

Identification of Somatic Mutations in Plasma Cell-Free DNA from Patients with Metastatic Oral Squamous Cell Carcinoma

Abstract
:1. Introduction
2. Results
2.1. Characterization of Patients
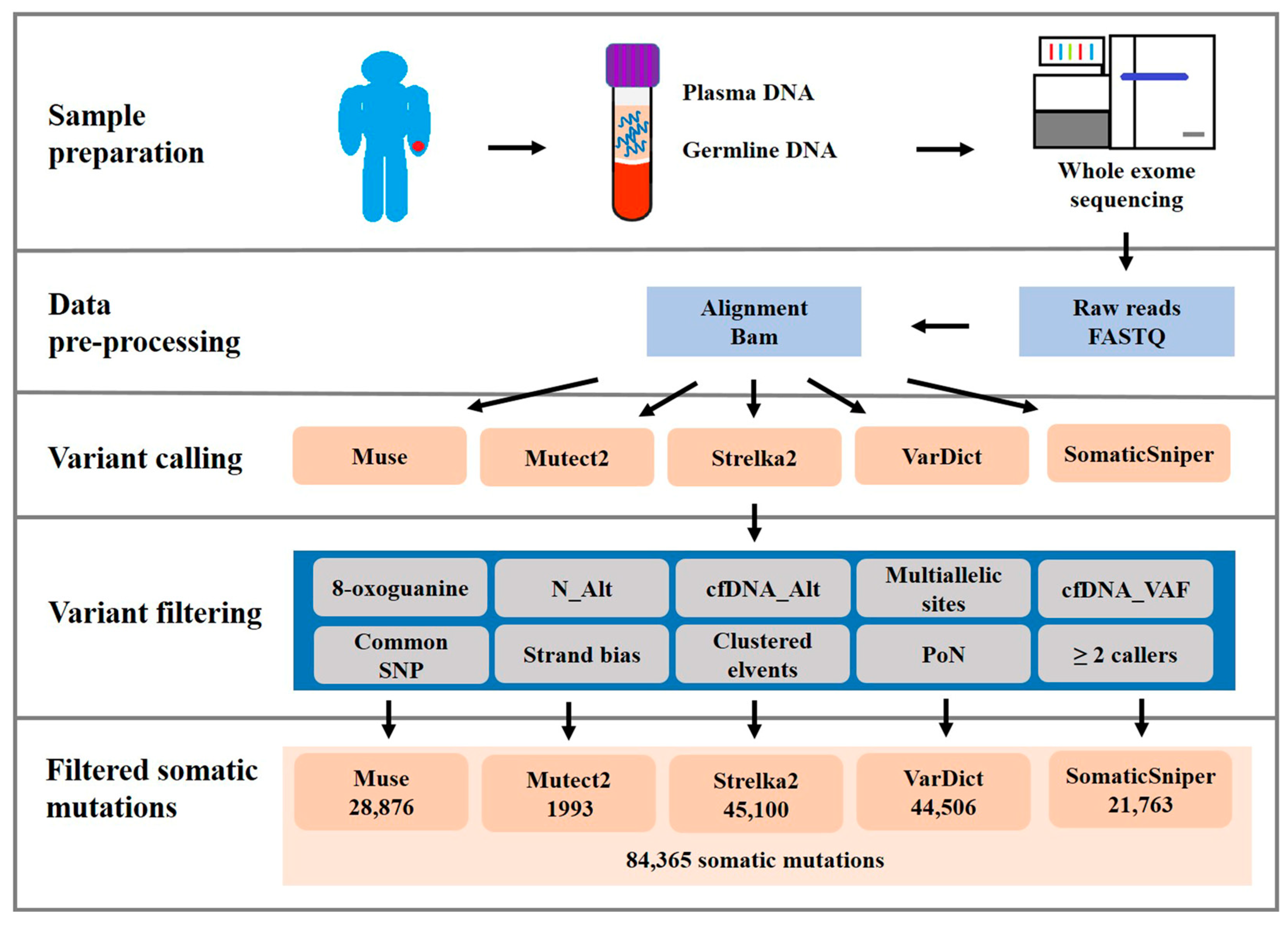
2.2. cfDNA WES
2.3. Validation and Assessment of Somatic Mutation Calling
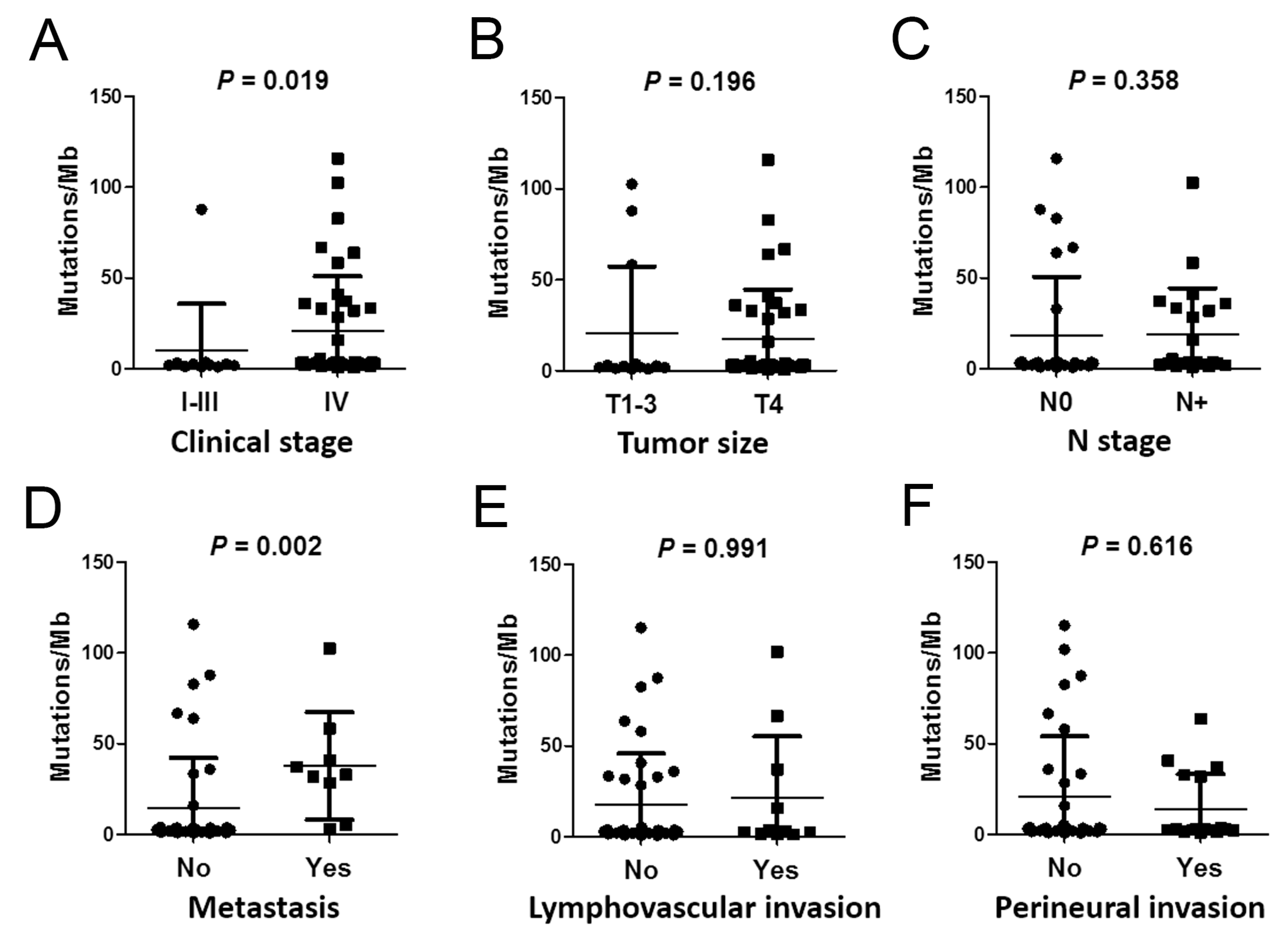
2.4. Correlations between Mutation Burden and Sequencing Quality and Clinical Parameters
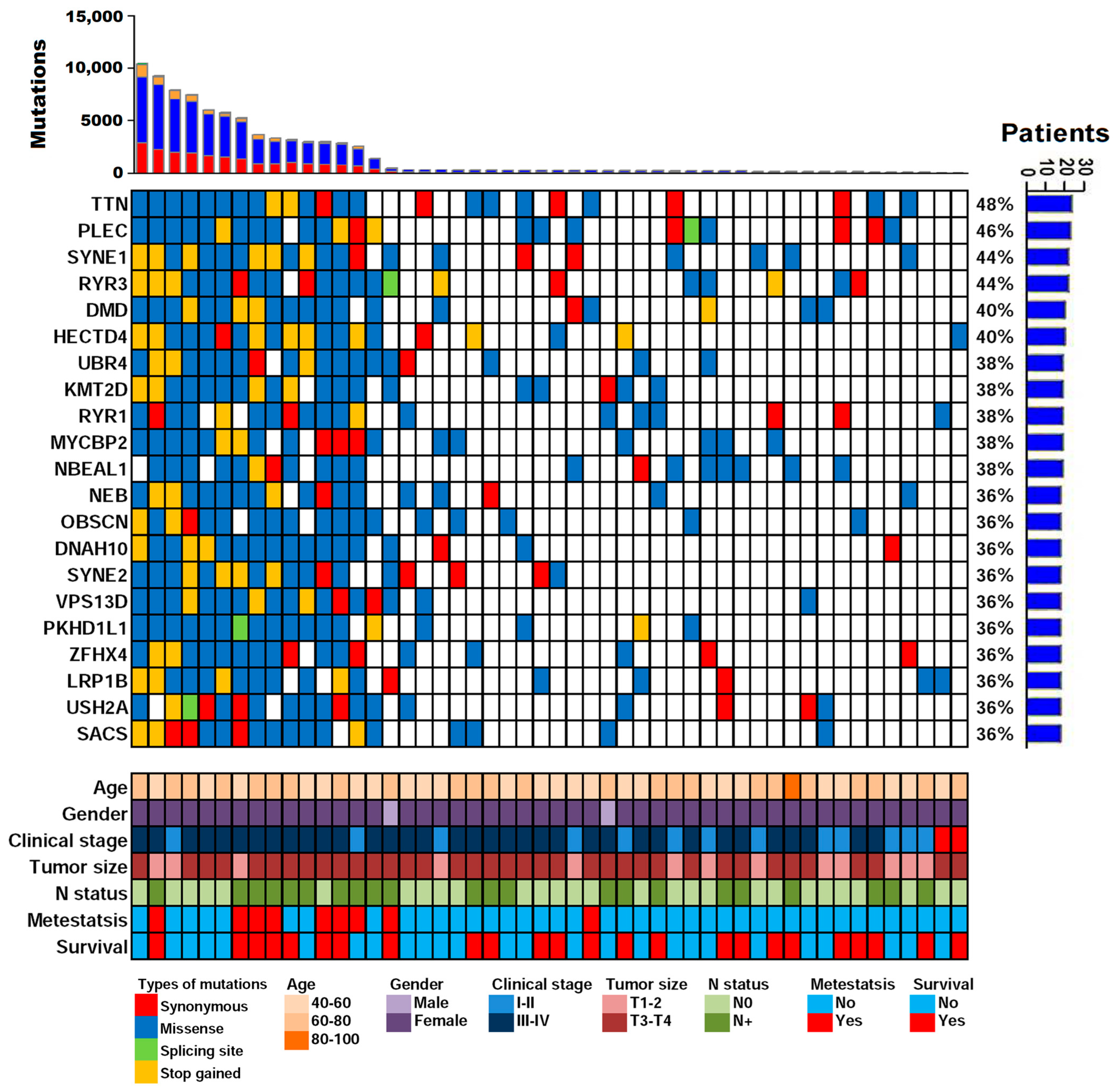
2.5. Analysis of Plasma cfDNA Revealed Clinically Actionable Mutations without Prior Knowledge of the Tumor
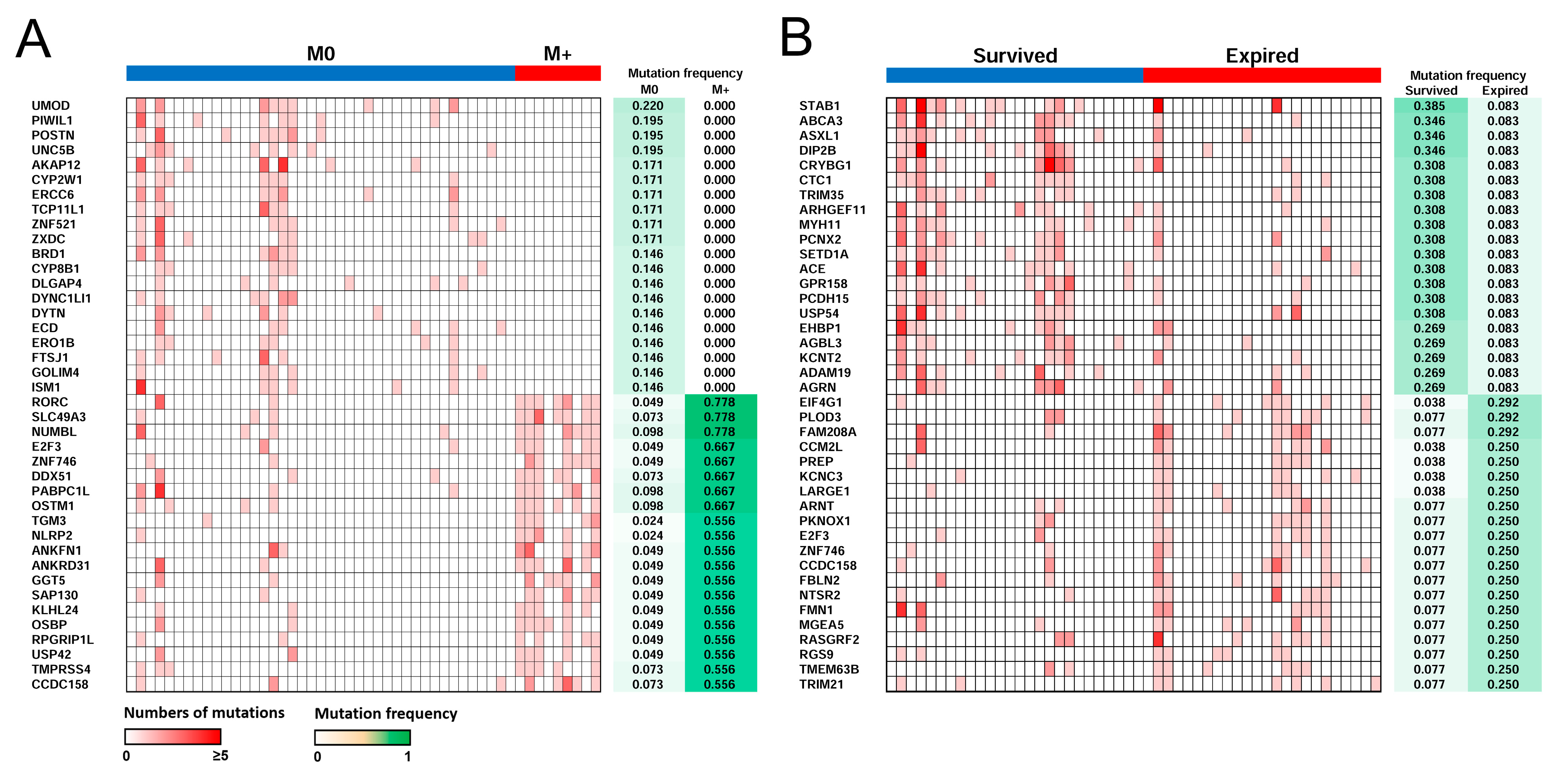
