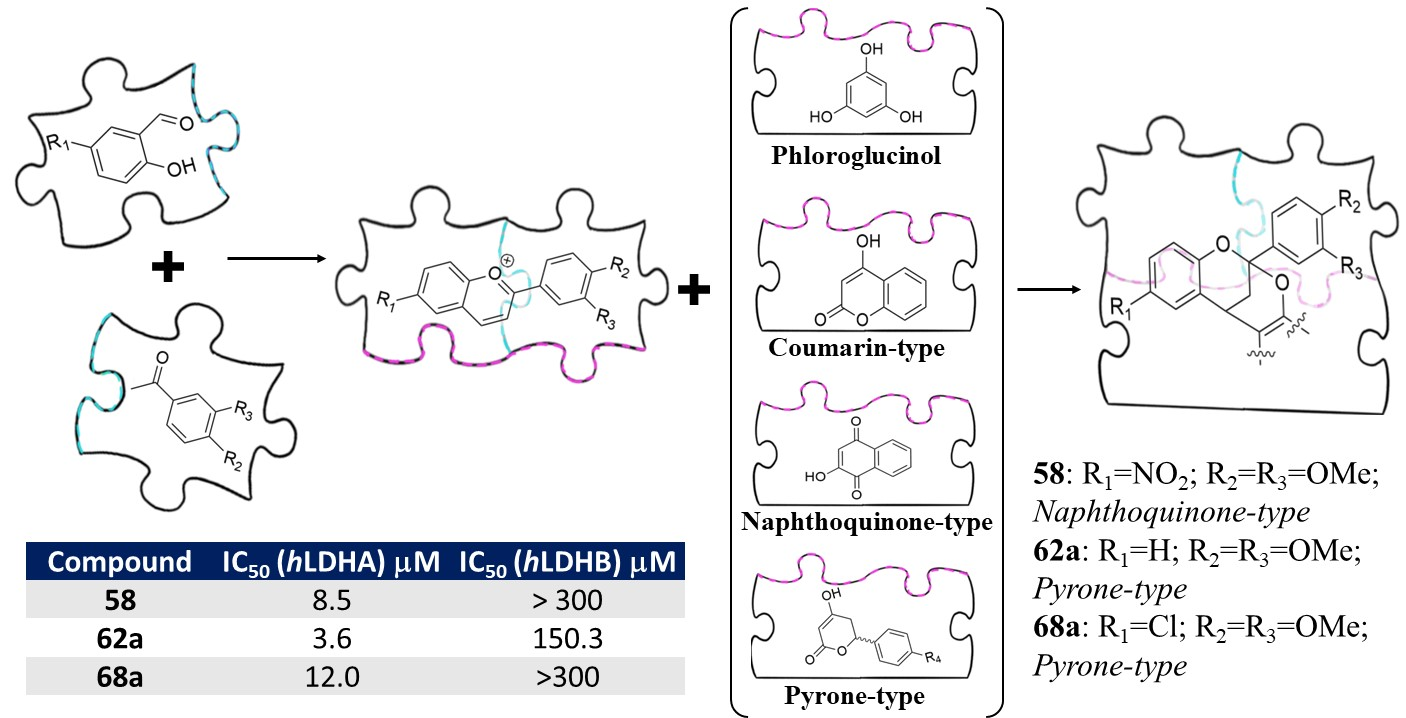
3.3. General Methodology B Applied to Synthesize 2,8-Dioxabicyclo[3.3.1]nonanes (1–6, 42–70)
In a round flask was added the flavylium salt species (
27–
35) (0.5 mmol), phloroglucinol (
36), 4-hydroxycoumarin (
37), 2-hydroxy-1,4-naphthoquinone (
38), 4-hydroxy-6-phenyl-5,6-dihydro-2H-pyran-2-one (
39), 6-(4-chlorophenyl)-4-hydroxy-5,6-dihydro-2H-pyran-2-one (
40) or 4-hydroxy-6-(4-methoxyphenyl)-5,6-dihydro-2
H-pyran-2-one (
41) (0.5 mmol), and absolute MeOH (8 mL). The mixture was stirred overnight at 50 °C in an oil bath using the method previously described by Kraus et al. [
52] and also used by us [
35,
45,
46]. Finally, the solvent was removed and the crude was purified by CC using Silica gel 60 as stationary phase.
The dioxabicyclic derivatives
1 (0.069 g, 34% yield from
21),
2 (0.170 g, 64% from
22),
3 (0.084 g, 43% from
23),
4 (0.032 g, 22% from
21),
5 (0.088 g, 45% from
22),
6 (0.077 g, 42% from
23),
48 (0.123 g, 55% from
21),
51 (0.129 g, 56% from
21), and
57 (0.053 g, 23% from
21) were previously described by us [
35] and others [
52,
53,
54] with comparable yields and their structures could be confirmed by analogy to their spectral data with those reported in the literature:
1-
2 [
35,
52],
3-6 [
35],
48 [
53],
51 [
53], and
57 [
54]. Analytical HPLC (
λ = 280 nm): compound
1 (purity: 98%;
tR = 15.9 min); compound
2 (purity: 97%;
tR = 21.2 min); compound
3 (purity: 98%;
tR = 18.8 min); compound
4 (purity: 97%;
tR = 18.0 min); compound
5 (purity: >99%;
tR = 14.9 min); compound
6 (purity: 98%;
tR = 20.7 min); compound
48 (purity 98%;
tR = 21.2 min); compound
51 (purity >99%;
tR = 19.0 min); and compound
57 (purity 97%;
tR = 21.6 min).
3.3.1. 2-(3′,4′-Dimethoxyphenyl)-chromane-(4→4″, 2→O-5″)-phloroglucinol (42)
General methodology B was applied using the flavylium salt 30 (0.182 g) and phloroglucinol (36, 0.063 g, 0.5 mmol). The purification step was performed by CC using mixtures of hexane-EtOAc (75:25) as mobile phase. Pure compound 42 was obtained as a pale orange amorphous solid (0.07 g, 38% from 21). Melting point: 77–80 °C (decomposes); Compound 42: 1H NMR (400 MHz, CDCl3) δ 7.28 (dd, J = 7.5, 1.7 Hz, 1H, H-5), 7.15 (ov, 1H, H-5′), 7.13 (br s, 1H, H-2′), 6.97 (ddd, J = 8.1, 7.5, 1.7 Hz, 1H, H-7), 6.87 (d, J = 8.9 Hz, 1H, H-6′), 6.81 (dd, J = 8.1, 1.2 Hz, 1H, H-8), 6.75 (td, J = 7.5, 1.2 Hz, 1H, H-6), 5.84 (br s, 2H, H-2″, H-6″), 4.26 (t, J = 3.0 Hz, 1H, H-4), 3.74 (s, 6H, 3′-OMe, 4′-OMe), 2.29 (d, J = 3.0 Hz, 2H, H-3); 13C NMR (100 MHz, CDCl3) δ 158.2 * (C-1″ (D)), 156.3 * (C-3″ (D)), 154.6 (C-9 (A)), 153.7 (C-5″ (D)), 150.8 (C-4′ (B)), 150.2 (C-3′ (B)), 136.4 (C-1′ (B)), 129.3 (C-10 (A)), 129.0 (C-5 (A)), 128.5 (C-7(A)), 122.1 (C-6(A)), 119.7 (C-6′ (B)), 117.0 (C-8 (A)), 112.5 (C-5′ (B)), 111.0 (C-2′ (B)), 107.3 (C-4″ (D)), 100.0 (C-2 (C)), 96.9 # (C-2″ (D)), 95.8 # (C-6″ (D)), 56.6 (3′-OMe, 4′-OMe), 34.8 (C-3 (C)), 28.2 (C-4 (C)); *, # these signals may be interchangeable. HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C23H20O6 392.126, found 392.1265. Analytical HPLC (λ = 280 nm): purity: >99%; tR = 15.9 min.
3.3.2. 2-(3′,4′-Dimethoxyphenyl)-6-nitrochromane-(4→4″, 2→O-5″)-phloroglucinol (43)
General methodology B was applied using the flavylium salt 31 (0.205 g) and phloroglucinol (36, 0.063 g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (95:5) as mobile phase. Pure compound 43 was obtained as a pale yellow amorphous solid (0.116 g, 52% from 22). Melting point: 193–195 °C; Compound 43: 1H NMR (400 MHz, DMSO-d6) δ 8.20 (d, J = 2.8 Hz, 1H, H-5), 8.04 (dd, J = 9.0, 2.8 Hz, 1H, H-7), 7.03 (d, J = 8.3 Hz, 1H, H-5′), 7.27–7.22 (ov, 2H, H-2′, H-6′), 7.17 (d, J = 9.0 Hz, 1H, H-8), 5.97 (d, J = 2.2 Hz, 1H, H-2″), 5.89 (d, J = 2.2 Hz, H-6″), 4.44 (br s, 1H, H-4), 3.80 (s, 3H, 3′-OMe), 3.78 (s, 3H, 4′-OMe), 2.45–2.36 (m, 2H, H-3); 13C NMR (100 MHz, DMSO-d6) δ 158.5 (C-9 (A)), 158.0 (C-1″ (D)), 155.5 (C-3″ (D)), 152.8 (C-5″ (D)), 149.9 (C-4′ (B)), 149.1 (C-3′ (B)), 141.4 (C-6(A)), 133.4 (C-1′ (B)), 129.6 (C-10 (A)), 124.2 (C-7(A)), 123.4 (C-5 (A)), 118.6 (C-6′ (B)), 117.5 (C-8 (A)), 111.9 (C-5′ (B)), 111.1 (C-2′ (B)), 104.3 (C-4″ (D)), 100.1 (C-2 (C)), 96.8 (C-2″ (D)), 95.0 (C-6″ (D)), 56.2 (3′-OMe, 4′-OMe), 32.0 (C-3 (C)), 26.6 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C23H19NO8 437.1111, found 437.1108. Analytical HPLC (λ = 280 nm): purity: 98%; tR = 16.7 min.
3.3.3. 2-(3′,4′-Dimethoxyphenyl)-6-chlorochromane-(4→4″, 2→O-5″)-phloroglucinol (44)
General methodology B was applied using the flavylium salt 32 (0.199 g) and phloroglucinol (36, 0.063 g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-MeOH (97:3) as mobile phase. Pure compound 44 was obtained as a pale pink amorphous solid (0.135 g, 62% from 23). Melting point: 257–259 °C; Compound 44: 1H NMR (400 MHz, MeOD) δ 7.37 (d, J = 2.6 Hz, 1H, H-5), 7.26–7.23 (ov, 2H, H-2′, H-6′), 7.07 (dd, J = 8.6, 2.6 Hz, 1H, H-7), 7.00 (d, J = 8.9 Hz, 1H, H-5′), 6.90 (d, J = 8.6 Hz, 1H, H-8), 5.98-5.96 (ov, 2H, H-2″, H-6″), 4.365 (t, J = 3.0 Hz, 1H, H-4), 3.87 (s, 3H, 4′-OMe), 3.86 (s, 3H, 3′-OMe), 2.30–2.19 (m, 2H, H-3); 13C NMR (100 MHz, MeOD) δ 158.5 (C-3″ (D)), 156.3 (C-1″ (D)), 154.5 (C-5″ (D)), 152.5 (C-9 (A)), 150.9 (C-4′ (B)), 150.2 (C-3′ (B)), 136.0 (C-1′ (B)), 131.2 (C-10 (A)), 128.4 (C-5 (A)), 128.2 (C-7(A)), 126.7 (C-6(A)), 119.7 (C-6′ (B)), 118.5 (C-8 (A)), 112.4 (C-5′ (B)), 111.0 (C-2′ (B)), 106.5 (C-4″ (D)), 100.2 (C-2 (C)), 97.0 (C-2″ (D)), 95.9 (C-6″ (D)), 55.1 (3′-OMe), 55.0 (4′-OMe), 32.8 (C-3 (C)), 26.6 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C23H19ClO6 426.087, found 426.0866. Analytical HPLC (λ = 280 nm): purity: >99%; tR = 17.6 min.
