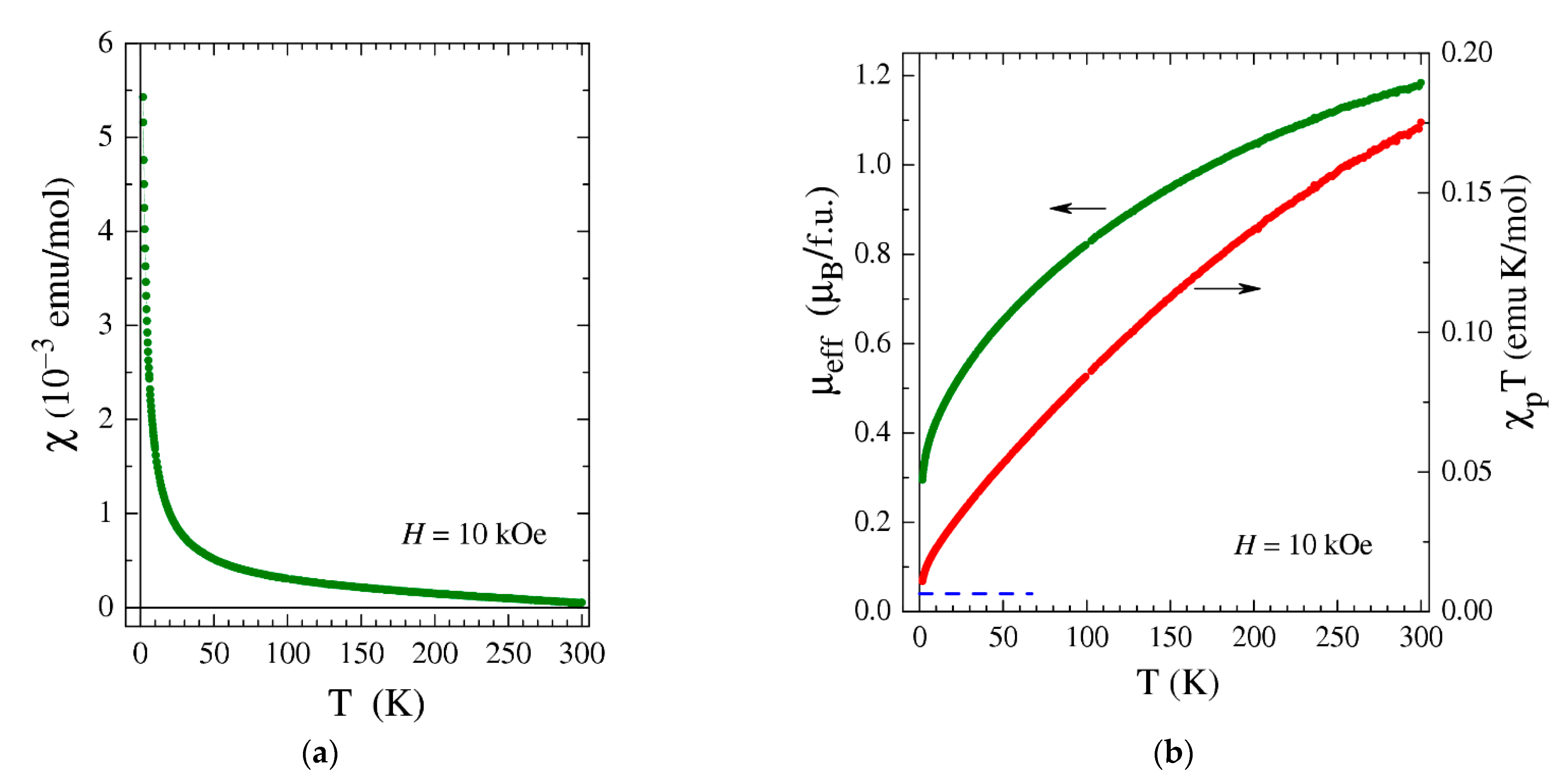
2.5. Magnetic Measurements
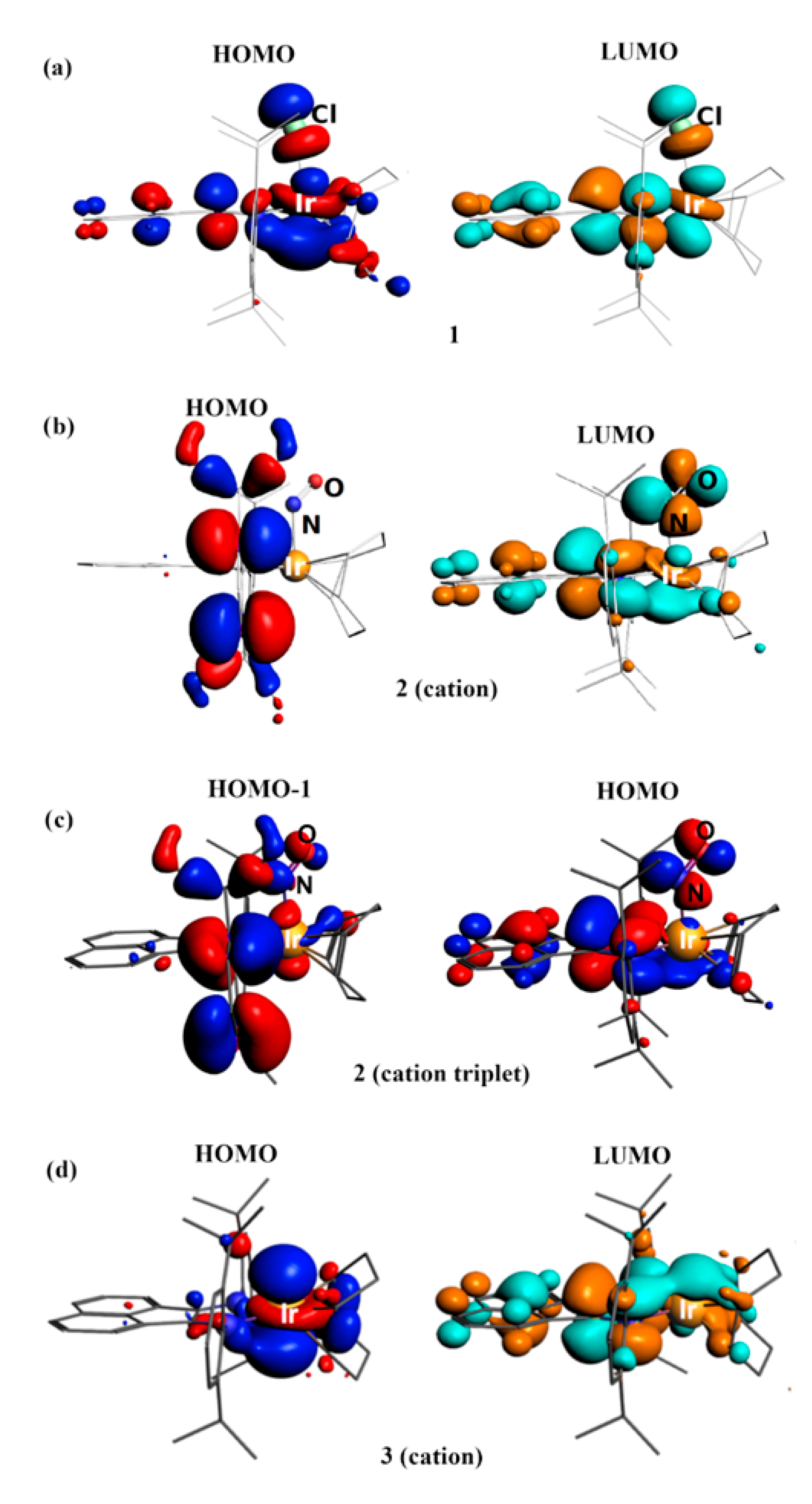
2.6. Computational Studies
3. Materials and Methods
3.1. Physical Measurements
3.2. X-ray Crystallography
3.3. Computational Details
3.4. Synthesis of [Ir(cod)(dpp-bian)Cl] (1)
3.5. Synthesis of [Ir(cod)(NO)(dpp-bian)](BF4)2 (2)
3.6. Synthesis of [Ir(cod)(dpp-bian)](BF4) (3)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernauer, J.; Pölker, J.; Jacobi von Wangelin, A. Redox-Active BIAN-Based Diimine Ligands in Metal-Catalyzed Small Molecule Syntheses. ChemCatChem 2022, 14, e202101182. [Google Scholar] [CrossRef]
- Abakumov, G.A.; Piskunov, A.V.; Cherkasov, V.K.; Fedushkin, I.L.; Ananikov, V.P.; Eremin, D.B.; Gordeev, E.G.; Beletskaya, I.P.; Averin, A.D.; Bochkarev, M.N.; et al. Organoelement Chemistry: Promising Growth Areas and Challenges. Russ. Chem. Rev. 2018, 87, 393–507. [Google Scholar] [CrossRef]
- Kaim, W. Chelate Rings of Different Sizes with Non-Innocent Ligands. Dalton Trans. 2019, 48, 8521–8529. [Google Scholar] [CrossRef] [PubMed]
- Fomenko, I.S.; Gushchin, A.L. Mono- and Binuclear Complexes of Group 5 Metals with Diimine Ligands: Synthesis, Reactivity and Prospects for Application. Russ. Chem. Rev. 2020, 89, 966–998. [Google Scholar] [CrossRef]
- Hill, N.J.; Vargas-Baca, I.; Cowley, A.H. Recent Developments in the Coordination Chemistry of Bis(Imino)Acenaphthene (BIAN) Ligands with s- and p-Block Elements. Dalton Trans. 2009, 2, 240–253. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Moskalev, M.V.; Lukoyanov, A.N.; Tishkina, A.N.; Baranov, E.V.; Abakumov, G.A. Dialane with a Redox-Active Bis-Amido Ligand: Unique Reactivity towards Alkynes. Chem. Eur. J. 2012, 18, 11264–11276. [Google Scholar] [CrossRef] [PubMed]
- Fedushkin, I.L.; Skatova, A.A.; Bazyakina, N.L.; Chudakova, V.A.; Khvoinova, N.M.; Nikipelov, A.S.; Eremenko, O.V.; Piskunov, A.V.; Fukin, G.K.; Lyssenko, K.A. Syntheses and Structures of Magnesium, Calcium, Europium, Gallium, and Zinc Complexes with Bis(Imino)Acenaphthene Ligands. Russ. Chem. Bull. 2013, 62, 1815–1828. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Hill, M.S.; Kociok-Köhn, G. Dearomatized BIAN Alkaline-Earth Alkyl Catalysts for the Intramolecular Hydroamination of Hindered Aminoalkenes. Organometallics 2014, 33, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ganguly, R.; Yongxin, L.; Díaz, J.; Soo, H.S.; García, F. Synthesis and the Optical and Electrochemical Properties of Indium(III) Bis(Arylimino)Acenaphthene Complexes. Inorg. Chem. 2017, 56, 7811–7820. [Google Scholar] [CrossRef] [PubMed]
- Lukina, D.A.; Skatova, A.A.; Sokolov, V.G.; Baranov, E.V.; Demeshko, S.; Ketkov, S.Y.; Fedushkin, I.L. Low-Coordinate Sm(II) and Yb(II) Complexes Derived from Sterically-Hindered 1,2-Bis(Imino)Acenaphthene (ArBIG-Bian). Dalton Trans. 2020, 49, 14445–14451. [Google Scholar] [CrossRef] [PubMed]
- Komlyagina, V.I.; Romashev, N.F.; Kokovkin, V.V.; Gushchin, A.L.; Benassi, E.; Sokolov, M.N.; Abramov, P.A. Trapping of Ag+ into a Perfect Six-Coordinated Environment: Structural Analysis, Quantum Chemical Calculations and Electrochemistry. Molecules 2022, 27, 6961. [Google Scholar] [CrossRef]
