
The VEGA Tool to Check the Applicability Domain Gives Greater Confidence in the Prediction of In Silico Models

 , , , ,
, , , ,
Abstract
:1. Introduction
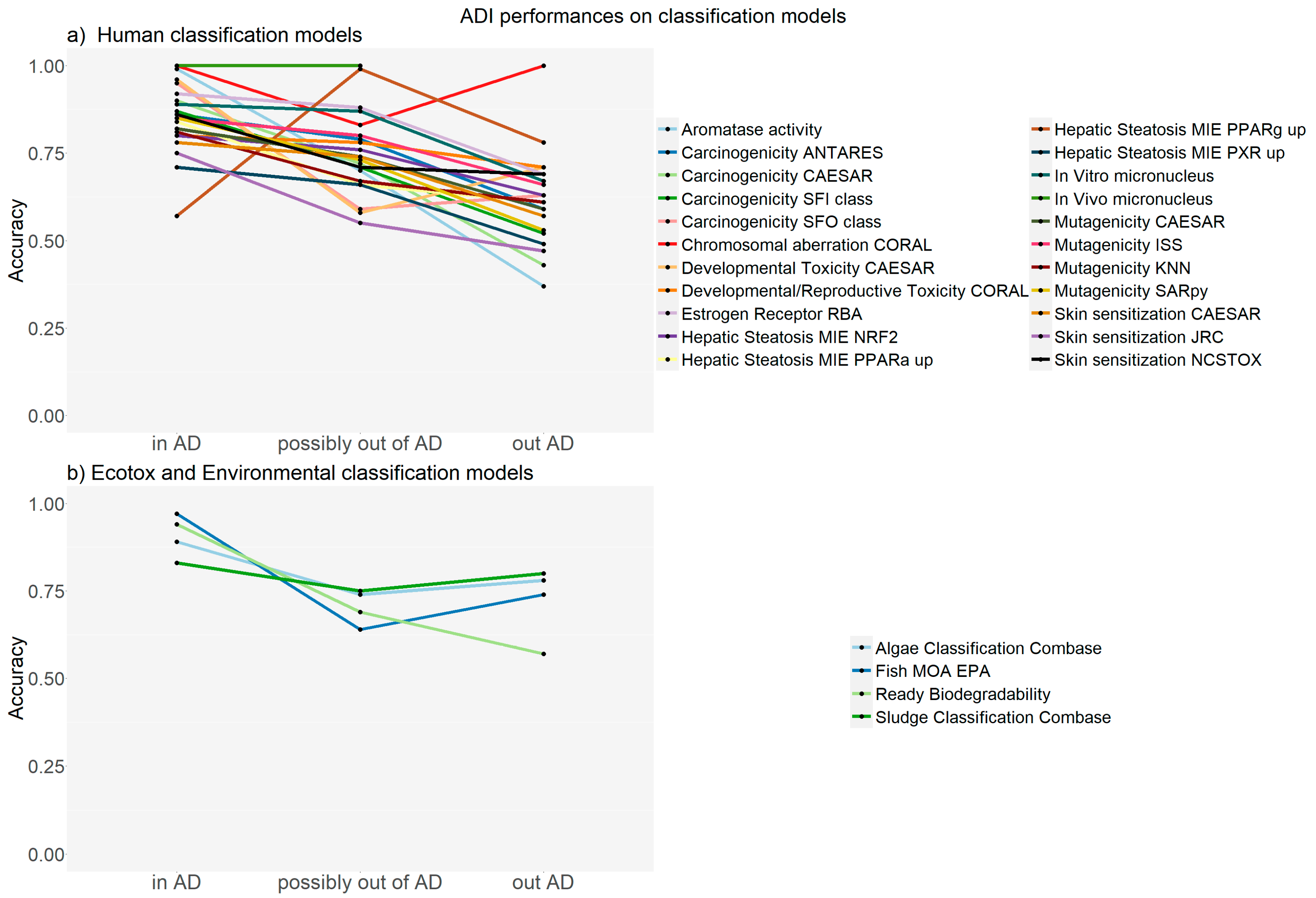