3.3.4. 2-(3′,4′-Dihydroxyphenyl)-chromane-(4→3″, 2→O-4″)-4″-hydroxycoumarin (45)
General methodology B was applied using the flavylium salt 27 (0.168 g) and 4-hydroxycoumarin (37, 0.081g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (95:5) as mobile phase. Pure compound 45 was obtained as a pink amorphous solid (0.159 g, 72% from 21). Melting point: 128–130 °C (decomposes); Compound 45: 1H NMR (400 MHz, DMSO-d6) δ 7.79 (dd, J = 7.9, 1.6 Hz, 1H, H-5″), 7.62 (ddd, J = 8.5, 7.3, 1.6, 1H, H-7″), 7.43–7.33 (ov, 3H, H-5, H-6″, H-8″), 7.18 (ov, 2H, H-7, H-2′), 7.05 (dd, J = 8.3, 2.3 Hz, 1H, H-6′), 7.00 (dd, J = 8.2, 1.2 Hz, 1H, H-8), 6.96 (td, J = 7.4, 1.2 Hz, 1H, H-6), 6.85 (d, J = 8.3 Hz, 1H, H-5′), 4.21 (d, J = 3.0 Hz, 1H, H-4), 2.54–2.38 (m, 2H, H-3); 13C NMR (100 MHz, DMSO-d6) δ 160.5 (C-2″ (D)), 157.5 (C-4″ (D)), 151.8 (C-9″ (D)), 151.2 (C-9 (A)), 146.3 (C-4′ (B)), 145.1 (C-3′ (B)), 132.5 (C-7″ (D)), 130.3 (C-1′ (B)), 128.3 (C-7(A)), 127.8 * (C-6″ (D)), 125.6 (C-10 (A)), 124.6 (C-5 (A)), 122.4 (C-5″ (D)), 121.7 (C-6(A)), 116.6 (C-8″ (D), C-6′ (B)), 116.1 (C-8 (A)), 115.4 (C-5′ (B)), 114.4 (C-10″ (D)), 113.3 (C-2′ (B)), 105.9 (C-3″ (D)), 100.5 (C-2 (C)), 31.6 (C-3 (C)), 26.8 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C26H20O6 428.126, found 428.1261. Analytical HPLC (λ = 280 nm): purity: >99%; tR = 17.2 min.
3.3.5. 2-(3′,4′-Dihydroxyphenyl)-6-nitrochromane-(4→3″, 2→O-4″)-4″-hydroxycoumarin (46)
General methodology B was applied using the flavylium salt 28 (0.191 g) and 4-hydroxycoumarin (37, 0.081g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (95:5). Pure compound 46 was obtained as a pale grey amorphous solid (0.228 g, 79% from 22). Melting point: 128–130 °C (decomposes); Compound 46: 1H NMR (400 MHz, MeOD) δ 8.23 (d, J = 2.8 Hz, 1H, H-5), 7.99 (dd, J = 9.0, 2.8 Hz 1H, H-7), 7.74 (dd, J = 8.5, 1.6 Hz, 1H, H-5″), 7.50 (ddd, J = 8.2, 7.5, 1.6, 1H, H-7″), 7.26–7.20 (ov, 2H, H-6″, H-8″), 7.11 (d, J = 2.3 Hz, 1H, H-2′), 7.05 (d, J = 9.0 Hz, 1H, H-8), 7.00 (dd, J = 8.3, 2.3 Hz, 1H, H-6′), 6.78 (d, J = 8.3 Hz, 1H, H-5′), 4.30 (t, J = 3.0 Hz, 1H, H-4), 2.50 (dd, J = 14.0, 3.0 Hz, 1H, H-3), 2.39 (dd, J = 14.0, 3.0 Hz, 1H, H-3); 13C NMR (100 MHz, MeOD) δ 163.3 (C-2″ (D)), 159.9 (C-4″ (D)), 158.5 (C-9 (A)), 153.9 (C-9″ (D)), 148.0 (C-4′ (B)), 146.7 (C-3′ (B)), 143.6 (C-6(A)), 134.0 (C-7″ (D)), 131.6 (C-1′ (B)), 128.3 (C-10 (A)), 125.9 (C-6″ (D)), 125.5 (C-7(A)), 124.8 (C-5 (A)), 123.9 (C-5″ (D)), 118.5 (C-6′ (B)), 118.3 (C-8 (A)), 117.9 (C-8″ (D)), 116.4 (C-5′ (B)), 116.1 (C-10″ (D)), 114.2 (C-2′ (B)), 106.4 (C-3″ (D)), 102.6 (C-2 (C)), 33.0 (C-3 (C)), 28.8 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C24H15NO8 445.0798, found 445.0795. Analytical HPLC (λ = 280 nm): purity: 99%; tR = 17.8 min.
3.3.6. 2-(3′,4′-Dihydroxyphenyl)-6-chlorochromane-(4→3″, 2→O-4″)-4″-hydroxycoumarin (47)
General methodology B was applied using the flavylium salt 29 (0.185 g) and 4-hydroxycoumarin (37, 0.081g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (95:5). Pure compound 47 was obtained as a pale pink amorphous solid (0.225 g, 81% from 23). Melting point: 98–100 °C (decomposes); Compound 47: 1H NMR (400 MHz, DMSO-d6) δ 7.81 (dd, J = 7.9, 1.6 Hz, 1H, H-5″), 7.65 (ddd, J = 8.7, 7.3, 1.6, 1H, H-7″), 7.44–7.37 (ov, 2H, H-6″, H-8″), 7.36 (d, J = 2.7 Hz, 1H, H-5), 7.24 (dd, J = 8.7, 2.6 Hz 1H, H-7), 7.17 (d, J = 2.3 Hz, 1H, H-2′), 7.05 (ov, 2H, H-8, H-6′), 6.85 (d, J = 8.3 Hz, 1H, H-5′), 4.23 (br t, J = 3.0 Hz, 1H, H-4), 2.55 (dd, J = 13.9, 3.2 Hz, 1H, H-3), 2.44 (dd, J = 13.9, 2.9 Hz, 1H, H-3); 13C NMR (100 MHz, DMSO-d6) δ 160.5 (C-2″ (D)), 157.5 (C-4″ (D)), 151.8 (C-9″ (D)), 150.2 (C-9 (A)), 146.4 (C-4′ (B)), 145.1 (C-3′ (B)), 132.7 (C-7″ (D)), 129.9 (C-1′ (B)), 128.0 (C-7(A)), 127.6 (C-10 (A)), 127.1 (C-5 (A)), 125.1 (C-6(A)), 124.6 (C-8″ (D)), 122.4 (C-5″ (D)), 117.9 (C-8 (A)), 116.8 (C-6′ (B)), 116.6 (C-6″ (D)), 115.4 (C-5′ (B)), 114.3 (C-10″ (D)), 113.2 (C-2′ (B)), 105.3 (C-3″ (D)), 100.6 (C-2 (C)), 30.7 (C-3 (C)), 26.7 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C24H15ClO6 434.0557, found 434.0557. Analytical HPLC (λ = 280 nm): purity: 98%; tR = 19.0 min.
3.3.7. 2-(3′,4′-Dimethoxyphenyl)-6-nitrochromane-(4→3″, 2→O-4″)-4″-hydroxycoumarin (49)
General methodology B was applied using the flavylium salt 31 (0.205 g) and 4-hydroxycoumarin (37, 0.081g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (97:3). Pure compound 49 was obtained as a white amorphous solid (0.126 g, 54% from 22). Melting point: 142–144 °C; Compound 49: 1H NMR (400 MHz, MeOD) δ 8.43 (d, J = 2.7 Hz, 1H, H-5), 8.08 (dd, J = 9.0, 2.7 Hz 1H, H-7), 7.84 (dd, J = 7.9, 1.6 Hz, 1H, H-5″), 7.54 (ddd, J = 8.7, 7.3, 1.6, 1H, H-7″), 7.33-7.26 (ov, 3H, H-6′, H-6″, H-8″), 7.21 (d, J = 2.2 Hz, 1H, H-2′), 7.11 (d, J = 9.0 Hz, 1H, H-8), 6.98 (d, J = 8.5 Hz, 1H, H-5′), 4.48 (t, J = 3.0 Hz, 1H, H-4), 3.95 (s, 3H, 3′-OMe), 3.94 (s, 3H, 4′-OMe), 2.51 (qd, J = 13.8, 3.0 Hz, 2H, H-3); 13C NMR (100 MHz, MeOD) δ 161.3 (C-2″ (D)), 158.3 (C-4″ (D)), 156.9 (C-9 (A)), 152.8 (C-9″ (D)), 152.8 (C-4′ (B)), 150.5 (C-3′ (B)), 142.6 (C-6(A)), 132.8 (C-7″ (D)), 131.2 (C-1′ (B)), 126.4 (C-10 (A)), 124.7 * (C-8″ (D)), 124.6 (C-7(A)), 124.3 (C-5 (A)), 122.9 (C-5″ (D)), 118.4 * (C-6′ (B)), 117.3 (C-8 (A)), 117.3 * (C-6″ (D)), 114.9 (C-10″ (D)), 111.1 (C-5′ (B)), 109.0 (C-2′ (B)), 105.3 (C-3″ (D)), 100.8 (C-2 (C)), 56.4 (3′-OMe), 56.3 (4′-OMe), 32.4 (C-3 (C)), 27.4 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C26H19NO8 473.1110, found 473.1111. Analytical HPLC (λ = 280 nm): purity: 97%; tR = 21.3 min.
3.3.8. 2-(3′,4′-Dimethoxyphenyl)-6-chlorochromane-(4→3″, 2→O-4″)-4″-hydroxycoumarin (50)
General methodology B was applied using the flavylium salt 32 (0.199 g) and 4-hydroxycoumarin (37, 0.081g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-MeOH (98:2). Pure compound 50 was obtained as a white amorphous solid (0.168 g, 81% from 23). Melting point: 130–133 °C; Compound 50: 1H NMR (400 MHz, CDCl3) δ 7.84 (dd, J = 7.9, 1.3 Hz, 1H, H-5″), 7.53–7.49 (ov, 2H, H-5, H-8″), 7.28–7.25 (ov, 3H, H-6′, H-6″, 7″), 7.21 (d, J = 2.2 Hz, 1H, H-2′), 7.13 (dd, J = 8.7, 2.5 Hz 1H, H-7), 6.96 (d, J = 8.5 Hz, 2H, H-8, H-5′), 4.33 (t, J = 3.0 Hz, 1H, H-4), 3.93 (s, 6H, 3′-OMe, 4′-OMe), 2.42 (d, J = 3.0 Hz, 2H, H-3); 13C NMR (100 MHz, CDCl3) δ 161.6 (C-2″ (D)), 158.6 (C-9″ (D)), 152.7 (C-4″ (D)), 150.2 (C-9 (A), C-4′ (B)), 149.2 (C-3′ (B)), 124.3 (C-7″ (D)), 132.6 (C-8″ (D)), 132.0 * (C-1′ (B)), 128.6 (C-7(A)), 128.1 (C-5 (A)), 127.2 * (C-6(A)), 126.9 * (C-10 (A)), 122.9 (C-5″ (D)), 118.4 (C-6′ (B)), 117.8 (C-8 (A)), 117.2 (C-6″ (D)), 115.7 (C-3″ (D)), 115.1 (C-10″ (D)), 111.1 (C-5′ (B)), 109.1 (C-2′ (B)), 100.5 (C-2 (C)), 56.3 (3′-OMe), 56.2 (4′-OMe), 32.8 (C-3 (C)), 27.4 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C26H19ClO6 462.087, found 462.0868. Analytical HPLC (λ = 280 nm): purity: >99%; tR = 22.9 min.