- Rosa, V.; Santos, C.I.M.; Welter, R.; Aullón, G.; Lodeiro, C.; Avilés, T. Comparison of the Structure and Stability of New α-Diimine Complexes of Copper(I) and Silver(I): Density Functional Theory versus Experimental. Inorg. Chem. 2010, 49, 8699–8708. [Google Scholar] [CrossRef]
- Yambulatov, D.S.; Nikolaevskii, S.A.; Kiskin, M.A.; Kholin, K.V.; Khrizanforov, M.N.; Budnikova, Y.G.; Babeshkin, K.A.; Efimov, N.N.; Goloveshkin, A.S.; Imshennik, V.K.; et al. Generation of a Hetero Spin Complex from Iron(II) Iodide with Redox Active Acenaphthene-1,2-Diimine. Molecules 2021, 26, 2998. [Google Scholar] [CrossRef] [PubMed]
- Fedushkin, I.L.; Makarov, V.M.; Sokolov, V.G.; Fukin, G.K.; Maslov, M.O.; Ketkov, S.Y. Compounds of Chromium, Titanium, and Zirconium with Different Reduced Forms of Acenaphthene-1,2-Diimine. Russ. Chem. Bull. 2014, 63, 870–882. [Google Scholar] [CrossRef]
- Villa, M.; Miesel, D.; Hildebrandt, A.; Ragaini, F.; Schaarschmidt, D.; Jacobi von Wangelin, A. Synthesis and Catalysis of Redox-Active Bis(Imino)Acenaphthene (BIAN) Iron Complexes. ChemCatChem 2017, 9, 3203–3209. [Google Scholar] [CrossRef]
- Fomenko, I.S.; Gongola, M.I.; Shul’pina, L.S.; Ikonnikov, N.S.; Komarovskikh, A.Y.; Nadolinny, V.A.; Kozlov, Y.N.; Gushchin, A.L.; Shul’pin, G.B. Mononuclear Oxidovanadium(IV) Complexes with BIAN Ligands: Synthesis and Catalytic Activity in the Oxidation of Hydrocarbons and Alcohols with Peroxides. Catalysts 2022, 12, 1168. [Google Scholar] [CrossRef]
- Romashev, N.F.; Abramov, P.A.; Bakaev, I.V.; Fomenko, I.S.; Samsonenko, D.G.; Novikov, A.S.; Tong, K.K.H.; Ahn, D.; Dorovatovskii, P.V.; Zubavichus, Y.V.; et al. Heteroleptic Pd(II) and Pt(II) Complexes with Redox-Active Ligands: Synthesis, Structure, and Multimodal Anticancer Mechanism. Inorg. Chem. 2022, 61, 2105–2118. [Google Scholar] [CrossRef]
- Johnson, L.K.; Killian, C.M.; Brookhart, M. New Pd(II)- and Ni(II)-Based Catalysts for Polymerization of Ethylene and α-Olefins. J. Am. Chem. Soc. 1995, 117, 6414–6415. [Google Scholar] [CrossRef]
- Romashev, N.F.; Bakaev, I.V.; Komlyagina, V.I.; Sokolov, M.N.; Gushchin, A.L. Synthesis and Structure of Palladacyclopentadienyl Complex With Acenaphthene-1,2-Diimine Ligand. J. Struct. Chem. 2022, 63, 1304–1312. [Google Scholar] [CrossRef]
- Yambulatov, D.S.; Nikolaevskii, S.A.; Kiskin, M.A.; Magdesieva, T.V.; Levitskiy, O.A.; Korchagin, D.V.; Efimov, N.N.; Vasil’ev, P.N.; Goloveshkin, A.S.; Sidorov, A.A.; et al. Complexes of Cobalt(II) Iodide with Pyridine and Redox Active 1,2-Bis(Arylimino)Acenaphthene: Synthesis, Structure, Electrochemical, and Single Ion Magnet Properties. Molecules 2020, 25, 2054. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Maslova, O.V.; Baranov, E.V.; Shavyrin, A.S. Redox Isomerism in the Lanthanide Complex [(Dpp-Bian)Yb(DME)(μ-Br)]2(Dpp-Bian ) 1,2-Bis[(2,6-Diisopropylphenyl)Imino]Acenaphthene). Inorg. Chem. 2009, 48, 2355–2357. [Google Scholar] [CrossRef]
- Bendix, J.; Clark, K.M. Delocalization and Valence Tautomerism in Vanadium Tris(Iminosemiquinone) Complexes. Angew. Chem. Int. Ed. 2016, 55, 2748–2752. [Google Scholar] [CrossRef] [PubMed]
- Saini, A.; Smith, C.R.; Wekesa, F.S.; Helms, A.K.; Findlater, M. Conversion of Aldimines to Secondary Amines Using Iron-Catalysed Hydrosilylation. Org. Biomol. Chem. 2018, 16, 9368–9372. [Google Scholar] [CrossRef]