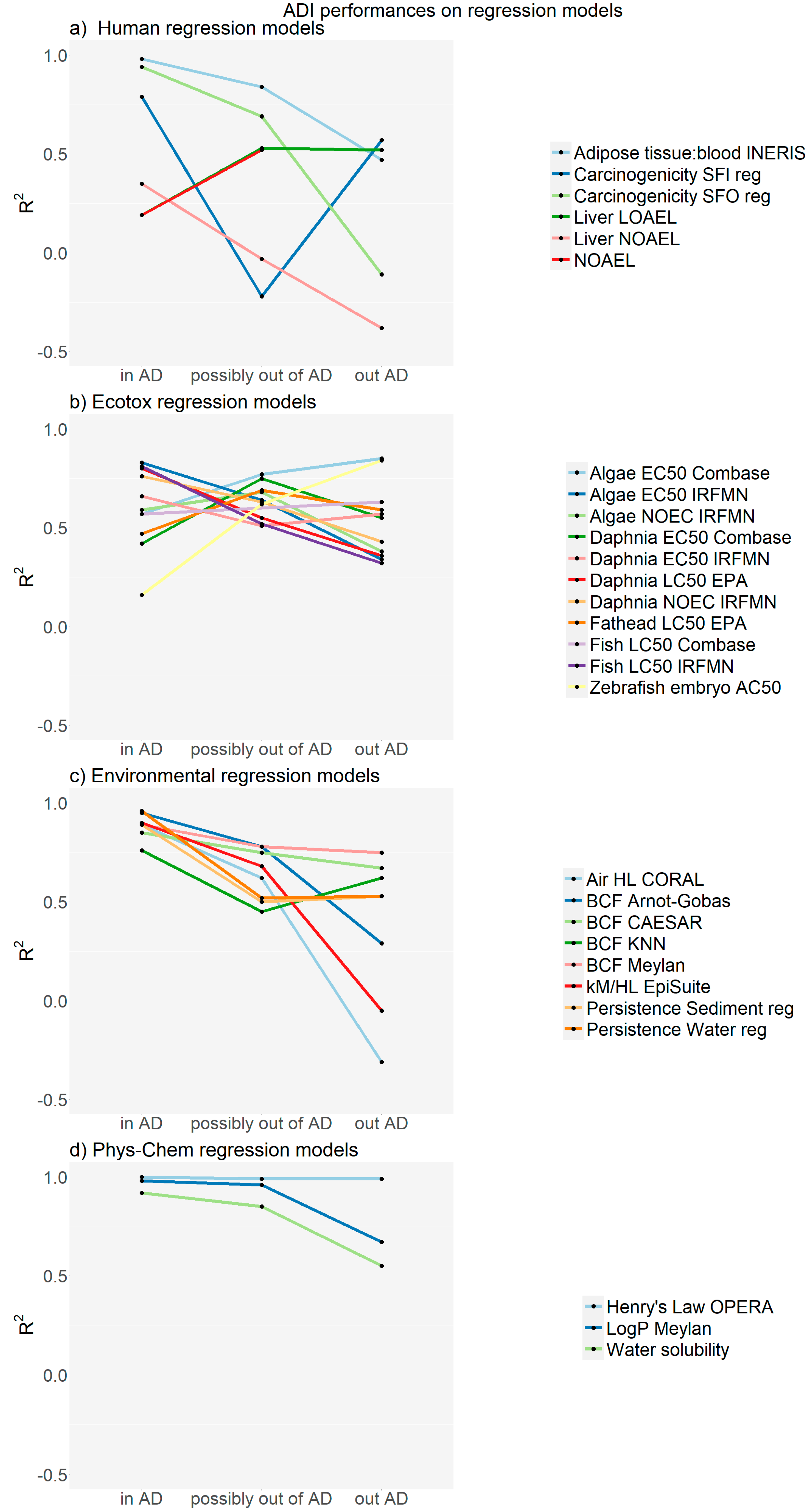
2. Results
- Allowing the identification of issues related to prediction accuracy and providing the user with an opportunity for thorough analysis.