3.3.9. 2-(4′-Hydroxyphenyl)-6-nitrochromane-(4→3″, 2→O-4″)-4″-hydroxycoumarin (52)
General methodology B was applied using the flavylium salt 34 (0.183 g) and 4-hydroxycoumarin (37, 0.081g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (95:5). Pure compound 52 was obtained as a white amorphous solid (0.158 g, 58% from 22). Melting point: 275–278 °C; Compound 52: 1H NMR (400 MHz, DMSO-d6) δ 8.23 (d, J = 2.9 Hz, 1H, H-5), 8.10 (dd, J = 9.0, 2.9 Hz 1H, H-7), 7.84 (dd, J = 8.0, 1.7 Hz, 1H, H-5″), 7.68–7.63 (ov, 3H, H-2′, H-6′, H-7″), 7.43 (dd, J = 8.5, 1.1 Hz, 1H, H-8″), 7.39 (td, J = 7.6, 1.1 Hz, 1H, H-6″), 7.26 (d, J = 9.0 Hz, 1H, H-8), 6.95-6.89 (m, 2H, H-3′, H-5′), 4.42 (t, J = 3.0 Hz, 1H, H-4), 2.67 (dd, J = 13.9, 3.0 Hz, 1H, H-3), 2.58 (dd, J = 13.9, 3.0 Hz, 1H, H-3); 13C NMR (100 MHz, DMSO-d6) δ 160.4 (C-2″ (D)), 158.6 (C-4′ (B)), 157.3 (C-4″ (D)), 156.9 (C-9 (A)), 151.9 (C-9″ (D)), 141.4 (C-6(A)), 132.8 (C-7″ (D)), 128.1 (C-1′ (B)), 127.2 (C-2′ (B), C-6′ (B)), 126.7 (C-10 (A)), 124.7 (C-6″ (D)), 124.4 (C-7(A)), 123.3 (C-5 (A)), 122.6 (C-5″ (D)), 117.3 (C-8 (A)), 116.7 (C-8″ (D)), 115.2 (C-3′ (B), C-5′ (B)), 114.2 (C-10″ (D)), 105.3 (C-3″ (D)), 101.1 (C-2 (C)), 30.3 (C-3 (C)), 26.6 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C24H15ClO6 429.0849, found 429.0847. Analytical HPLC (λ = 280 nm): purity: 98%; tR = 19.3 min.
3.3.10. 2-(4′-Hydroxyphenyl)-6-chlorochromane-(4→3″, 2→O-4″)-4″-hydroxycoumarin (53)
General methodology B was applied using the flavylium salt 35 (0.177 g) and 4-hydroxycoumarin (37, 0.081g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (95:5). Pure compound 53 was obtained as a white amorphous solid (0.190 g, 57% from 23). Melting point: 250–252 °C (decomposes); Compound 53: 1H NMR (400 MHz, DMSO-d6) δ 7.81 (dd, J = 7.9, 1.6 Hz, 1H, H-5″), 7.67–7.59 (ov, 3H, H-2′, H-6′, H-7″), 7.42 (dd, J = 8.4, 1.0 Hz, 1H, H-8″), 7.40–7.35 (ov, 2H, H-5, H-6″), 7.24 (dd, J = 8.7, 2.6 Hz 1H, H-7), 7.06 (d, J = 8.7 Hz, 1H, H-8), 6.92–6.87 (m, 2H, H-3′, H-5′), 4.25 (t, J = 3.0 Hz, 1H, H-4), 2.59 (dd, J = 13.8, 3.2 Hz, 1H, H-3), 2.49–2.42 (ov, 1H, H-3); 13C NMR (100 MHz, DMSO-d6) δ 160.5 (C-2″ (D)), 158.4 (C-4′ (B)), 157.6 (C-4″ (D)), 151.8 (C-9″ (D)), 150.2 (C-9 (A)), 132.7 (C-7″ (D)), 129.4 (C-1′ (B)), 128.0 (C-7(A)), 127.6 (C-10 (A)), 127.1 (C-2′ (B), C-6′ (B)), 127.0 (C-5 (A)), 125.1 (C-6(A)), 124.6 (C-6″ (D)), 122.5 (C-5″ (D)), 118.0 (C-8 (A)), 116.6 (C-8″ (D)), 115.1 (C-3′ (B), C-5′ (B)), 114.3 (C-10″ (D)), 105.2 (C-3″ (D)), 100.7 (C-2 (C)), 30.8 (C-3 (C)), 26.7 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C24H15ClO6 418.0608, found 418.0606. Analytical HPLC (λ = 280 nm): purity: 98%; tR = 20.7 min.
3.3.11. 2-(3′,4′-Dihydroxyphenyl)-chromane-(4→3″, 2→O-2″)-2″-hydroxy-1″,4″-naphthoquinone (54)
General methodology B was applied using the flavylium salt 27 (0.168 g) and 2-hydroxy-1,4-naphthoquinone (38, 0.087g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (90:10). Pure compound 54 was obtained as a yellow amorphous solid (0.023 g, 10% from 21). Melting point: 190–192 °C (decomposes); Compound 54: 1H NMR (400 MHz, DMSO-d6) δ 8.01–7.94 (m, 2H, H-5″, H-8″), 7.86–7.76 (m, 2H, H-6″, H-7″), 7.35 (dd, J = 7.6, 1.7 Hz, 1H, H-5), 7.02–6.99 (m, 2H, H-8, H-6′), 7.14 (d, J = 2.3 Hz, 1H, H-2′), 7.18 (ddd, J = 8.2, 7.4, 1.7 Hz, 1H, H-7), 6.95 (td, J = 7.4, 1.2 Hz, 1H, H-6), 6.83 (d, J = 8.3 Hz, 1H, H-5′), 4.41 (t, J = 3.0 Hz, 1H, H-4), 2.43–2.34 (m, 2H, H-3); 13C NMR (100 MHz, DMSO-d6) δ 182.2 (C-1″ (D)), 178.5 (C-4″ (D)), 152.8 (C-3″ (D)), 151.6 (C-9 (A)), 146.3 (C-4′ (B)), 145.0 (C-3′ (B)), 134.5 # (C-7″ (D)), 133.9 # (C-6″ (D)), 131.1$ (C-9″ (D)), 130.6$ (C-10″ (D)), 130.3 (C-1′ (B)), 128.3 (C-5 (A), C-7 (A)), 126.0 * (C-8″ (D)), 125.8 * (C-5″ (D)), 124.5 (C-2″ (D)), 124.8 (C-10 (A)), 121.6 (C-6(A)), 116.8 (C-6′ (B)), 116.2 (C-8 (A)), 115.2 (C-5′ (B)), 113.4 (C-2′ (B)), 100.1 (C-2 (C)), 30.6 (C-3 (C)), 25.7 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C25H16O6 412.0947, found 412.0942. Analytical HPLC (λ = 280 nm): purity: 97%; tR = 18.6 min.
3.3.12. 2-(3′.4′-Dihydroxyphenyl)-6-nitrochromane-(4→3″, 2→O-2″)-2″-hydroxy-1″,4″-naphthoquinone (55)
General methodology B was applied using the flavylium salt 28 (0.191 g) and 2-hydroxy-1,4-naphthoquinone (38, 0.087g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (95:5). Pure compound 55 was obtained as a yellow amorphous solid (0.140 g, 47% from 22). Melting point: 247–250 °C; Compound 55: 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 2.8 Hz, 1H, H-5), 8.08 (dd, J = 9.0, 2.8 Hz 1H, H-7), 8.02–7.96 (m, 2H, H-5″, H-8″), 7.86–7.78 (m, 2H, H-6″, H-7″), 7.24 (d, J = 9.0 Hz, 1H, H-8), 7.17 (d, J = 2.3 Hz, 1H, H-2′), 7.03 (dd, J = 8.3, 2.2 Hz 1H, H-6′), 6.85 (d, J = 8.2 Hz, 1H, H-5′), 4.58 (t, J = 2.9 Hz, 1H, H-4), 2.54-2.46 (m, 2H, H-3); 13C NMR (100 MHz, CDCl3) δ 182.2 (C-1″ (D)), 178.2 (C-4″ (D)), 157.3 (C-9 (A)), 152.5 (C-3″ (D)), 146.6 (C-4′ (B)), 145.1 (C-3′ (B)), 141.3 (C-6(A)), 134.5 # (C-6″ (D)), 134.1 # (C-7″ (D)), 131.1 (C-10″ (D)), 130.7 (C-9″ (D)), 129.2 (C-1′ (B)), 126.4 (C-2″ (D)), 126.0 * (C-10 (A), C-8″ (D)), 125.8 * (C-5″ (D)) 124.3 (C-7(A)), 123.9 (C-5 (A)), 117.3 (C-8 (A)), 116.8 (C-6′ (B)), 115.3 (C-5′ (B)), 113.4 (C-2′ (B)), 100.6 (C-2 (C)), 29.7 (C-3 (C)), 25.6 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C25H15NO8 457.0798, found 457.0803. Analytical HPLC (λ = 280 nm): purity: 98%; tR = 18.2 min.
3.3.13. 2-(3′,4′-Dihydroxyphenyl)-6-chlorochromane-(4→3″, 2→O-2″)-2″-hydroxy-1″,4″-naphthoquinone (56)
General methodology B was applied using the flavylium salt 29 (0.185 g) and 2-hydroxy-1,4-naphthoquinone (38, 0.087g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (90:10). Pure compound 56 was obtained as a yellow amorphous solid (0.052 g, 19% from 23). Melting point: 300–302 °C (decomposes); Compound 56: 1H NMR (400 MHz, DMSO-d6) δ 8.03-7.95 (m, 2H, H-5″, H-8″), 7.87–7.77 (m, 2H, H-6″, H-7″), 7.35–7.33 (m, 1H, H-5), 7.22 (dd, J = 8.7, 2.6 Hz 1H, H-7), 7.13 (d, J = 2.3 Hz, 1H, H-2′), 7.04 (d, J = 8.7 Hz, 1H, H-8), 7.00 (dd, J = 8.3, 2.3 Hz 1H, H-6′), 6.85–6.81 (m, 1H, H-5′), 4.41 (t, J = 3.1 Hz, 1H, H-4), 2.40 (ov, 1H, H-3); 13C NMR (100 MHz, DMSO-d6) δ 182.2 (C-1″ (D)), 178.3 (C-4″ (D)), 152.9 (C-3″ (D)), 150.6 (C-9 (A)), 146.4 (C-4′ (B)), 145.1 (C-3′ (B)), 134.5 # (C-7″ (D)), 133.9 # (C-6″ (D)), 131.1$ (C-9″ (D)), 130.7$ (C-10″ (D)), 129.9 (C-1′ (B)), 128.0 (C-7(A)), 127.6 (C-5 (A)), 126.8 (C-10 (A)), 126.0 * (C-8″ (D)), 125.8 * (C-5″ (D)), 125.0 (C-6(A)), 123.8 (C-2″ (D)), 118.0 (C-8 (A)), 116.8 (C-6′ (B)), 115.2 (C-5′ (B)), 113.4 (C-2′ (B)), 100.2 (C-2 (C)), 30.1 (C-3 (C)), 25.7 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C25H15ClO6 446.0557, found 446.0557. Analytical HPLC (λ = 280 nm): purity: 95%; tR = 19.7 min.