- Maier, T.M.; Gawron, M.; Coburger, P.; Bodensteiner, M.; Wolf, R.; Van Leest, N.P.; De Bruin, B.; Demeshko, S.; Meyer, F. Low-Valence Anionic α-Diimine Iron Complexes: Synthesis, Characterization, and Catalytic Hydroboration Studies. Inorg. Chem. 2020, 59, 16035–16052. [Google Scholar] [CrossRef] [PubMed]
- Kluwer, A.M.; Koblenz, T.S.; Jonischkeit, T.; Woelk, K.; Elsevier, C.J. Kinetic and Spectroscopic Studies of the [Palladium(Ar-Bian)]-Catalyzed Semi-Hydrogenation of 4-Octyne. J. Am. Chem. Soc. 2005, 127, 15470–15480. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zheng, Z.; Yu, F.; Ma, S.; Holuigue, A.; Tromp, D.S.; Elsevier, C.J.; Yu, Y. [Pd(Ar-BIAN)(Alkene)]-Catalyzed Highly Chemo-, Regio-, and Stereoselective Semihydrogenation of 1,2-Allenyl Phosphonates and Related Compounds. Angew. Chem. Int. Ed. 2006, 118, 5119–5122. [Google Scholar] [CrossRef]
- Sandl, S.; Maier, T.M.; Van Leest, N.P.; Kröncke, S.; Chakraborty, U.; Demeshko, S.; Koszinowski, K.; De Bruin, B.; Meyer, F.; Bodensteiner, M.; et al. Cobalt-Catalyzed Hydrogenations via Olefin Cobaltate and Hydride Intermediates. ACS Catal. 2019, 9, 7596–7606. [Google Scholar] [CrossRef]
- Maier, T.M.; Sandl, S.; Shenderovich, I.G.; Jacobi von Wangelin, A.; Weigand, J.J.; Wolf, R. Amine-Borane Dehydrogenation and Transfer Hydrogenation Catalyzed by α-Diimine Cobaltates. Chem. Eur. J. 2019, 25, 238–245. [Google Scholar] [CrossRef]
- Ferretti, F.; Ramadan, D.R.; Ragaini, F. Transition Metal Catalyzed Reductive Cyclization Reactions of Nitroarenes and Nitroalkenes. ChemCatChem 2019, 11, 4450–4488. [Google Scholar] [CrossRef]
- Viganò, M.; Ragaini, F.; Buonomenna, M.G.; Lariccia, R.; Caselli, A.; Gallo, E.; Cenini, S.; Jansen, J.C.; Drioli, E. Catalytic Polymer Membranes under Forcing Conditions: Reduction of Nitrobenzene by CO/H2O Catalyzed by Ruthenium Bis(Arylimino)Acenaphthene Complexes. ChemCatChem 2010, 2, 1150–1164. [Google Scholar] [CrossRef]
- Ragaini, F.; Cenini, S.; Borsani, E.; Dompé, M.; Gallo, E.; Moret, M. Synthesis of N-Arylpyrroles, Hetero-Diels-Alder Adducts, and Allylic Amines by Reaction of Unfunctionalized Dienes with Nitroarenes and Carbon Monoxide, Catalyzed by Ru(CO)3(Ar-BIAN). Organometallics 2001, 20, 3390–3398. [Google Scholar] [CrossRef]
- Yakub, A.M.; Moskalev, M.V.; Bazyakina, N.L.; Fedushkin, I.L. Carbon—Carbon and Carbon—Nitrogen Bond Formation Reactions Catalyzed by the Magnesium and Calcium Acenaphthene-1,2-Diimine Complexes. Russ. Chem. Bull. 2018, 67, 473–478. [Google Scholar] [CrossRef]
- Li, L.; Lopes, P.S.; Figueira, C.A.; Gomes, C.S.B.; Duarte, M.T.; Rosa, V.; Fliedel, C.; Avilés, T.; Gomes, P.T. Cationic and Neutral (Ar-BIAN)Copper(I) Complexes Containing Phosphane and Arsane Ancillary Ligands: Synthesis, Molecular Structure and Catalytic Behaviour in Cycloaddition Reactions of Azides and Alkynes. Eur. J. Inorg. Chem. 2013, 2013, 1404–1417. [Google Scholar] [CrossRef]
- Li, L.; Lopes, P.S.; Rosa, V.; Figueira, C.A.; Lemos, M.A.N.D.A.; Duarte, M.T.; Avilés, T.; Gomes, P.T. Synthesis and Structural Characterisation of (Aryl-BIAN)Copper(I) Complexes and Their Application as Catalysts for the Cycloaddition of Azides and Alkynes. Dalton Trans. 2012, 41, 5144–5154. [Google Scholar] [CrossRef] [PubMed]