- Allowing identification of mechanisms associated with structural features of the substances.
- Analyzing similar substances through a read-across approach.
- Filtering substances with more reliable predictions, to be used in batch mode for a range of substances.
2.1. Examples
2.1.1. Trifluralin
2.1.2. Diethyl(nitroso)amine
3. Discussion
4. Materials and Methods
4.1. Applicability Domain Index within VEGA
- VEGA checks the accuracy of the predictions for similar substances. In this case, the predicted value of the similar substance is compared with the experimental value. If the value is a label, such as mutagenic or not, the comparison is provided instantly. In the case of quantitative values, the software considers the quantitative differences across substances and an additional factor reports whether the difference in the prediction is very large or not.
- Concordance between the predicted value for the target substance and the experimental value of the similar compound is another very important parameter for the ADI. In this case, the prediction (i.e., the prediction accuracy of the in silico model) can be related to the “read-across” use of the VEGA output, showing the most similar substances. In particular, if predictions are different from the experimental values of similar substances, this poses a question, while if there is agreement, this increases the ADI. If the model provides structural alerts, VEGA provides an additional check and indicates whether for a similar substance, one or more structural alerts are present, and if such a structural alert is present in the target substance too. This is a valuable piece of information, highlighting to the user, for instance, that there is a structural alert only for a similar substance. Thus, the user can decide to disregard such a similar substance as non-relevant.
- The last component of the ADI regarding the specific endpoint is the presence of fragments associated with outliers for that endpoint. This component is present only for a few models, where the model poorly predicted a particular chemical family.
4.2. Categories of ADI Values
4.3. Modified ADI
4.4. Test Set
4.5. Performance Parameters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- REACH Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 Concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), Establishing a European Chemicals Agency, Amending Directive 1999/45/EC and Repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as Well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC. 2006. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A02006R1907-20140410 (accessed on 6 February 2023).
- OECD, Organisation for Economic Co-operation and Development. “OECD Principles for the Validation, for Regulatory Purpose, of (Q)SAR Models. 2004”. Available online: https://www.oecd.org/chemicalsafety/riskassessment/37849783.pdf (accessed on 6 February 2023).
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of different approaches to define the applicability domain of QSAR models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Roy, K.; Leszczynski, J. Applicability domain: A step toward confident predictions and decidability for QSAR modeling. In Computational Toxicology: Methods and Protocols, 1st ed.; Nicolotti, O., Ed.; Humana Press: New York, NY, USA, 2018; Volume 1800, pp. 141–169. [Google Scholar] [CrossRef]
- Fjodorova, N.; Novič, M.; Roncaglioni, A.; Benfenati, E. Evaluating the applicability domain in the case of classification predictive models for carcinogenicity based on the counter propagation artificial neural network. J. Comput. Aided Mol. Des. 2011, 25, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Toropov, A.A.; Toropova, A.P.; Lombardo, A.; Roncaglioni, A.; Benfenati, E.; Gini, G.I. CORAL: Building up the model for bioconcentration factor and defining it’s applicability domain. Eur. J. Med. Chem. 2011, 46, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, D.; Mangiatordi, G.F.; Catto, M.; Carotti, A.; Nicolotti, O. Applicability domain for QSAR models: Where theory meets reality. Int. J. Quant. Struct. Prop. Relatsh. 2016, 1, 45–63. [Google Scholar] [CrossRef]
- Roy, K.; Ambure, P.; Aher, R.B. How important is to detect systematic error in predictions and understand statistical applicability domain of QSAR models? Chemom. Intell. Lab. Syst. 2017, 162, 44–54. [Google Scholar] [CrossRef]
- Toropova, A.P.; Toropov, A.A.; Lombardo, A.; Lavado, G.; Benfenati, E. Paradox of ‘ideal correlations’: Improved model for air half-life of persistent organic pollutants. Environ. Technol. 2021, 43, 2510–2515. [Google Scholar] [CrossRef]
- Toropov, A.A.; Benfenati, E. Additive SMILES-based optimal descriptors in QSAR modelling bee toxicity: Using rare SMILES attributes to define the applicability domain. Bioorg. Med. Chem. 2008, 16, 4801–4809. [Google Scholar] [CrossRef]
- DTU Food. National Food Institute (Q)SAR Tool. Available online: https://qsar.food.dtu.dk/ (accessed on 8 February 2023).