3.3.14. 2-(3′.4′-Dimethoxyphenyl)-6-nitrochromane-(4→3″, 2→O-2″)-2″-hydroxy-1″,4″-naphthoquinone (58)
General methodology B was applied using the flavylium salt 31 (0.205 g) and 2-hydroxy-1,4-naphthoquinone (38, 0.087g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (98:2). Pure compound 58 was obtained as a yellow amorphous solid (0.100 g, 42% from 22). Melting point: 148–150 °C; Compound 58: 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 2.7 Hz, 1H, H-5), 8.13-8.04 (ov, 3H, H-7, H-5″, H-8″), 7.77–7.64 (m, 2H, H-6″, H-7″), 7.27 (dd, J = 8.3, 2.1 Hz 1H, H-6′), 7.24 (d, J = 2.1 Hz, 1H, H-2′), 7.10 (d, J = 9.0 Hz, 1H, H-8), 6.94 (d, J = 8.3 Hz, 1H, H-5′), 4.63 (t, J = 2.7 Hz, 1H, H-4), 3.94 (s, 3H, 3′-OMe), 3.92 (s, 3H, 3′-OMe), 2.49 (dd, J = 13.8, 3.0 Hz, 1H, H-3), 2.40 (dd, J = 13.8, 3.0 Hz, 1H, H-3); 13C NMR (100 MHz, CDCl3) δ 182.5 (C-1″ (D)), 178.7 (C-4″ (D)), 157.4 (C-9 (A)), 153.0 (C-3″ (D)), 150.4 (C-4′ (B)), 149.2 (C-3′ (B)), 142.5 (C-6(A)), 134.6 # (C-6″ (D)), 134.0 # (C-7″ (D)), 131.7 (C-10″ (D)), 131.4 (C-1′ (B)), 130.8 (C-9″ (D)), 126.8 * (C-5″ (D)), 126.7 * (C-8″ (D)), 125.4 (C-10 (A)), 123.4 (C-2″ (D)), 124.7 (C-5 (A), C-7 (A)), 118.6 (C-6′ (B)), 117.4 (C-8 (A)), 111.1 (C-5′ (B)), 109.1 (C-2′ (B)), 100.5 (C-2 (C)), 56.3 (3′-OMe), 56.2 (4′-OMe), 31.5 (C-3 (C)), 26.3 (C-4 (C)); *, #these signals may be interchangeable. HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C27H19NO8 485.1111, found 485.1112. Analytical HPLC (λ = 280 nm): purity: >99%; tR = 21.3 min.
3.3.15. 2-(3′,4′-Dimethoxyphenyl)-6-chlorochromane-(4→3″, 2→O-2″)-2″-hydroxy-1″,4″-naphthoquinone (59)
General methodology B was applied using the flavylium salt 32 (0.199 g) and 2-hydroxy-1,4-naphthoquinone (38, 0.087g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-MeOH (98:2). Pure compound 59 was obtained as a yellow amorphous solid (0.150 g, 67% from 23). Melting point: 124–127 °C.; Compound 59: 1H NMR (400 MHz, DMSO-d6) δ 8.06-7.99 (m, 2H, H-5″, H-8″), 7.71–7.64 (m, 2H, H-6″, H-7″), 7.42 (d, J = 2.6 Hz, 1H, H-5), 7.29 (dd, J = 8.4, 2.1 Hz 1H, H-6′), 7.23 (d, J = 2.1 Hz, 1H, H-2′), 7.10 (dd, J = 8.7, 2.6 Hz 1H, H-7), 6.94 (d, J = 8.7 Hz, 1H, H-8), 6.91 (d, J = 8.4 Hz, 1H, H-5′), 4.48 (t, J = 3.0 Hz, 1H, H-4), 3.92 (s, 3H, 3′-OMe), 3.90 (s, 3H, 4′-OMe), 2.43 (dd, J = 13.7, 3.0 Hz, 1H, H-3), 2.29 (dd, J = 13.7, 3.0 Hz, 1H, H-3); 13C NMR (100 MHz, CDCl3) δ 182.4 (C-1″ (D)), 178.6 (C-4″ (D)), 153.2 (C-3″ (D)), 150.5 (C-9 (A)), 149.8 (C-4′ (B)), 148.9 (C-3′ (B)), 134.2 (C-6″ (D)), 133.5 (C-7″ (D)), 131.5 (C-10″ (D)), 131.4 (C-1′ (B)), 130.9 (C-9″ (D)), 128.3 (C-7(A)), 128.0 (C-5 (A)), 126.7 (C-2″ (D)), 126.5 * (C-8″ (D)), 126.3 * (C-5″ (D)), 125.8 (C-6(A)), 124.1 (C-10 (A)), 118.3 (C-6′ (B)), 117.9 (C-8 (A)), 110.8 (C-5′ (B)), 108.9 (C-2′ (B)), 100.0 (C-2 (C)), 56.0 (3′-OMe, 4′-OMe)31.6 (C-3 (C)), 26.1 (C-4 (C)). HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C27H19ClO6 474.087, found 474.087. Analytical HPLC (λ = 280 nm): purity: 98%; tR = 23.2 min.
3.3.16. 2-(4′-Hydroxyphenyl)-6-nitrochromane-(4→3″, 2→O-2″)-2″-hydroxy-1″,4″-naphthoquinone (60)
General methodology B was applied using the flavylium salt 34 (0.183 g) and 2-hydroxy-1,4-naphthoquinone (38, 0.087 g, 0.5 mmol). Compound 60 was recovered by filtration as a yellow amorphous solid (0.134 g, 46% from 22). Melting point: 270–273 °C (decomposes); Compound 60: 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 2.8 Hz, 1H, H-5), 8.08 (dd, J = 9.0, 2.9 Hz 1H, H-7), 8.00–7.95 (m, 2H, H-5″, H-8″), 7.86–7.79 (m, 2H, H-6″, H-7″), 7.59 (d, J = 8.3 Hz, 2H, H-2′, H-6′), 7.25 (d, J = 9.0 Hz, 1H, H-8), 6.89 (d, J = 8.3 Hz, 2H, H-3′, H-5′), 4.60 (t, J = 3.0 Hz, 1H, H-4), 2.53-2.49 (m, 2H, H-3); 13C NMR (100 MHz, CDCl3) δ 182.2 (C-1″ (D)), 178.0 (C-4″ (D)), 158.5 (C-4′ (B)), 157.3 (C-9 (A)), 152.6 (C-3″ (D)), 141.3 (C-6(A)), 134.6 # (C-7″ (D)), 134.0 # (C-6″ (D)), 131.1 * (C-9″ (D)), 130.7 * (C-10″ (D)), 128.8 (C-1′ (B)), 127.2 (C-2′ (B), C-6′ (B)), 126.1 (C-5″ (D)), 126.0 (C-10 (A)), 125.9 (C-8″ (D)), 124.3 (C-7(A)), 123.9 (C-5 (A)), 123.4 (C-2″ (D)), 117.3 (C-8 (A)), 115.1 (C-3′ (B), C-5′ (B)), 100.6 (C-2 (C)), 29.5 (C-3 (C)), 25.6 (C-4 (C)); *these signals may be interchangeable. HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C25H15NO7 441.0849, found 441.0855. Analytical HPLC (λ = 280 nm): purity: >99%; tR = 19.6 min.
3.3.17. 2-(4′-Hydroxyphenyl)-6-chlorochromane-(4→3″, 2→O-2″)-2″-hydroxy-1″,4″-naphthoquinone (61)
General methodology B was applied using the flavylium salt 35 (0.177 g) and 2-hydroxy-1,4-naphthoquinone (38, 0.087g, 0.5 mmol). The purification step was performed by CC using mixtures of DCM-acetone (98:2). Pure compound 61 was obtained as a red-orange amorphous solid (0.050 g, 15% from 23). Melting point: 249–251 °C; Compound 61: 1H NMR (400 MHz, DMSO-d6) δ 8.03-7.96 (m, 2H, H-5″, H-8″), 7.89–7.78 (m, 2H, H-6″, H-7″), 7.59–7.53 (m, 2H, H-2′, H-6′), 7.34 (d, J = 2.6 Hz, 1H, H-5), 7.23 (dd, J = 8.7, 2.6 Hz 1H, H-7), 7.06 (d, J = 8.7 Hz, 1H, H-8), 6.92-6.85 (m, 2H, H-3′, H-5′), 4.45-4.42 (m, 1H, H-4), 2.48-2.39 (m, 2H, H-3); 13C NMR (100 MHz, CDCl3) δ 182.2 (C-1″ (D)), 178.3 (C-4″ (D)), 158.3 (C-4′ (B)), 152.9 (C-3″ (D)), 150.0 (C-9 (A)), 134.5 # (C-7″ (D)), 133.9 # (C-6″ (D)), 131.1 * (C-9″ (D)), 130.7 * (C-10″ (D)), 129.4 (C-1′ (B)), 128.0 (C-7(A)), 127.6 (C-5 (A)), 127.1 (C-2′ (B), C-6′ (B)), 126.7 (C-10 (A)), 126.0 (C-5″ (D)), 125.8 (C-8″ (D)), 125.0 (C-6(A)), 123.7 (C-2″ (D)), 118.1 (C-8 (A)), 115.2 (C-3′ (B), C-5′ (B)), 100.3 (C-2 (C)), 30.0 (C-3 (C)), 25.6 (C-4 (C)); *these signals may be interchangeable. HRMS (ESI-TOF) m/z [M+H]+ Calcd. For C25H15ClO5 430.0608, found 430.0615. Analytical HPLC (λ = 280 nm): purity: 99%; tR = 21.3 min.