- Cenini, S.; Ragaini, F.; Tollari, S.; Paone, D. Allylic Amination of Cyclohexene Catalyzed by Ruthenium Complexes. A New Reaction Involving an Intermolecular C-H Functionalization. J. Am. Chem. Soc. 1996, 118, 11964–11965. [Google Scholar] [CrossRef]
- Fomenko, I.S.; Gushchin, A.L.; Shul’pina, L.S.; Ikonnikov, N.S.; Abramov, P.A.; Romashev, N.F.; Poryvaev, A.S.; Sheveleva, A.M.; Bogomyakov, A.S.; Shmelev, N.Y.; et al. New Oxidovanadium(IV) Complex with a BIAN Ligand: Synthesis, Structure, Redox Properties and Catalytic Activity. New J. Chem. 2018, 42, 16200–16210. [Google Scholar] [CrossRef]
- Lukoyanov, A.N.; Fomenko, I.S.; Gongola, M.I.; Shul’pina, L.S.; Ikonnikov, N.S.; Shul’pin, G.B.; Ketkov, S.Y.; Fukin, G.K.; Rumyantcev, R.V.; Novikov, A.S.; et al. Novel Oxidovanadium Complexes with Redox-Active r-Mian and r-Bian Ligands: Synthesis, Structure, Redox and Catalytic Properties. Molecules 2021, 26, 5706. [Google Scholar] [CrossRef]
- Romashev, N.F.; Gushchin, A.L.; Fomenko, I.S.; Abramov, P.A.; Mirzaeva, I.V.; Kompan’kov, N.B.; Kal’nyi, D.B.; Sokolov, M.N. A New Organometallic Rhodium(I) Complex with Dpp-Bian Ligand: Synthesis, Structure and Redox Behaviour. Polyhedron 2019, 173, 114110. [Google Scholar] [CrossRef]
- Gushchin, A.L.; Romashev, N.F.; Shmakova, A.A.; Abramov, P.A.; Ryzhikov, M.R.; Fomenko, I.S.; Sokolov, M.N. Novel Redox Active Rhodium(III) Complex with Bis(Arylimino)Acenaphthene Ligand: Synthesis, Structure and Electrochemical Studies. Mendeleev Commun. 2020, 30, 81–83. [Google Scholar] [CrossRef]
- Romashev, N.F.; Mirzaeva, I.V.; Bakaev, I.V.; Komlyagina, V.I.; Komarov, V.Y.; Fomenko, I.S.; Gushchin, A.L. Structure of a Binuclear Rhodium(I) Complex With the Acenaphthene- 1,2-Diimine Ligand. J. Struct. Chem. 2022, 63, 242–251. [Google Scholar] [CrossRef]
- Kennedy, D.F.; Messerle, B.A.; Smith, M.K. Synthesis of Cp* Iridium and Rhodium Complexes Containing Bidentate sp2-N-Donor Ligands and Counter-Anions [Cp*MCl3]-. Eur. J. Inorg. Chem. 2007, 2007, 80–89. [Google Scholar] [CrossRef]
- Singh, S.K.; Dubey, S.K.; Pandey, R.; Mishra, L.; Zou, R.Q.; Xu, Q.; Pandey, D.S. Ruthenium(II), Rhodium(III) and Iridium(III) Based Effective Catalysts for Hydrogenation under Aerobic Conditions. Polyhedron 2008, 27, 2877–2882. [Google Scholar] [CrossRef]
- Gray, K.; Page, M.J.; Wagler, J.; Messerle, B.A. Iridium(III) Cp* Complexes for the Efficient Hydroamination of Internal Alkynes. Organometallics 2012, 31, 6270–6277. [Google Scholar] [CrossRef]
- Hasan, K.; Zysman-Colman, E. Panchromic Cationic Iridium(III) Complexes. Inorg. Chem. 2012, 51, 12560–12564. [Google Scholar] [CrossRef]
- Salohiddinov, S.; Vignesh, A.; Li, Z.; Ma, Y.; Sun, W.-H. Cationic iridium (III) complexes bearing fluorinated Ar-BIAN ligands: Synthesis, structure, electronic, and electrochemical properties. J. Organomet. Chem. 2021, 951, 122002. [Google Scholar] [CrossRef]
- Hattori, T.; Matsukawa, S.; Kuwata, S.; Ishii, Y.; Hidai, M. Mono(Sulfido)-Bridged Mixed-Valence Nitrosyl Complex: Protonation and Oxidative Addition of Iodine across the Ir(II)-Ir(0) Bond. Chem. Commun. 2003, 3, 510–511. [Google Scholar] [CrossRef]
- Tiripicchio, A.; Camellini, M.T.; Neve, F.; Ghedini, M. Heterobinuclear Nitrosyl Complexes. Part 2. Crystal Structures of [(Ph3P)2(NO)Ir(μ-Dppn)(μ-Cl)PdCl][PF6]2 and [(Ph3P)Cl2Irμ-NO)-μ-Dppn)PdCl]PF6 [Dppn = 3,6-Bis(2′-Pyridyl)Pyridazine]. Dalton Trans. 1990, 5, 1651–1656. [Google Scholar] [CrossRef]