- Cross, K.; Johnson, C.; Myatt, G.J. Implementation of In Silico Toxicology Protocols in Leadscope. Methods Mol. Biol. 2022, 2425, 419–434. [Google Scholar] [CrossRef]
- Benfenati, E. In Silico Methods for Predicting Drug Toxicity, 1st ed.; Humana Press: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Chakravarti, S.K.; Saiakhov, R.D.; Klopman, G. Optimizing Predictive Performance of CASE Ultra Expert System Models Using the Applicability Domains of Individual Toxicity Alerts. J. Chem. Inf. Model. 2012, 52, 2609–2618. [Google Scholar] [CrossRef]
- Ponting, D.J.; Burns, M.J.; Foster, R.S.; Hemingway, R.; Kocks, G.; MacMillan, D.S.; Shannon-Little, A.L.; Tennant, R.E.; Tidmarsh, J.R.; Yeo, D.J. Use of Lhasa Limited Products for the In Silico Prediction of Drug Toxicity. Methods Mol. Biol. 2022, 2425, 435–478. [Google Scholar] [CrossRef]
- VEGAHUB. Available online: www.vegahub.eu (accessed on 8 February 2023).
- ECHA. Preparation of an Inventory of Substances Suspected to Meet REACH Annex III Criteria. Technical Documentation. 2016. Available online: https://echa.europa.eu/documents/10162/1819180/annex_iii_preparation_inventory_en.pdf/e42ea5b1-28f0-4390-8d34-6d09e2875ebd?t=1463575533871 (accessed on 15 February 2023).
- AMBIT—Cheminformatics Data Management System. Available online: http://cefic-lri.org/toolbox/ambit/ (accessed on 15 February 2023).
- Chemical Life Cycle Collaborative—CLCC. Available online: https://clicc.ucsb.edu/ (accessed on 20 February 2023).
- Golbamaki, A.; Benfenati, E. In Silico Methods for Carcinogenicity Assessment. In In Silico Methods for Predicting Drug Toxicity, 2nd ed.; Benfenati, E., Ed.; Humana Press: New York, NY, USA, 2022; Volume 2425, pp. 201–216. [Google Scholar] [CrossRef]
- Manganelli, S.; Gamba, A.; Colombo, E.; Benfenati, E. Using VEGAHUB Within a Weight-of-Evidence Strategy. In In Silico Methods for Predicting Drug Toxicity, 2nd ed.; Benfenati, E., Ed.; Humana Press: New York, NY, USA, 2022; Volume 2425, pp. 479–495. [Google Scholar] [CrossRef]
- Marzo, M.; Roncaglioni, R.; Kulkarni, S.; Barton-Maclaren, T.S.; Benfenati, E. In silico model for developmental toxicity. In In Silico Methods for Predicting Drug Toxicity, 2nd ed.; Benfenati, E., Ed.; Humana Press: New York, NY, USA, 2022; Volume 2425, pp. 217–240. [Google Scholar] [CrossRef]
- Mombelli, E.; Raitano, G.; Benfenati, E. In silico prediction of chemically induced mutagenicity: A weight of evidence approach integrating information from QSAR and read-across predictions. In In Silico Methods for Predicting Drug Toxicity, 2nd ed.; Benfenati, E., Ed.; Humana Press: New York, NY, USA, 2022; Volume 2425, pp. 149–184. [Google Scholar] [CrossRef]
- Pizzo, F.; Gadaleta, D.; Benfenati, E. In silico models for repeated-dose toxicity (RTD): Prediction of the No Observed Adverse Effect Level (NOAEL) and Lowest Observed Adverse Effect Level (LOAEL). In In Silico Methods for Predicting Drug Toxicity, 2nd ed.; Benfenati, E., Ed.; Humana Press: New York, NY, USA, 2022; Volume 2425, pp. 241–258. [Google Scholar] [CrossRef]