3.3.18. 2-(3′,4′-Dimethoxyphenyl)-chromane-(4→3″, 2→O-4″)-4″-hydroxy-6″-phenyl-5″,6″-dihydro-2H-pyran-2″-one (62a and 62b)
General methodology B was applied using the flavylium salt
30 (0.182 g) and 4-hydroxy-6-phenyl-5,6-dihydro-2
H-pyran-2-one (
39, 0.095 g, 0.5 mmol), previously prepared according to the methodology described by Andersh et al. [
48,
49]. The purification step was performed by CC using mixtures of hexane-EtOAc (75:25). Pure compound
62a was obtained as a pale yellow amorphous solid (0.011 g, 5.0% from
21) and pure compound
62b as a white amorphous solid (0.019 g, 9.0% from
21); Compound
62a:
1H NMR (400 MHz, CDCl
3) δ 7.38–7.24 (
m, 5H, H-5, H-2‴, H-3‴), 7.23–7.16 (ov, 3H, H-7, H-6′, H-4‴), 7.13 (
d,
J = 2.2 Hz, 1H, H-2′), 7.06 (
dd,
J = 8.1, 1.2 Hz, 1H, H-8), 6.97 (
td,
J = 7.4, 1.2 Hz, 1H, H-6), 6.90 (
d,
J = 8.5 Hz, 1H, H-5′), 5.31 (
dd,
J = 12.3, 4.0 Hz, 1H, H-6″), 4.30 (
t,
J = 3.0 Hz, 1H, H-4), 3.90 (
s, 6H, 3′-O
Me, 4′-O
Me), 3.03-2.96 (
m, 1H, H-5″), 2.70 (
dd,
J = 17.2, 4.0 Hz, 1H, H-5″), 2.35-2.22 (
m, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.8 (C-2″ (D)), 163.3 (C-4″ (D)), 151.6 (C-9 (A)), 150.0 (C-4′ (B)), 148.0 (C-3′ (B)), 138.4 (C-1‴ (E)), 132.2 (C-1′ (B)), 128.9 (C-5 (A), C-3‴ (E)), 128.3 * (C-7(A)), 128.1 * (C-4‴ (E)), 126.6 (C-2‴ (E)), 126.4 (C-10 (A)), 122.2 (C-6(A)), 118.3 (C-6′ (B)), 116.5 (C-8 (A)), 110.9 (C-5′ (B)), 109.1 (C-2′ (B)), 106.6 (C-3″ (D)), 100.6 (C-2 (C)), 76.6 (C-6″ (D)), 56.0 (3′-O
Me, 4′-O
Me), 34.3 (C-5″ (D)), 33.2 (C-3 (C)), 26.5 (C-4 (C)); *these signals could be interchangeable. Melting point: 94–96 °C. HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. For C
28H
24O
6 456.1573, found 456.1571. Analytical HPLC (
λ = 280 nm): purity: 96%;
tR = 20.7 min. Compound
62b:
1H NMR (400 MHz, CDCl
3) δ 7.56 (
dd,
J = 7.5, 1.6 Hz, 1H, H-5), 7.35–7.30 (
m, 5H, H-2‴, H-3‴, H-4‴), 7.21 (
dd,
J = 8.4, 2.1 Hz, 1H, H-6′), 7.19-7.16 (ov, 2H, H-2′, H-7), 7.01 (
dd,
J = 8.2, 1.1 Hz, 1H, H-8), 7.01 (
td,
J = 7.4, 1.1 Hz, 1H, H-6), 6.91 (
d,
J = 8.4, 1H, H-5′), 5.49 (
dd,
J = 12.2, 4.1 Hz, 1H, H-6″), 4.04 (
t,
J = 3.1 Hz, 1H, H-4), 3.92 (
s, 3H, 4′-O
Me), 3.90 (
s, 3H, 3′-O
Me), 2.84 (
dd,
J = 17.5, 12.3 Hz, 1H, H-5″), 2.68 (
ddd,
J = 17.5, 4.1, 1.1 Hz, 1H, H-5″), 2.38 (
qd,
J = 13.5, 3.1, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.7 (C-2″ (D)), 163.6 (C-4″ (D)), 151.5 (C-9 (A)), 150.0 (C-4′ (B)), 149.0 (C-3′ (B)), 138.3 (C-1‴ (E)), 132.4 (C-1′ (B)), 129.1 (C-5 (A)), 128.9 (C-3‴ (E), C-4‴ (E)), 128.2 (C-7(A)), 126.2 (C-2‴ (E)), 126.0 (C-10 (A)), 122.0 (C-6(A)), 118.3 (C-6′ (B)), 116.5 (C-8 (A)), 110.9 (C-5′ (B)), 109.1 (C-2′ (B)), 106.7 (C-3″ (D)), 100.4 (C-2 (C)), 77.5 (C-6″ (D)), 56.3 (4′-O
Me), 56.2 (3′-O
Me), 34.3 (C-5″ (D)), 33.3 (C-3 (C)), 27.6 (C-4 (C)). Melting point: 172–174 °C. HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. For C
28H
24O
6 456.1573, found 456.1573. Analytical HPLC (
λ = 280 nm): purity: >99%;
tR = 20.7 min.
3.3.19. 2-(3′,4′-Dimethoxyphenyl)-chromane-(4→3″, 2→O-4″)-6″-(4‴-chlorophenyl)-4″-hydroxy-5″,6″-dihydro-2H-pyran-2″-one (63a and 63b)
General methodology B was applied using the flavylium salt
30 (0.182 g) and 6-(4-chlorophenyl)-4-hydroxy-5,6-dihydro-2
H-pyran-2-one (
40, 0.122 g, 0.5 mmol), previously prepared according to the methodology described by Andersh et al. [
48,
49]. The purification step was performed by CC using mixtures of hexane-EtOAc (75:25). Pure compound
63a was obtained as a pale yellow amorphous solid (0.031 g, 12% from
21) and pure compound
63b as a pale yellow amorphous solid (0.050 g, 20% from
21); Compound
63a:
1H NMR (400 MHz, CDCl
3) δ 7.37–7.28 (ov, 5H, H-5, H-2‴, H-3‴), 7.28–7.16 (ov, 2H, H-7, H-6′), 7.12 (
d,
J = 2.1 Hz, 1H, H-2′), 7.05 (
dd,
J = 7.7, 1.1 Hz, 1H, H-8), 6.96 (
td,
J = 7.4, 1.2 Hz, 1H, H-6), 6.90 (
d,
J = 8.5 Hz, 1H, H-5′), 5.29 (
dd,
J = 12.3, 4.1 Hz, 1H, H-6″), 4.28 (
t,
J = 2.8 Hz, 1H, H-4), 3.90 (
s, 6H, 3′-O
Me, 4′-O
Me), 2.84 (
ddd,
J = 17.2, 12.3, 1.0 Hz, 1H, H-5″), 2.68 (
dd,
J = 17.2, 4.1 Hz, 1H, H-5″), 2.30 (
qd,
J = 13.5, 2.8 Hz, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.5 (C-2″ (D)), 163.1 (C-4″ (D)), 151.6 (C-9 (A)), 150.0 (C-4′ (B)), 149.0 (C-3′ (B)), 136.1 (C-1‴ ©), 134.7 (C-4‴ (E)), 132.1 (C-1′ (B)), 129.1 (C-3‴ (E)), 128.4 (C-7(A)), 128.1 (C-5 (A)), 127.6 (C-2‴ (E)), 126.3 (C-10 (A)), 122.2 (C-6(A)), 118.3 (C-6′ (B)), 116.5 (C-8 (A)), 110.9 (C-5′ (B)), 109.0 (C-2′ (B)), 106.6 (C-3″ (D)), 100.6 (C-2 (C)), 76.0 (C-6″ (D)), 56.2 (3′-O
Me, 4′-O
Me), 34.3 (C-5″ (D)), 33.1 (C-3 (C)), 26.8 (C-4 (C)). Melting point: 110–112 °C HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
28H
23ClO
6 490.1183, found 490.1177. Analytical HPLC (
λ = 280 nm): purity: 97%;
tR = 21.8 min. Compound
63b:
1H NMR (400 MHz, CDCl
3) δ 7.53 (
dd,
J = 7.5, 1.2 Hz, 1H, H-5), 7.31–7.25 (
m, 4H, H-2‴, H-3‴), 7.22-7.16 (ov, 3H, H-7, H-2′, H-6′), 7.01 (
dd,
J = 8.2, 1.2 Hz, 1H, H-8), 6.97 (
td,
J = 7.4, 1.2 Hz, 1H, H-6), 6.91 (
d,
J = 8.4 Hz, 1H, H-5′), 5.47 (
dd,
J = 12.0, 4.4 Hz, 1H, H-6″), 4.04 (
t,
J = 3.1 Hz, 1H, H-4), 3.92 (
s, 3H, 3′-O
Me), 3.90 (
s, 3H, 4′-O
Me), 2.79 (
ddd,
J = 17.5, 12.2, 0.8 Hz, 1H, H-5″), 2.67 (
ddd,
J = 17.5, 4.4, 1.2 Hz, 1H, H-5″), 2.39 (
qd,
J = 13.5, 3.1 Hz, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.4 (C-2″ (D)), 163.4 (C-4″ (D)), 151.5 (C-9 (A)), 150.1 (C-4′ (B)), 149.1 (C-3′ (B)), 136.8 (C-1‴ (E)), 134.7 (C-4‴ (E)), 132.3 (C-1′ (B)), 129.1 (C-3‴ (E)), 128.9 (C-5 (A)), 128.3 (C-7(A)), 127.6 (C-2‴ (E)), 125.9 (C-10 (A)), 122.1 (C-6(A)), 118.3 (C-6′ (B)), 116.2 (C-8 (A)), 110.9 (C-5′ (B)), 109.1 (C-2′ (B)), 106.8 (C-3″ (D)), 100.5 (C-2 (C)), 76.7 (C-6″ (D)), 56.3 (3′-O
Me), 56.0 (4′-O
Me), 34.2 (C-5″ (D)), 33.3 (C-3 (C)), 27.5 (C-4 (C)). Melting point: 173–176 °C HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
28H
23ClO
6 490.1183, found 490.1164. Analytical HPLC (
λ = 280 nm): purity: >99%;
tR = 21.9 min.
3.3.20. 2-(3′,4′-Dimethoxyphenyl)-chromane-(4→3″, 2→O-4″)-4″-hydroxy-6″-(4‴-methoxyphenyl)-5″,6″-dihydro-2H-pyran-2″-one (64a and 64b)
General methodology B was applied using the flavylium salt
30 (0.182 g) and 4-hydroxy-6-(4-methoxyphenyl)-5,6-dihydro-2
H-pyran-2-one (
41, 0.110 g, 0.5 mmol), previously prepared according to the methodology described by Andersh et al. [
48,
49] (spectroscopic data agree with those reported in the literature [
50]). The purification step was performed by CC using mixtures of hexane-EtOAc (75:25). Pure compound
64a was obtained as a white amorphous solid (0.044 g, 17% from
21) and pure compound
64b as a white amorphous solid (0.052 g, 21% from
21); Compound
64a:
1H NMR (400 MHz, CDCl
3) δ 7.36 (
dd,
J = 7.5, 1.6 Hz, 1H, H-5), 7.30–7.26 (
m, 2H, H-2‴), 7.23–7.16 (ov, 2H, H-7, H-6′), 7.14 (
d,
J = 2.2 Hz, 1H, H-2′), 7.05 (
dd,
J = 8.2, 1.2 Hz, 1H, H-8), 6.96 (
td,
J = 7.4, 1.1 Hz, 1H, H-6), 6.90 (
d,
J = 8.4 Hz, 1H, H-5′), 6.89-6.85 (
m, 2H, H-3‴), 5.25 (
dd,
J = 12.3, 4.0 Hz, 1H, H-6″), 4.28 (
t,
J = 3.1 Hz, 1H, H-4), 3.92 (
s, 3H, 4′-O
Me), 3.90 (
s, 3H, 3′-O
Me), 3.81 (
s, 3H, 4‴-O
Me), 3.00 (
ddd,
J = 17.2, 12.4, 4.0 Hz, 1H, H-5″), 2.64 (
dd,
J = 17.2, 4.0 Hz, 1H, H-5″), 2.29 (
qd,
J = 13.5, 3.2 Hz, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.9 (C-2″ (D)), 163.3 (C-4″ (D)), 160.0 (C-4‴ (E)), 151.6 (C-9 (A)), 150.0 (C-4′ (B)), 149.0 (C-3′ (B)), 132.3 (C-1′ (B)), 130.4 (C-1‴ (E)), 128.3 (C-7(A)), 128.1 (C-5 (A)), 128.0 (C-2‴ (E)), 126.5 (C-10 (A)), 122.2 (C-6(A)), 118.3 (C-6′ (B)), 116.5 (C-8 (A)), 114.2 (C-3‴ (E)), 110.9 (C-5′ (B)), 109.1 (C-2′ (B)), 106.5 (C-3″ (D)), 100.5 (C-2 (C)), 76.7 (C-6″ (D)), 56.2 (3′-O
Me, 4′-O
Me), 55.4 (4‴-O
Me), 34.2 (C-5″ (D)), 33.1 (C-3 (C)), 26.7 (C-4 (C)). Melting point: 172–175 °C. HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. For C
29H
26O
7 486.1679, found 486.1658. Analytical HPLC (
λ = 280 nm): purity: 98%;
tR = 20.7 min. Compound
64b:
1H NMR (400 MHz, CDCl
3) δ 7.59 (
dd,
J = 7.6, 1.7 Hz, 1H, H-5), 7.32–7.28 (
m, 2H, H-2‴), 7.28-7.20 (ov, 3H, H-7, H-2′, H-6′), 7.06 (
dd,
J = 8.2, 1.2 Hz, 1H, H-8), 7.01 (
td,
J = 7.5, 1.2 Hz, 1H, H-6), 6.95 (
d,
J = 8.4 Hz, 1H, H-5′), 6.92-6.86 (
m, 2H, H-3‴), 5.48 (
dd,
J = 12.4, 4.0 Hz, 1H, H-6″), 4.08 (
t,
J = 3.2 Hz, 1H, H-4), 3.97 (
s, 3H, 3′-O
me), 3.95 (
s, 3H, 4′-O
Me), 3.81 (
s, 3H, 4‴-O
Me), 2.89 (
dd,
J = 17.2, 12.4 Hz, 1H, H-5″), 2.68 (
ddd,
J = 17.2, 4.0, 1.2 Hz, 1H, H-5″), 2.42 (
qd,
J = 13.5, 3.2 Hz, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.8 (C-2″ (D)), 163.6 (C-4″ (D)), 160.0 (C-4‴ (E)), 151.5 (C-9 (A)), 149.9 (C-4′ (B)), 149.0 (C-3′ (B)), 132.4 (C-1′ (B)), 130.3 (C-1‴ (E)), 128.9 (C-7(A)), 128.1 (C-5 (A)), 127.8 (C-2‴ (E)), 126.0 (C-10 (A)), 121.9 (C-6(A)), 118.3 (C-6′ (B)), 116.1 (C-8 (A)), 114.1 (C-3‴ (E)), 110.9 (C-5′ (B)), 109.1 (C-2′ (B)), 106.6 (C-3″ (D)), 100.3 (C-2 (C)), 77.4 (C-6″ (D)), 56.2 (3′-O
Me, 4′-O
Me), 55.4 (4‴-O
Me), 34.2 (C-5″ (D)), 33.3 (C-3 (C)), 27.5 (C-4 (C)). Melting point: 170–173 °C (decomposes). HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. For C
29H
26O
7 486.1679, found 486.1663. Analytical HPLC (
λ = 280 nm): purity: 98%;
tR = 20.6 min.