- Seechurn, C.C.C.J.; Sivakumar, V.; Satoskar, D.; Colacot, T.J. Iridium-Catalyzed C-H Borylation of Heterocycles Using an Overlooked 1,10-Phenanthroline Ligand: Reinventing the Catalytic Activity by Understanding the Solvent-Assisted Neutral to Cationic Switch. Organometallics 2014, 33, 3514–3522. [Google Scholar] [CrossRef]
- Crotti, C.; Farnetti, E.; Filipuzzi, S.; Stener, M.; Zangrando, E.; Moras, P. Evaluation of the Donor Ability of Phenanthrolines in Iridium Complexes by Means of Synchrotron Radiation Photoemission Spectroscopy and DFT Calculations. Dalton Trans. 2006, 1, 133–142. [Google Scholar] [CrossRef]
- Emerson-King, J.; Knighton, R.C.; Gyton, M.R.; Chaplin, A.B. Rotaxane Synthesis Exploiting the M(I)/M(III) Redox Couple. Dalton Trans. 2017, 46, 11645–11655. [Google Scholar] [CrossRef] [Green Version]
- Matsukawa, S.; Kuwata, S.; Hidai, M. Syntheses, Structures, and Reactivities of Mono- and Dinuclear Iridium Thiolato Complexes Containing Nitrosyl Ligands. Inorg. Chem. 2000, 39, 791–798. [Google Scholar] [CrossRef]
- Hetterscheid, D.G.H.; Kaiser, J.; Reijerse, E.; Peters, T.P.J.; Thewissen, S.; Blok, A.N.J.; Smits, J.M.M.; De Gelder, R.; De Bruin, B. IrII(Ethene): Metal or Carbon Radical? J. Am. Chem. Soc. 2005, 127, 1895–1905. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Kirina, Y.V.; Novikov, A.S.; Starova, G.L.; Kukushkin, V.Y. Efficient π-Stacking with Benzene Provides 2D Assembly of Trans-[PtCl2(p-CF3C6H4CN)2]. J. Mol. Struct. 2016, 1104, 19–23. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Boyarskiy, V.P.; Boyarskaya, I.A.; Avdontceva, M.S.; Kukushkin, V.Y. Ligation-Enhanced π-Hole···π Interactions Involving Isocyanides: Effect of π-Hole···π Noncovalent Bonding on Conformational Stabilization of Acyclic Diaminocarbene Ligands. Inorg. Chem. 2018, 57, 6722–6733. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Novikov, A.S.; Ivanov, D.M.; Bolotin, D.S.; Bokach, N.A.; Kukushkin, V.Y. Structure-Directing Weak Interactions with 1,4-Diiodotetrafluorobenzene Convert One-Dimensional Arrays of [MII(acac)2] Species into Three-Dimensional Networks. Cryst. Growth Des. 2018, 18, 3626–3636. [Google Scholar] [CrossRef]
- Novikov, A.S.; Ivanov, D.M.; Bikbaeva, Z.M.; Bokach, N.A.; Kukushkin, V.Y. Noncovalent Interactions Involving Iodofluorobenzenes: The Interplay of Halogen Bonding and Weak Lp(O)···π-Holearene Interactions. Cryst. Growth Des. 2018, 18, 7641–7654. [Google Scholar] [CrossRef]
- Eliseeva, A.A.; Ivanov, D.M.; Novikov, A.S.; Kukushkin, V.Y. Recognition of the π-Hole Donor Ability of Iodopentafluorobenzene-a Conventional σ-Hole Donor for Crystal Engineering Involving Halogen Bonding. CrystEngComm 2019, 21, 616–628. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Krykova, M.A.; Ivanov, D.M.; Novikov, A.S.; Sinelshchikova, A.A.; Volostnykh, M.V.; Konovalov, M.A.; Grigoriev, M.S.; Gorbunova, Y.G.; Kukushkin, V.Y. Reverse Arene Sandwich Structures Based upon Π-Hole⋅⋅⋅[M II ] (d 8 M=Pt, Pd) Interactions, Where Positively Charged Metal Centers Play the Role of a Nucleophile. Angew. Chem. Int. Ed. 2019, 131, 4208–4212. [Google Scholar] [CrossRef]