- Roncaglioni, A.; Lombardo, A.; Benfenati, E. The VEGAHUB Platform: The philosophy and the tools. Altern. Lab. Anim. 2022, 50, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Selvestrel, G.; Lavado, G.J.; Toropova, A.P.; Toropov, A.A.; Gadaleta, D.; Marzo, M.; Baderna, D.; Benfenati, E. Monte Carlo Models for Sub-Chronic Repeated-Dose Toxicity: Systemic and Organ-Specific Toxicity. Int. J. Mol. Sci. 2022, 23, 6615. [Google Scholar] [CrossRef] [PubMed]
- Benfenati, E.; Lombardo, A. VEGAHUB for Ecotoxicological QSAR Modeling. In Ecotoxicological QSARs, 1st ed.; Roy, K., Ed.; Humana Press: New York, NY, USA, 2020; pp. 759–787. [Google Scholar] [CrossRef]
- Benfenati, E.; Chaudhry, Q.; Gini, G.; Dorne, J.L. Integrating in silico models and read-across methods for predicting toxicity of chemicals: A step-wise strategy. Environ. Int. 2019, 131, 105060. [Google Scholar] [CrossRef]
- Cassano, A.; Raitano, G.; Mombelli, E.; Fernández, A.; Cester, J.; Roncaglioni, A.; Benfenati, E. Evaluation of QSAR models for the prediction of ames genotoxicity: A retrospective exercise on the chemical substances registered under the EU REACH regulation. J. Environ. Sci. Health 2014, 32, 273–298. [Google Scholar] [CrossRef] [PubMed]
- EFSA Scientific Committee; Hardy, A.; Benford, D.; Halldorsson, T.; Jeger, M.J.; Knutsen, H.K.; More, S.; Naegeli, H.; Noteborn, H.; Ockleford, C.; et al. Scientific Opinion on the guidance on the use of the weight of evidence approach in scientific assessments. EFSA J. 2017, 15, 497. [Google Scholar] [CrossRef] [Green Version]
- Benfenati, E.; Roncaglioni, A.; Lombardo, A.; Manganaro, A. Integrating QSAR, read-across, and screening tools: The VEGAHUB platform as an example. In Advances in Computational Toxicology: Methodologies and Applications in Regulatory Science, 1st ed.; Hong, H., Ed.; Springer: Cham, Switzerland, 2019; Volume 30, pp. 365–381. [Google Scholar] [CrossRef]
- Viganò, E.L.; Colombo, E.; Raitano, G.; Manganaro, A.; Sommovigo, A.; Dorne, J.L.C.; Benfenati, E. Virtual Extensive Read-Across: A New Open-Access Software for Chemical Read-Across and Its Application to the Carcinogenicity Assessment of Botanicals. Molecules 2022, 27, 6605. [Google Scholar] [CrossRef]
- Floris, M.; Manganaro, A.; Nicolotti, O.; Medda, R.; Mangiatordi, G.F.; Benfenati, E. A generalizable definition of chemical similarity for read-across. J. Cheminform. 2014, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lunghini, F.; Marcou, G.; Azam, P.; Patoux, R.; Enrici, M.H.; Bonachera, F.; Horvath, D.; Varnek, A. QSPR models for bioconcentration factor (BCF): Are they able to predict data of industrial interest? SAR QSAR Environ. Res. 2019, 30, 507–524. [Google Scholar] [CrossRef]
- SKINSENS DB. Available online: https://cwtung.kmu.edu.tw/skinsensdb/search (accessed on 20 February 2023).