3.3.21. 2-(3′,4′-Dimethoxyphenyl)-6-nitrochromane-(4→3″, 2→O-4″)-4″-hydroxy-6″-phenyl-5″,6″-dihydro-2H-pyran-2″-one (65a and 65b)
General methodology B was applied using the flavylium salt
31 (0.205 g) and 4-hydroxy-6-phenyl-5,6-dihydro-2
H-pyran-2-one (
39, 0.095 g, 0.5 mmol), previously prepared according to the methodology described by Andersh et al. [
48,
49]. The purification step was performed by CC using mixtures of hexane-EtOAc (75:25). Pure compound
65a was obtained as a pale yellow amorphous solid (0.015 g, 6% from
22) and pure compound
65b as a pale orange amorphous solid (0.081 g, 33% from
22); Compound
65a:
1H NMR (400 MHz, CDCl
3) δ 8.28 (
d,
J = 2.7 Hz, 1H, H-5), 8.11 (
dd,
J = 9.0, 2.8 Hz, 1H, H-7), 7.37–7.33 (
m, 5H, H-2‴, H-3‴, H-4‴), 7.16 (
dd,
J = 8.5, 2.2 Hz, 1H, H-6′), 7.12 (
d,
J = 9.0 Hz, 1H, H-8), 7.10 (
d,
J = 2.2 Hz, 1H, H-2′), 6.92 (
d,
J = 8.5 Hz, 1H, H-5′), 5.32 (
dd,
J = 12.0, 4.0 Hz, 1H, H-6″), 4.40 (
t,
J = 3.2 Hz, 1H, H-4), 3.91 (
s, 6H, 3′-O
Me, 4′-O
Me), 3.03 (
ddd,
J = 17.2, 12.0, 1.1 Hz, 1H, H-5″), 2.74 (
dd,
J = 17.2, 4.1 Hz, 1H, H-5″), 2.36 (
d,
J = 3.2 Hz, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.5 (C-2″ (D)), 163.2 (C-4″ (D)), 157.0 (C-9 (A)), 150.4 (C-4′ (B)), 149.3 (C-3′ (B)), 142.5 (C-6(A)), 138.0 (C-1‴ (E)), 130.9 (C-1′ (B)), 129.0 (C-3‴ (E)), 129.1 (C-4‴ (E)), 127.3 (C-10 (A)), 126.2 (C-2‴ (E)), 124.5 (C-7(A)), 123.4 (C-5 (A)), 118.2 (C-6′ (B)), 117.1 (C-8 (A)), 111.0 (C-5′ (B)), 108.9 (C-2′ (B)), 106.1 (C-3″ (D)), 100.8 (C-2 (C)), 76.9 (C-6″ (D)), 56.3 (3′-O
Me), 56.2 (4′-O
Me), 34.0 (C-5″ (D)), 32.3 (C-3 (C)), 26.9 (C-4 (C)). Melting point: 198–200 °C. HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
28H
23NO
8 501.1424, found 501.1425. Analytical HPLC (
λ = 280 nm): purity: 99%;
tR = 20.4 min. Compound
65b:
1H NMR (400 MHz, CDCl
3) δ 8.44 (
d,
J = 2.7 Hz, 1H, H-5), 8.07 (
dd,
J = 9.0, 2.8 Hz, 1H, H-7), 7.34–7.31 (
m, 5H, H-2‴, H-3‴, H-4‴), 7.19 (
dd,
J = 8.4, 2.2 Hz, 1H, H-6′), 7.14 (
d,
J = 2.2 Hz, 1H, H-2′), 7.07 (
d,
J = 9.0 Hz, 1H, H-8), 6.92 (
d,
J = 8.5 Hz, 1H, H-5′), 5.51 (
dd,
J = 12.3, 4.1 Hz, 1H, H-6″), 4.14 (
br s, 1H, H-4), 3.93 (
s, 3H, 3′-O
Me), 3.91 (
s, 3H, 4′-O
Me), 2.85 (
dd,
J = 17.7, 12.3 Hz, 1H, H-5″), 2.71 (
ddd,
J = 17.6, 4.1, 1.1 Hz, 1H, H-5″), 2.53 (
dd,
J = 13.7, 3.3 Hz, 1H, H-3), 2.38 (
dd,
J = 13.7, 2.8 Hz, 1H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.3 (C-2″ (D)), 163.5 (C-4″ (D)), 156.8 (C-9 (A)), 150.3 (C-4′ (B)), 149.2 (C-3′ (B)), 142.3 (C-6(A)), 137.9 (C-1‴ (E)), 131.0 (C-1′ (B)), 129.0 (C-4‴ (E)), 128.9 (C-3‴ (E)), 126.9 (C-10 (A)), 126.1 (C-2‴ (E)), 124.8 (C-5 (A)), 124.4 (C-7(A)), 118.2 (C-6′ (B)), 116.7 (C-8 (A)), 111.0 (C-5′ (B)), 108.9 (C-2′ (B)), 106.1 (C-3″ (D)), 100.7 (C-2 (C)), 77.5 (C-6″ (D)), 56.3 (3′-O
Me), 56.2 (4′-O
Me), 34.2 (C-5″ (D)), 32.4 (C-3 (C)), 27.5 (C-4 (C)). Melting point: 143–145 °C. HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
28H
23NO
8 501.1424, found 501.1421. Analytical HPLC (
λ = 280 nm): purity: >99%;
tR = 20.6 min.
3.3.22. 2-(3′,4′-Dimethoxyphenyl)-6-nitrochromane-(4→3″, 2→O-4″)-6″-(4‴-chlorophenyl)-4″-hydroxy-5″,6″-dihydro-2H-pyran-2″-one (66a and 66b)
General methodology B was applied using the flavylium salt
31 (0.205 g) and 6-(4-chlorophenyl)-4-hydroxy-5,6-dihydro-2
H-pyran-2-one (
40, 0.122 g, 0.5 mmol), previously prepared according to the methodology described by Andersh et al. [
48,
49]. The purification step was performed by CC using mixtures of hexane-EtOAc (75:25). Pure compound
66a was obtained as a white amorphous solid (0.051 g, 19 % from
22) and pure compound
66b as a pale yellow amorphous solid (0.071 g, 26% from
22); Compound
66a:
1H NMR (400 MHz, CDCl
3) δ 8.27 (
d,
J = 2.7 Hz, 1H, H-5), 8.10 (
dd,
J = 9.0, 2.7 Hz, 1H, H-7), 7.37–7.28 (
m, 4H, H-2‴, H-3‴), 7.16 (
dd,
J = 8.5, 2.2 Hz, 1H, H-6′), 7.12 (
d,
J = 9.0 Hz, 1H, H-8), 7.09 (
d,
J = 2.22 Hz, 1H, H-2′), 6.91 (
d,
J = 8.5 Hz, 1H, H-5′), 5.30 (
dd,
J = 12.1, 4.1 Hz, 1H, H-6″), 4.38 (
t,
J = 3.1 Hz, 1H, H-4), 3.91 (
s, 3H, 3′-O
Me), 3.90 (
s, 3H, 4′-O
Me), 2.98 (
ddd,
J = 17.2, 12.1, 1.0 Hz, 1H, H-5″), 2.72 (
dd,
J = 17.2, 4.1 Hz, 1H, H-5″), 2.36 (
d,
J = 3.1 Hz, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 164.9 (C-2″ (D)), 163.0 (C-4″ (D)), 156.9 (C-9 (A)), 150.4 (C-4′ (B)), 149.2 (C-3′ (B)), 142.5 (C-6(A)), 136.5 (C-1‴ (E)), 134.9 (C-4‴ (E)), 130.7 (C-1′ (B)), 129.2 (C-3‴ (E)), 127.1 (C-2‴ (E), C-10 (A)), 124.5 (C-7(A)), 123.9 (C-5 (A)), 118.2 (C-6′ (B)), 117.1 (C-8 (A)), 111.0 (C-5′ (B)), 108.9 (C-2′ (B)), 106.1 (C-3″ (D)), 100.9 (C-2 (C)), 76.1 (C-6″ (D)), 56.3 (3′-O
Me), 56.2 (4′-O
Me), 34.0 (C-5″ (D)), 32.5 (C-3 (C)), 26.6 (C-4 (C)). Melting point: 194–196 °C. HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
28H
22NO
8 535.1034, found 535.1032. Analytical HPLC (
λ = 280 nm): purity: 97%;
tR = 21.7 min. Compound (+)-
66a: Chiral HPLC (
λ = 280 nm): purity: >99%;
tR = 19.7 min. [α]
D: +34 (c. 0.4, CHCl
3). Compound (−)-
66a: Chiral HPLC (
λ = 280 nm): purity: >99%;
tR = 25.5 min. [α]
D: −26 (c. 0.2, CHCl
3). Compound
66b:
1H NMR (400 MHz, CDCl
3) δ 8.42 (
d,
J = 2.7 Hz, 1H, H-5), 8.08 (
dd,
J = 8.9, 2.7 Hz, 1H, H-7), 7.31–7.26 (
m, 4H, H-2‴, H-3‴), 7.18 (
dd,
J = 8.4, 2.1 Hz, 1H, H-6′), 7.13 (
d,
J = 2.1 Hz, 1H, H-2′), 7.06 (
d,
J = 9.0 Hz, 1H, H-8), 6.91 (
d,
J = 8.5 Hz, 1H, H-5′), 5.49 (
dd,
J = 12.1, 4.2 Hz, 1H, H-6″), 4.12 (
br s, 1H, H-4), 3.92 (
s, 3H, 3′-O
Me), 3.91 (
s, 3H, 4′-O
Me), 2.80 (
dd,
J = 17.6, 12.1 Hz, 1H, H-5″), 2.70 (
dd,
J = 17.6, 4.2 Hz, 1H, H-5″), 2.52 (
dd,
J = 13.8, 3.3 Hz, 1H, H-3), 2.38 (
dd,
J = 13.8, 2.8 Hz, 1H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.1 (C-2″ (D)), 163.4 (C-4″ (D)), 156.8 (C-9 (A)), 150.4 (C-4′ (B)), 149.2 (C-3′ (B)), 142.4 (C-6(A)), 136.4 (C-1‴ (E)), 134.9 (C-4‴ (E)), 130.9 (C-1′ (B)), 129.1 (C-3‴ (E)), 127.6 (C-2‴ (E)), 126.8 (C-10 (A)), 124.8 (C-5 (A)), 124.4 (C-7(A)), 118.2 (C-6′ (B)), 116.8 (C-8 (A)), 111.0 (C-5′ (B)), 108.9 (C-2′ (B)), 106.1 (C-3″ (D)), 100.7 (C-2 (C)), 76.7 (C-6″ (D)), 56.3 (3′-O
Me), 56.2 (4′-O
Me), 34.1 (C-5″ (D)), 32.4 (C-3 (C)), 27.5 (C-4 (C)). Melting point: 154–157 °C. HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
28H
22NO
8 535.1034, found 535.1030. Analytical HPLC (
λ = 280 nm): purity: >99%;
tR = 21.8 min.