- Katkova, S.; Mikherdov, A.; Kinzhalov, M.; Novikov, A.; Zolotarev, A.; Boyarskiy, V.; Kukushkin, V. (Isocyano Group π-Hole)⋅⋅⋅[dz2 -MII] Interactions of (Isocyanide)[MII] Complexes, in Which Positively Charged Metal Centers (d8-M=Pt, Pd) Act as Nucleophiles. Chem. Eur. J. 2019, 25, 8590–8598. [Google Scholar] [CrossRef]
- Afanasenko, A.M.; Novikov, A.S.; Chulkova, T.G.; Grigoriev, Y.M.; Kolesnikov, I.E.; Selivanov, S.I.; Starova, G.L.; Zolotarev, A.A.; Vereshchagin, A.N.; Elinson, M.N. Intermolecular Interactions-Photophysical Properties Relationships in Phenanthrene-9,10-Dicarbonitrile Assemblies. J. Mol. Struct. 2020, 1199, 126789. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X-H⋯F-Y Systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Lanfranchi, M.; Tiripicchio, A.; Giuliano, G.; Ghedini, M. The Crystal Structure of Tris(Acetonitrile)Nitrosylbis(Triphenylphosphine)Iridium(III) Dihexafluorophosphate. An Unusual Iridium Nitrosyl Hexacoordinate Complex. Transit. Met. Chem. 1980, 5, 21–25. [Google Scholar] [CrossRef]
- Popp, J.; Klüfers, P. Bond Strength of a Diatomic Acceptor Ligand: A Reliable Measure of Its Antibond Occupation and Its Charge? Eur. J. Inorg. Chem. 2022, 2022, e202200374. [Google Scholar] [CrossRef]
- Ampßler, T.; Monsch, G.; Popp, J.; Riggenmann, T.; Salvador, P.; Schröder, D.; Klüfers, P. Not Guilty on Every Count: The “Non-Innocent” Nitrosyl Ligand in the Framework of IUPAC′s Oxidation-State Formalism. Angew. Chem. Int. Ed. 2020, 59, 12381–12386. [Google Scholar] [CrossRef]
- Hasil, A.; Beck, D.; Schröder, D.; Pillet, S.; Wenger, E.; Woike, T.; Klüfers, P.; Schaniel, D. Pas de Deux of an NO Couple: Synchronous Photoswitching from a Double-Linear to a Double-Bent Ru(NO)2 Core under Nitrosyl Charge Conservation. Angew. Chem. Int. Ed. 2022, 61, e202210671. [Google Scholar] [CrossRef]
- Fuchigami, K.; Rath, N.P.; Mirica, L.M. Mononuclear Rhodium(II) and Iridium(II) Complexes Supported by Tetradentate Pyridinophane Ligands. Inorg. Chem. 2017, 56, 9404–9408. [Google Scholar] [CrossRef]
- Hetterscheid, D.G.H.; Klop, M.; Kicken, R.J.N.A.M.; Smits, J.M.M.; Reijerse, E.J.; De Bruin, B. Hydrogen-Atom Transfer in Open-Shell Organometallic Chemistry: The Reactivity of RhII(cod) and IrII(cod) Radicals. Chem. Eur. J. 2007, 13, 3386–3405. [Google Scholar] [CrossRef]
- De Bruin, B.; Peters, T.P.J.; Thewissen, S.; Blok, A.N.J.; Wilting, J.B.M.; de Gelder, R.; Smits, J.M.M.; Gal, A.W. Dioxygen Activation by a Mononuclear IrII–Ethene Complex. Angew. Chem. Int. Ed. 2002, 114, 2239. [Google Scholar] [CrossRef]
- Empsall, H.D.; Hyde, E.M.; Shaw, B.L. Some Unusual Iridium-(I), -(II), and -(III) Complexes Formed from 2-Methoxyphenyl- or 2-Hydroxyphenyl-Di-t-Butylphosphine. Dalton Trans. 1975, 16, 1690–1696. [Google Scholar] [CrossRef]
- Figgis, B.N.; Lewis, J. The Magnetic Properties of Transition Metal Complexes. Prog. Inorg. Chem. 2007, 6, 37–239. [Google Scholar] [CrossRef]
- Norman, V.; Morrow, J.C. Magnetic Properties of Some Indium Complexes. J. Chem. Phys. 1959, 31, 455–459. [Google Scholar] [CrossRef]
- Figgis, B.N. Ligand Field Theory. In Comprehensive Coordination Chemistry; Wilkinson, G., Gillard, R.D., McCleverty, J.A., Eds.; Elsevier Ltd.: Pergamon, Turkey, 1987; Volume 1, pp. 213–279. [Google Scholar]
- Mason, R.; Thomas, K.M.; Empsall, H.D.; Fletcher, S.R.; Heys, P.N.; Hyde, E.M.; Jones, C.E.; Shaw, B.L. Synthesis and Structural Characteristics of Planar Iridium(II) Complexes. Chem. Commun. 1974, 15, 612–614. [Google Scholar] [CrossRef]