- Alves, V.M.; Muratov, E.; Fourches, D.; Strickland, J.; Kleinstreuer, N.; Andrade, C.H.; Tropsha, A. Predicting chemically-induced skin reactions. Part I: QSAR models of skin sensitization and their application to identify potentially hazardous compounds. Toxicol. Appl. Pharmacol. 2015, 284, 262–272. [Google Scholar] [CrossRef] [Green Version]
- NICEATM LLNA Database. National Toxicology Program. Available online: https://ntp.niehs.nih.gov/iccvam/methods/immunotox/niceatm-llnadatabase-23dec2013.xls (accessed on 20 February 2023).
- Jaworska, J.S.; Natsch, A.; Ryan, C.; Strickland, J.; Ashikaga, T.; Miyazawa, M. Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: A decision support system for quantitative weight of evidence and adaptive testing strategy. Arch. Toxicol. 2015, 89, 2355–2383. [Google Scholar] [CrossRef] [PubMed]
- Natsch, A.; Emter, R.; Gfeller, H.; Haupt, T.; Ellis, G. Predicting skin sensitizer potency based on in vitro data from KeratinoSens and kinetic peptide binding: Global versus domain-based assessment. Toxicol. Sci. 2015, 143, 319–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickland, J.; Zang, Q.; Paris, M.; Lehmann, D.M.; Allen, D.; Choksi, N.; Matheson, J.; Jacobs, A.; Casey, W.; Kleinstreuer, N. Multivariate models for prediction of human skin sensitization hazard. J. Appl. Toxicol. 2017, 37, 347–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- QSAR Toolbox. Available online: https://qsartoolbox.org/ (accessed on 20 February 2023).
- Honma, M.; Kitazawa, A.; Cayley, A.; Williams, R.V.; Barber, C.; Hanser, T.; Saiakhov, R.; Chakravarti, S.; Myatt, G.J.; Cross, K.P.; et al. Improvement of quantitative structure–activity relationship (QSAR) tools for predicting Ames mutagenicity: Outcomes of the Ames/QSAR International Challenge Project. Mutagenesis 2019, 34, 3–16. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Parameters | Trifluralin | Diethyl(nitroso)amine |
|---|---|---|
| ADI | 1.000 | 0.309 |
| Similarity index | 0.975 | 0.773 |
| Accuracy index | 0.486 | 0.452 |
| Concordance index | 0.470 | 1.860 |
| Max error index | 0.507 | 0.765 |
| Descriptors range check | True | - |
| ACF index | 1.000 | 0.400 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danieli, A.; Colombo, E.; Raitano, G.; Lombardo, A.; Roncaglioni, A.; Manganaro, A.; Sommovigo, A.; Carnesecchi, E.; Dorne, J.-L.C.M.; Benfenati, E. The VEGA Tool to Check the Applicability Domain Gives Greater Confidence in the Prediction of In Silico Models. Int. J. Mol. Sci. 2023, 24, 9894. https://doi.org/10.3390/ijms24129894
Danieli A, Colombo E, Raitano G, Lombardo A, Roncaglioni A, Manganaro A, Sommovigo A, Carnesecchi E, Dorne J-LCM, Benfenati E. The VEGA Tool to Check the Applicability Domain Gives Greater Confidence in the Prediction of In Silico Models. International Journal of Molecular Sciences. 2023; 24(12):9894. https://doi.org/10.3390/ijms24129894
Chicago/Turabian StyleDanieli, Alberto, Erika Colombo, Giuseppa Raitano, Anna Lombardo, Alessandra Roncaglioni, Alberto Manganaro, Alessio Sommovigo, Edoardo Carnesecchi, Jean-Lou C. M. Dorne, and Emilio Benfenati. 2023. "The VEGA Tool to Check the Applicability Domain Gives Greater Confidence in the Prediction of In Silico Models" International Journal of Molecular Sciences 24, no. 12: 9894. https://doi.org/10.3390/ijms24129894