3.3.23. 2-(3′,4′-Dimethoxyphenyl)-6-nitrochromane-(4→3″, 2→O-4″)-4″-hydroxy-6″-(4‴-methoxyphenyl)-5″,6″-dihydro-2H-pyran-2″-one (67a and 67b)
General methodology B was applied using the flavylium salt
31 (0.205 g) and 4-hydroxy-6-(4-methoxyphenyl)-5,6-dihydro-2
H-pyran-2-one (
41, 0.110 g, 0.5 mmol), previously prepared according to the methodology described by Andersh et al. [
48,
49] (spectroscopic data agree with those reported in the literature [
50]). The purification step was performed by CC using mixtures of hexane-EtOAc (75:25). Pure compound
67a was obtained as a yellow foam (0.040 g, 14% from
22) and pure compound
67b as a pale yellow amorphous solid (0.064 g, 24% from
22); Compound
67a:
1H NMR (400 MHz, CDCl
3) δ 8.28 (
d,
J = 2.7 Hz, 1H, H-5), 8.10 (
dd,
J = 9.0, 2.7 Hz, 1H, H-7), 7.30–7.25 (
m, 2H, H-2‴), 7.16 (
dd,
J = 8.5, 2.2 Hz, 1H, H-6′), 7.13ߝ7.09 (ov, 2H, H-8, H-2′), 6.92 (
d,
J = 8.5 Hz, 1H, H-5′), 6.90–6.84 (
m, 2H, H-3‴), 5.26 (
dd,
J = 12.1, 4.1 Hz, 1H, H-6″), 4.39 (
t,
J = 3.0 Hz, 1H, H-4), 3.91 (
s, 6H, 3′-O
Me, 4′-O
Me), 3.78 (
s, 3H, 4‴-O
Me), 3.03 (
ddd,
J = 17.3, 12.2, 1.0 Hz, 1H, H-5″), 2.69 (
dd,
J = 17.3, 4.1 Hz, 1H, H-5″), 2.40-2.32 (
m, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.3 (C-2″ (D)), 163.3 (C-4″ (D)), 160.1 (C-4‴ (E)), 156.9 (C-9 (A)), 150.4 (C-4′ (B)), 149.2 (C-3′ (B)), 142.5 (C-6(A)), 138.0 (C-1‴ (E)), 130.9 (C-1′ (B)), 127.7 (C-2‴ (E)), 127.0 (C-10 (A)), 124.4 (C-7(A)), 123.7 (C-5 (A)), 118.2 (C-6′ (B)), 117.1 (C-8 (A)), 114.3 (C-3‴ (E)), 111.1 (C-5′ (B)), 108.9 (C-2′ (B)), 105.9 (C-3″ (D)), 100.8 (C-2 (C)), 76.7 (C-6″ (D)), 56.3 (3′-O
Me), 56.2 (4′-O
Me), 55.5 (4‴-O
Me), 33.9 (C-5″ (D)), 32.3 (C-3 (C)), 26.6 (C-4 (C)). HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
29H
25NO
9 531.1529, found 531.1532. Analytical HPLC (
λ = 280 nm): purity: >99%;
tR = 20.6 min. Compound
67b:
1H NMR (400 MHz, CDCl
3) δ 8.44 (
d,
J = 2.7 Hz, 1H, H-5), 8.08 (
dd,
J = 8.9, 2.8 Hz, 1H, H-7), 7.27–7.24 (
m, 2H, H-2‴), 7.19 (
dd,
J = 8.4, 2.2 Hz, 1H, H-6′), 7.13 (
d,
J = 2.2 Hz, 1H, H-2′), 7.07 (
d,
J = 8.9 Hz, 1H, H-8), 6.92 (
d,
J = 8.5 Hz, 1H, H-5′), 6.87-6.82 (
m, 2H, H-3‴), 5.45 (
dd,
J = 12.5, 4.0 Hz, 1H, H-6″), 4.13 (
br s, 1H, H-4), 3.93 (
s, 3H, 3′-O
Me), 3.91 (
s, 3H, 4′-O
Me), 3.77 (
s, 3H, 4‴-O
Me), 2.86 (
dd,
J = 17.4, 12.5 Hz, 1H, H-5″), 2.67 (
ddd,
J = 17.4, 4.0, 1.2 Hz, 1H, H-5″), 2.51 (
dd,
J = 13.7, 3.3 Hz, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.5 (C-2″ (D)), 163.6 (C-4″ (D)), 160.1 (C-4‴ (E)), 156.9 (C-9 (A)), 150.4 (C-4′ (B)), 149.2 (C-3′ (B)), 142.4 (C-6(A)), 131.0 (C-1′ (B)), 129.9 (C-1‴ (E)), 127.8 (C-2‴ (E)), 126.9 (C-10 (A)), 125.0 (C-5 (A)), 124.4 (C-7(A)), 118.2 (C-6′ (B)), 116.7 (C-8 (A)), 114.2 (C-3‴ (E)), 111.0 (C-5′ (B)), 108.9 (C-2′ (B)), 106.1 (C-3″ (D)), 100.6 (C-2 (C)), 77.6 (C-6″ (D)), 56.3 (3′-O
Me), 56.2 (4′-O
Me), 55.6 (4‴-O
Me), 34.1 (C-5″ (D)), 32.5 (C-3 (C)), 27.5 (C-4 (C)). Melting point: 162–164 °C. HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
29H
25NO
9 531.1529, found 531.1529. Analytical HPLC (
λ = 280 nm): purity: >99%;
tR = 20.6 min.
3.3.24. 2-(3′,4′-Dimethoxyphenyl)-6-chlorochromane-(4→3″, 2→O-4″)-4″-hydroxy-6″-phenyl-5″,6″-dihydro-2H-pyran-2″-one (68a and 68b)
General methodology B was applied using the flavylium salt
32 (0.199 g) and 4-hydroxy-6-phenyl-5,6-dihydro-2
H-pyran-2-one (
39, 0.095 g, 0.5 mmol), previously prepared according to the methodology described by Andersh et al. [
48,
49]. The purification step was performed by CC using mixtures of hexane-EtOAc (75:25). Pure compound
68a was obtained as a white amorphous solid (0.026 g, 12% from
23) and pure compound
68b as a white amorphous solid (0.066 g, 29% from
23); Compound
68a:
1H NMR (400 MHz, CDCl
3) δ 7.38–7.30 (
m, 6H, H-5, H-2‴, H-3‴, H-4‴), 7.17–7.13 (ov, 2H, H-7, H-6′), 7.10 (
d,
J = 2.2 Hz, 1H, H-2′), 6.97 (
d,
J = 8.7 Hz, 1H, H-8), 6.90 (
d,
J = 8.5 Hz, 1H, H-5′), 5.32 (
dd,
J = 12.2, 4.0 Hz, 1H, H-6″), 4.25 (
t,
J = 2.8 Hz, 1H, H-4), 3.90 (
s, 6H, 3′-O
Me, 4′-O
Me), 3.00 (
ddd,
J = 17.2, 12.3, 1.1 Hz, 1H, H-5″), 2.71 (
dd,
J = 17.2, 4.1 Hz, 1H, H-5″), 2.30 (
qd,
J = 13.5, 2.8 Hz, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.5 (C-2″ (D)), 163.4 (C-4″ (D)), 150.3 (C-9 (A)), 150.1 (C-4′ (B)), 149.1 (C-3′ (B)), 138.8 (C-1‴ (E)), 131.8 (C-1′ (B)), 128.9 (C-5 (A), C-3‴ (E)), 128.3 (C-7(A)), 127.9 (C-10 (A)), 127.7 (C-4‴ (E)), 127.0 (C-6(A)), 126.2 (C-2‴ (E)), 118.3 (C-6′ (B)), 117.8 (C-8 (A)), 110.9 (C-5′ (B)), 109.0 (C-2′ (B)), 106.1 (C-3″ (D)), 100.6 (C-2 (C)), 76.9 (C-6″ (D)), 56.2 (3′-O
Me, 4′-O
Me), 34.2 (C-5″ (D)), 32.7 (C-3 (C)), 26.7 (C-4 (C)). Melting point: 106–108 °C HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
28H
23ClO
6 490.1182, found 490.1183. Analytical HPLC (
λ = 280 nm): purity: >99%;
tR = 21.9 min. Compound (+)-
68a: Chiral HPLC (
λ = 280 nm): purity: >99%;
tR = 10.9 min. [α]
D: + 17 (c. 0.5, CHCl
3). Compound (−)-
68a: Chiral HPLC (
λ = 280 nm): purity: > 99%;
tR = 14.1 min. [α]
D: −16 (c. 0.2, CHCl
3). Compound
68b:
1H NMR (400 MHz, CDCl
3) δ 7.63 (
d,
J = 2.5 Hz, 1H, H-5), 7.47–7.40 (
m, 5H, H-2‴, H-3‴, H-4‴), 7.28 (
dd,
J = 8.4, 2.2 Hz, 1H, H-6′), 7.25-7.19 (ov, 1H, H-7), 7.24 (
d,
J = 2.2 Hz, 1H, H-2′), 7.04-6.98 (ov, 1H, H-8, H-5′), 5.59 (
dd,
J = 12.3, 4.1 Hz, 1H, H-6″), 4.25 (
br s, 1H, H-4), 3.90 (
s, 6H, 3′-O
Me, 4′-O
Me), 2.94 (
dd,
J = 17.5, 12.3 Hz, 1H, H-5″), 2.78 (
ddd,
J = 17.5, 4.1, 1.1 Hz, 1H, H-5″), 2.46 (
qd,
J = 13.5, 2.8 Hz, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.5 (C-2″ (D)), 163.7 (C-4″ (D)), 150.2 (C-9 (A)), 150.1 (C-4′ (B)), 149.1 (C-3′ (B)), 138.1 (C-1‴ (E)), 131.9 (C-1′ (B)), 129.0 (C-4‴ (E)), 128.9 (C-3‴ (E)), 128.6 (C-5 (A)), 128.2 (C-7(A)), 127.5 (C-10 (A)), 126.8 (C-6(A)), 126.2 (C-2‴ (E)), 118.2 (C-6′ (B)), 117.4 (C-8 (A)), 110.9 (C-5′ (B)), 109.0 (C-2′ (B)), 106.2 (C-3″ (D)), 100.4 (C-2 (C)), 77.5 (C-6″ (D)), 56.3 (3′-O
me, 4′-O
me), 34.3 (C-5″ (D)), 32.9 (C-3 (C)), 27.5 (C-4 (C)). Melting point: 178–180 °C. HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. For C
28H
23ClO
6 490.1182, found 490.1183. Analytical HPLC (
λ = 280 nm): purity: 97%;
tR = 21.8 min. Compound (+)-
68b: Chiral HPLC (
λ = 280 nm): purity: >99%;
tR = 8.2 min. [α]
D: + 236 (c. 0.5, CHCl
3). Compound (−)-
68b: Chiral HPLC (
λ = 280 nm): purity: > 99%;
tR = 20.9 min. [α]
D: −237 (c. 0.9, CHCl
3).