- Pandey, K.K. Mononuclear D7 Complexes of Platinum Metals. Coord. Chem. Rev. 1992, 121, 1–42. [Google Scholar] [CrossRef]
- Tezgerevska, T.; Alley, K.G.; Boskovic, C. Valence Tautomerism in Metal Complexes: Stimulated and Reversible Intramolecular Electron Transfer between Metal Centers and Organic Ligands. Coord. Chem. Rev. 2014, 268, 23–40. [Google Scholar] [CrossRef]
- Pierpont, C.G. Studies on Charge Distribution and Valence Tautomerism in Transition Metal Complexes of Catecholate and Semiquinonate Ligands. Coord. Chem. Rev. 2001, 216–217, 99–125. [Google Scholar] [CrossRef]
- Sato, O.; Cui, A.; Matsuda, R.; Tao, J.; Hayami, S. Photo-Induced Valence Tautomerism in Co Complexes. Acc. Chem. Res. 2007, 40, 361–369. [Google Scholar] [CrossRef]
- Zolotukhin, A.A.; Korshunova, A.A.; Bubnov, M.P.; Bogomyakov, A.S.; Baranov, E.V.; Cherkasov, V.K. Nickel(II) and Cobalt(III) Bis(Dioxolene) Complexes with Di(2-Pyridyl)Imine Ligands: Synthesis and Magnetic Properties. Inorg. Chim. Acta 2020, 512, 119869. [Google Scholar] [CrossRef]
- Zolotukhin, A.A.; Bubnov, M.P.; Skorodumova, N.A.; Kocherova, T.N.; Bogomyakov, A.S.; Kozlova, E.A.; Fukin, G.K.; Cherkasov, V.K. Valence Tautomerism in Cobalt Complexes Based on Isopropyl- and Cyclohexyl-Substituted o-Quinones. Inorg. Chim. Acta 2022, 534, 120811. [Google Scholar] [CrossRef]
- Mitsumi, M.; Komatsu, Y.; Hashimoto, M.; Toriumi, K.; Kitagawa, Y.; Miyazaki, Y.; Akutsu, H.; Akashi, H. Large-Amplitude Thermal Vibration-Coupled Valence Tautomeric Transition Observed in a Conductive One-Dimensional Rhodium–Dioxolene Complex. Chem. Eur. J. 2021, 27, 3074–3084. [Google Scholar] [CrossRef]
- Maity, S.; Kundu, S.; Bera, S.; Weyhermüller, T.; Ghosh, P. O-Iminobenzoquinone and o-Iminobenzosemiquinonate Anion Radical Complexes of Rhodium and Ruthenium. Eur. J. Inorg. Chem. 2016, 2016, 3691–3697. [Google Scholar] [CrossRef]
- Abakumov, G.A.; Cherkasov, V.K.; Bubnov, M.P.; Abakumova, L.G.; Zakharov, L.N.; Fukin, G.K. New Semiquinone-Catecholate Rhodium Complex with 2,2′-Dipyridyl. Russ. Chem. Bull. 1999, 48, 1762–1766. [Google Scholar] [CrossRef]
- Abakumov, G.A.; Cherkasov, V.K.; Nevodchikov, V.I.; Bubnov, M.P. Preparation of Iridium O-Semiquinone Complexes and a Study of Their Reactions with n-Donor Ligands. Bull. Acad. Sci. USSR Div. Chem. Sci. 1987, 36, 1725–1727. [Google Scholar] [CrossRef]
- Autschbach, J.; Pritchard, B. Calculation of Molecular G-Tensors Using the Zeroth-Order Regular Approximation and Density Functional Theory: Expectation Value versus Linear Response Approaches. Theor. Chem. Acc. 2011, 129, 453–466. [Google Scholar] [CrossRef]
- Paulovicova, A.; El-Ayaan, U.; Shibayama, K.; Morita, T.; Fukuda, Y. Mixed-Ligand Copper(II) Complexes with the Rigid Bidentate Bis(N-Arylimino)Acenaphthene Ligand: Synthesis, Spectroscopic-, and X-Ray Structural Characterization. Eur. J. Inorg. Chem. 2001, 2001, 2641–2646. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS Program for Scaling and Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. PLATON SQUEEZE: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Cryst. 2009, 65, 148–155. [Google Scholar] [CrossRef] [Green Version]
- ADF: SCM, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2019.
- Swart, M. A New Family of Hybrid Density Functionals. Chem. Phys. Lett. 2013, 580, 166–171. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-Type Basis Sets for the Elements 1-118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Van Lenthe, E. Geometry Optimizations in the Zero Order Regular Approximation for Relativistic Effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- Van Lenthe, E.; Van Leeuwen, R.; Baerends, E.J.; Snijders, J.G. Relativistic Regular Two-Component Hamiltonians. Int. J. Quantum Chem. 1996, 57, 281–293. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990; ISBN 9780198558651. [Google Scholar]
- Da Chai, J.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Barros, C.L.; De Oliveira, P.J.P.; Jorge, F.E.; Canal Neto, A.; Campos, M. Gaussian Basis Set of Double Zeta Quality for Atoms Rb through Xe: Application in Non-Relativistic and Relativistic Calculations of Atomic and Molecular Properties. Mol. Phys. 2010, 108, 1965–1972. [Google Scholar] [CrossRef]
- Jorge, F.E.; Canal Neto, A.; Camiletti, G.G.; MacHado, S.F. Contracted Gaussian Basis Sets for Douglas-Kroll-Hess Calculations: Estimating Scalar Relativistic Effects of Some Atomic and Molecular Properties. J. Chem. Phys. 2009, 130, 064108. [Google Scholar] [CrossRef]
- Canal Neto, A.; Jorge, F.E. All-Electron Double Zeta Basis Sets for the Most Fifth-Row Atoms: Application in DFT Spectroscopic Constant Calculations. Chem. Phys. Lett. 2013, 582, 158–162. [Google Scholar] [CrossRef]
- De Berrêdo, R.C.; Jorge, F.E. All-Electron Double Zeta Basis Sets for Platinum: Estimating Scalar Relativistic Effects on Platinum(II) Anticancer Drugs. J. Mol. Struct. Theochem. 2010, 961, 107–112. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Ayers, P.W.; Jenkins, S. Bond Metallicity Measures. Comput. Theor. Chem. 2015, 1053, 112–122. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | |||
|---|---|---|---|---|---|
| Experimental Structure | |||||
| Ir—N (dpp-bian) | 2.095 (3) | Ir—N (NO) | 2.02 (3) | Ir—N (dpp-bian) | 2.095 (3) |
| N—C (dpp-bian) | 1.324 (5); 1.448 (5) | Ir—N (dpp-bian) | 2.115 (11) | N—C (dpp-bian) | 1.298 (5); 1.452 (5) |
| Ir— Cl | 2.4751 (15) | N—C (dpp-bian) | 1.296 (16); 1.449 (17); | Ir—C (cod) | 2.127 (5) |
| Ir—C (cod) | 2.147 (5); 2.121 (4) | Ir—C (cod) | 2.181 (16); 2.241 (16); | ||
| Calculations for Ground State | |||||
| Ir—N (dpp-bian) | 2.103; 2.126 | Ir—N (NO) | 1.987 | Ir—N (dpp-bian) | 2.113; 2.109 |
| N—C (dpp-bian) | 1.322 | Ir—N (dpp-bian) | 2.164; 2.131 | N—C (dpp-bian) | 1.302 |
| Ir— Cl | 2.497 | N—C (dpp-bian) | 1.303; 1.309 | ||
| Ir—C (cod) | 2.127; 2.143; 2.126; 2.132 | Ir—C (cod) | 2.330; 2.250; 2.169; 2.221 | Ir—C (cod) | 2.172; 2.158; 2.176; 2.157 |
| [Ir(cod)(dpp-bian)Cl] (1) | |||||
|---|---|---|---|---|---|
| Orbital | E, eV | Ir | dpp-bian | cod | Cl |
| HOMO-2 | −5.460 | 14.6% | 2.9% | 3.9% | 78.6% |
| HOMO-1 | −5.275 | 23.0% | 15.2% | 8.2% | 53.6% |
| HOMO | −4.364 | 41.7% | 34.3% | 10.5% | 13.5% |
| LUMO | −3.601 | 22.7% | 64.2% | 0.0% | 13.1% |
| LUMO+1 | −3.032 | 0.0% | 100.0% | 0.0% | 0.0% |
| LUMO+2 | −1.700 | 0.0% | 100.0% | 0.0% | 0.0% |
| [Ir(cod)(NO)(dpp-bian)]2+ (Cation of 2) | |||||
| Orbital | E, eV | Ir | dpp-bian | cod | NO |
| HOMO-2 | −11.314 | 1.2% | 95.9% | 2.9% | 0.0% |
| HOMO-1 | −11.201 | 0.0% | 100.0% | 0.0% | 0.0% |
| HOMO | −11.157 | 0.0% | 100.0% | 0.0% | 0.0% |
| LUMO | −10.001 | 5.7% | 58.7% | 4.2% | 31.4% |
| LUMO+1 | −9.883 | 10.5% | 9.3% | 6.3% | 73.9% |
| LUMO+2 | −8.975 | 24.9% | 36.3% | 6.0% | 32.8% |
| [Ir(cod)(dpp-bian)]+ (Cation of 3) | |||||
| Orbital | E, eV | Ir | dpp-bian | cod | |
| HOMO-2 | −8.483 | 28.8% | 58.3% | 12.9% | |
| HOMO-1 | −8.318 | 80.8% | 14.4% | 4.8% | |
| HOMO | −7.651 | 86.5% | 10.7% | 2.8% | |
| LUMO | −6.804 | 0.0% | 97.6% | 2.4% | |
| LUMO + 1 | −5.911 | 0.0% | 99.3% | 0.7% | |
| LUMO + 2 | −4.493 | 5.2% | 86.7% | 8.1% | |
| gxx | gyy | gzz | giso | |
|---|---|---|---|---|
| Perturbational SO treatment | 2.917 | 2.757 | 2.029 | 2.568 |
| Two-component ZORA SO | 2.691 | 2.598 | 1.893 | 2.394 |
| Experiment | 2.393 | 2.393 | 1.88 | 2.222 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romashev, N.F.; Bakaev, I.V.; Komlyagina, V.I.; Abramov, P.A.; Mirzaeva, I.V.; Nadolinny, V.A.; Lavrov, A.N.; Kompan’kov, N.B.; Mikhailov, A.A.; Fomenko, I.S.; et al. Iridium Complexes with BIAN-Type Ligands: Synthesis, Structure and Redox Chemistry. Int. J. Mol. Sci. 2023, 24, 10457. https://doi.org/10.3390/ijms241310457
Romashev NF, Bakaev IV, Komlyagina VI, Abramov PA, Mirzaeva IV, Nadolinny VA, Lavrov AN, Kompan’kov NB, Mikhailov AA, Fomenko IS, et al. Iridium Complexes with BIAN-Type Ligands: Synthesis, Structure and Redox Chemistry. International Journal of Molecular Sciences. 2023; 24(13):10457. https://doi.org/10.3390/ijms241310457
Chicago/Turabian StyleRomashev, Nikolai F., Ivan V. Bakaev, Veronika I. Komlyagina, Pavel A. Abramov, Irina V. Mirzaeva, Vladimir A. Nadolinny, Alexander N. Lavrov, Nikolai B. Kompan’kov, Artem A. Mikhailov, Iakov S. Fomenko, and et al. 2023. "Iridium Complexes with BIAN-Type Ligands: Synthesis, Structure and Redox Chemistry" International Journal of Molecular Sciences 24, no. 13: 10457. https://doi.org/10.3390/ijms241310457