3.3.25. 2-(3′,4′-Dimethoxyphenyl)-6-chlorochromane-(4→3″, 2→O-4″)-6″-(4‴-chlorophenyl)-4″-hydroxy-5″,6″-dihydro-2H-pyran-2″-one (69a and 69b)
General methodology B was applied using the flavylium salt
32 (0.199 g) and 6-(4-chlorophenyl)-4-hydroxy-5,6-dihydro-2
H-pyran-2-one (
40, 0.122 g, 0.5 mmol), previously prepared according to the methodology described by Andersh et al. [
48,
49]. The purification step was performed by CC using mixtures of hexane-EtOAc (75:25). Pure compound
69a was obtained as a yellow amorphous solid (0.025 g, 10% from
23) and pure compound
69b as a pale orange amorphous solid (0.082 g, 33% from
23); Compound
69a:
1H NMR (400 MHz, CDCl
3) δ 7.36–7.29 (
m, 5H, H-5, H-2‴, H-3‴), 7.17–7.13 (ov, 2H, H-7, H-6′), 7.09 (
d,
J = 2.2 Hz, 1H, H-2′), 6.97 (
d,
J = 8.7 Hz, 1H, H-8), 6.90 (
d,
J = 8.5 Hz, 1H, H-5′), 5.30 (
dd,
J = 12.3, 4.1 Hz, 1H, H-6″), 4.24 (
br s, 1H, H-4), 3.90 (
s, 6H, 3′-O
Me, 4′-O
Me), 2.95 (
dd,
J = 17.2, 12.3 Hz, 1H, H-5″), 2.69 (
dd,
J = 17.2, 4.1 Hz, 1H, H-5″), 2.34-2.23 (
m, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.3 (C-2″ (D)), 163.3 (C-4″ (D)), 150.2 * (C-4′ (B)), 150.1 * (C-9 (A)), 149.1 (C-3′ (B)), 136.7 (C-1‴ (E)), 134.8 (C-4‴ (E)), 131.7 (C-1′ (B)), 129.2 (C-3‴ (E)), 128.3 (C-7(A)), 127.8 (C-6(A)), 127.6 (C-2‴ (E)), 127.7 (C-5 (A)), 127.1 (C-10 (A)), 118.2 (C-6′ (B)), 117.8 (C-8 (A)), 110.9 (C-5′ (B)), 109.0 (C-2′ (B)), 106.2 (C-3″ (D)), 100.6 (C-2 (C)), 76.1 (C-6″ (D)), 56.2 (3′-O
Me, 4′-O
Me), 34.2 (C-5″ (D)), 32.7 (C-3 (C)), 26.6 (C-4 (C)); *these signals could be interchangeable. Melting point: 172–175 °C. HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
28H
22Cl
2O
6 524.0793, found 524.0793. Analytical HPLC (
λ = 280 nm): purity: 97%;
tR = 22.9 min. Compound
69b:
1H NMR (400 MHz, CDCl
3) δ 7.51 (
d,
J = 2.5 Hz, 1H, H-5), 7.34–7.24 (
m, 4H, H-2‴, H-3‴), 7.17 (
dd,
J = 8.4, 2.2 Hz, 1H, H-6′), 7.16-7.09 (ov, 2H, H-2′, H-7), 6.93-6.91 (ov,
2H, H-8, H-5′), 5.47 (
dd,
J = 12.2, 4.3 Hz, 1H, H-6″), 4.00 (
d,
J = 3.0 Hz, 1H, H-4), 3.92 (
s, 3H, 3′-O
Me), 3.90 (
s, 3H, 4′-O
Me), 2.80 (
dd,
J = 17.5, 12.2 Hz, 1H, H-5″), 2.68 (
ddd,
J = 17.5, 4.3, 1.0 Hz, 1H, H-5″), 2.37 (
qd,
J = 13.6, 3.0, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 166.2 (C-2″ (D)), 163.3 (C-4″ (D)), 150.1 (C-4′ (B), C-9 (A)), 149.1 (C-3′ (B)), 136.7 (C-1‴ (E)), 134.8 (C-4‴ (E)), 131.8 (C-1′ (B)), 129.1 (C-3‴ (E)), 128.5 (C-5 (A)), 128.2 (C-7(A)), 127.5 (C-2‴ (E)), 127.4 (C-10 (A)), 126.9 (C-6(A)), 118.2 (C-6′ (B)), 117.4 (C-8 (A)), 110.9 (C-5′ (B)), 109.2 (C-2′ (B)), 106.3 (C-3″ (D)), 100.5 (C-2 (C)), 76.7 (C-6″ (D)), 56.3 (3′-O
Me), 56.2 (4′-O
Me), 34.2 (C-5″ (D)), 32.8 (C-3 (C)), 27.4 (C-4 (C)). Melting point: 110–113 °C. HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
28H
22Cl
2O
6 524.0793, found 524.0794. Analytical HPLC (
λ = 280 nm): purity: 98%;
tR = 23.2 min.
3.3.26. 2-(3′.4′-Dimethoxyphenyl)-6-chlorochromane-(4→3″, 2→O-4″)-4″-hydroxy-6″-(4‴-methoxyphenyl)-5″,6″-dihydro-2H-pyran-2″-one (70a and 70b)
General methodology B was applied using the flavylium salt
32 (0.199 g) and 4-hydroxy-6-(4-methoxyphenyl)-5,6-dihydro-2
H-pyran-2-one (
41, 0.110 g, 0.5 mmol), previously prepared according to the methodology described by Andersh et al. [
48,
49] (spectroscopic data agree with those reported in the literature [
50]). The purification step was performed by CC using mixtures of hexane-EtOAc (75:25). Pure compound
70a was obtained as a white foam (0.032 g, 12% from
23) and pure compound
70b as a yellow amorphous solid (0.075 g, 29% from
23); Compound
70a:
1H NMR (400 MHz, CDCl
3) δ 7.35 (
d,
J = 2.5 Hz, 1H, H-5), 7.29 (
d,
J = 8.7 Hz, 2H, H-2‴), 7.17–7.13 (ov, 2H, H-7, H-6′), 7.10 (
d, J = 2.1 Hz, 1H, H-2′), 6.97 (
d,
J = 8.7 Hz, 1H, H-8), 6.92-6.87 (ov, 3H, H-5′, H-3‴), 5.26 (
dd,
J = 12.4, 4.0 Hz, 1H, H-6″), 4.24 (
br s, 1H, H-4), 3.90 (
s, 6H, 3′-O
Me, 4′-O
Me), 3.78 (
s, 3H, 4‴-O
Me), 3.00 (
dd,
J = 17.0, 12.6 Hz, 1H, H-5″), 2.65 (
dd,
J = 17.2, 4.0 Hz, 1H, H-5″), 2.36-2.20 (
m, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.7 (C-2″ (D)), 163.5 (C-4″ (D)), 160.0 (C-4‴ (E)), 150.3 (C-9 (A)), 150.1 (C-4′ (B)), 149.1 (C-3′ (B)), 131.8 (C-1′ (B)), 130.2 (C-1‴ (E)), 127.8 (C-2‴ (E)), 128.2 (C-7(A)), 127.9 (C-10 (A)), 127.7 (C-5 (A)), 127.0 (C-6(A)), 118.2 (C-6′ (B)), 117.8 (C-8 (A)), 114.2 (C-3‴ (E)), 110.9 (C-5′ (B)), 109.0 (C-2′ (B)), 106.1 (C-3″ (D)), 100.5 (C-2 (C)), 76.7 (C-6″ (D)), 56.2 (3′-O
Me, 4′-O
Me), 55.3 (4‴-O
Me), 34.1 (C-5″ (D)), 32.7 (C-3 (C)), 26.6 (C-4 (C)). HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
29H
25ClO
7 520.1289, found 520.1280. Analytical HPLC (
λ = 280 nm): purity: 98%;
tR = 21.9 min. Compound
70b:
1H NMR (400 MHz, CDCl
3) δ 7.53 (
d,
J = 2.5 Hz, 1H, H-5), 7.27 (
d,
J = 8.7 Hz, 2H, H-2‴), 7.18 (
dd,
J = 8.4, 2.1 Hz, 1H, H-6′), 7.14-7.11 (ov, 2H, H-7, H-2′), 6.94-6.90 (ov, 2H, H-8, H-5′), 6.85 (
d,
J = 8.7 Hz, 2H, H-3‴), 5.44 (
dd,
J = 12.3, 3.8 Hz, 1H, H-6″), 4.00 (
br s, 1H, H-4), 3.92 (
s, 3H, 3′-O
Me), 3.90 (
s, 3H, 4′-O
Me), 3.78 (
s, 3H, 4‴-O
Me), 2.85 (
dd,
J = 17.5, 12.5 Hz, 1H, H-5″), 2.64 (
dd,
J = 17.5, 3.8 Hz, 1H, H-5″), 2.36 (
qd,
J = 13.6, 3.2 Hz, 2H, H-3);
13C NMR (100 MHz, CDCl
3) δ 165.7 (C-2″ (D)), 163.8 (C-4″ (D)), 160.1 (C-4‴ (E)), 150.2 (C-9 (A)), 150.1 (C-4′ (B)), 149.1 (C-3′ (B)), 132.0 (C-1′ (B)), 130.2 (C-1‴ (E)), 127.8 (C-2‴ (E)), 128.6 (C-5 (A)), 128.2 (C-7(A)), 127.5 (C-10 (A)), 126.8 (C-6(A)), 118.2 (C-6′ (B)), 117.4 (C-8 (A)), 114.2 (C-3‴ (E)), 110.9 (C-5′ (B)), 109.0 (C-2′ (B)), 106.2 (C-3″ (D)), 100.4 (C-2 (C)), 77.4 (C-6″ (D)), 56.3 (3′-O
Me), 56.2 (4′-O
Me), 55.5 (4‴-O
Me), 34.2 (C-5″ (D)), 32.9 (C-3 (C)), 27.5 (C-4 (C)). Melting point: 108-111 °C HRMS (ESI-TOF)
m/z [M+H]
+ Calcd. for C
29H
25ClO
7 520.1289, found 520.1289. Analytical HPLC (
λ = 280 nm): purity: 98%;
tR = 21.9 min.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