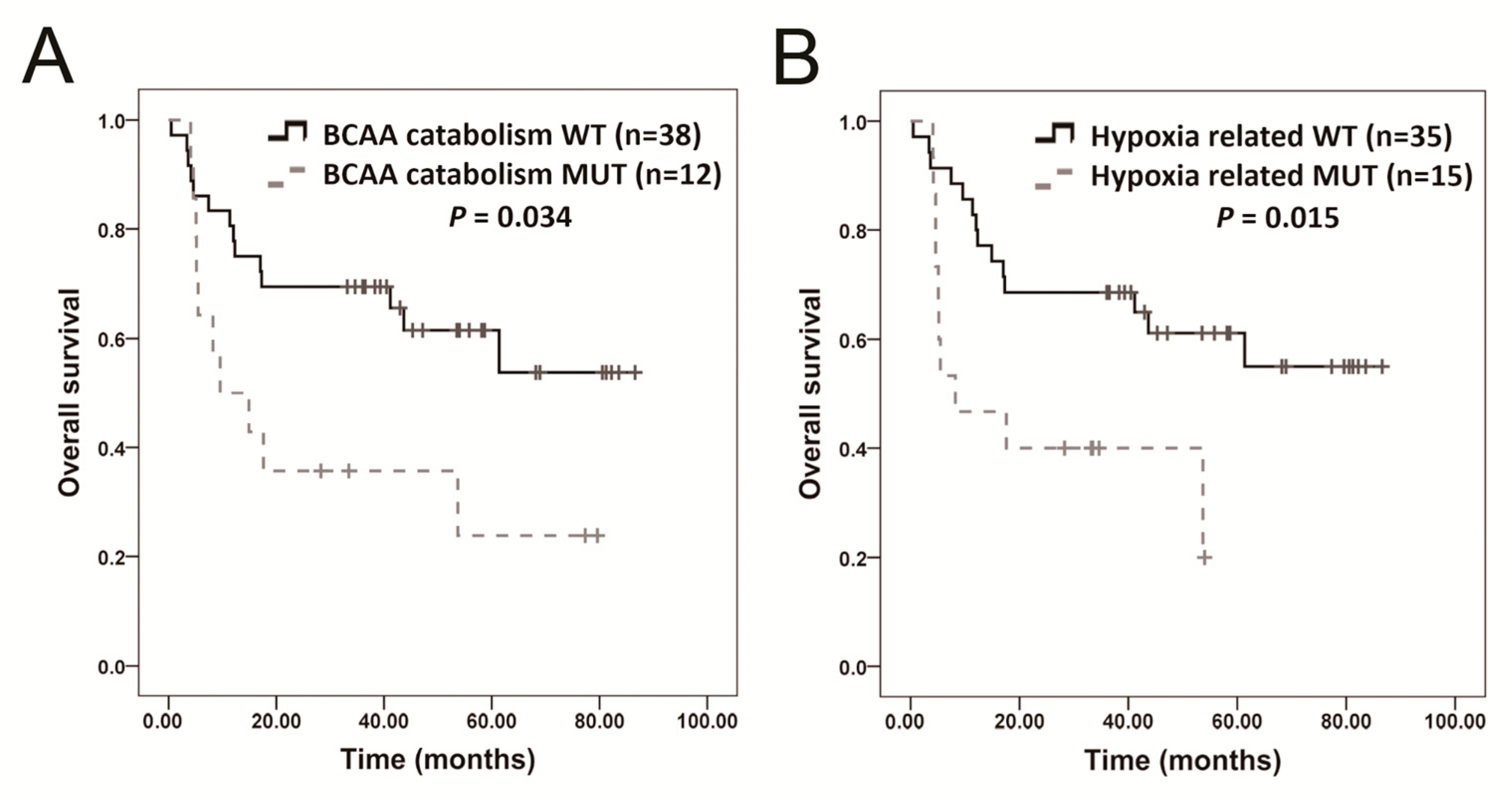
2.6. Distant Metastasis and Survival-Related Genes
2.7. Molecular Pathway Analysis
3. Discussion
4. Materials and Methods
4.1. Participants and Data Collection
4.2. DNA Extraction
4.3. Sequencing of Plasma cfDNA
4.4. Sequencing Data Processing
4.5. Validation of Mutations
4.6. Pathway Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Cancer Today, GLOBOCAN 2020th ed.; GLOBOCAN: Lyon, France, 2020; Volume 2020, pp. 1–2. [Google Scholar]
- Boute, P.; Page, C.; Biet, A.; Cuvelier, P.; Strunski, V.; Chevalier, D. Epidemiology, prognosis and treatment of simultaneous squamous cell carcinomas of the oral cavity and hypopharynx. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2014, 131, 283–287. [Google Scholar] [CrossRef] [Green Version]
- Gluckman, J.L. Synchronous multiple primary lesions of the upper aerodigestive system. Arch. Otolaryngol. 1979, 105, 597–598. [Google Scholar] [CrossRef]
- Gupta, S.; Kong, W.; Peng, Y.; Miao, Q.; Mackillop, W.J. Temporal trends in the incidence and survival of cancers of the upper aerodigestive tract in Ontario and the United States. Int. J. Cancer 2009, 125, 2159–2165. [Google Scholar] [CrossRef]
- Braakhuis, B.J.; Tabor, M.P.; Leemans, C.R.; van der Waal, I.; Snow, G.B.; Brakenhoff, R.H. Second primary tumors and field cancerization in oral and oropharyngeal cancer: Molecular techniques provide new insights and definitions. Head Neck 2002, 24, 198–206. [Google Scholar] [CrossRef]
- Kidess, E.; Jeffrey, S.S. Circulating tumor cells versus tumor-derived cell-free DNA: Rivals or partners in cancer care in the era of single-cell analysis? Genome Med. 2013, 5, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spellman, P.T.; Gray, J.W. Detecting cancer by monitoring circulating tumor DNA. Nat. Med. 2014, 20, 474–475. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Pantel, K. Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell 2017, 31, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venesio, T.; Siravegna, G.; Bardelli, A.; Sapino, A. Liquid Biopsies for Monitoring Temporal Genomic Heterogeneity in Breast and Colon Cancers. Pathobiology 2018, 85, 146–154. [Google Scholar] [CrossRef]
- Couraud, S.; Vaca-Paniagua, F.; Villar, S.; Oliver, J.; Schuster, T.; Blanche, H.; Girard, N.; Tredaniel, J.; Guilleminault, L.; Gervais, R.; et al. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: A proof-of-concept study from BioCAST/IFCT-1002. Clin. Cancer Res. 2014, 20, 4613–4624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.; Fleischhacker, M.; Rabien, A. Cell-free DNA in the blood as a solid tumor biomarker—A critical appraisal of the literature. Clin. Chim. Acta 2010, 411, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, A.M.; Rutkowski, T.; Fiszer-Kierzkowska, A.; Malusecka, E.; Skladowski, K. Assessment of the total cfDNA and HPV16/18 detection in plasma samples of head and neck squamous cell carcinoma patients. Oral Oncol. 2016, 54, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-H.; Chang, K.-W.; Kao, S.-Y.; Cheng, H.-W.; Liu, C.-J. Increased Plasma Circulating Cell-Free DNA Could Be a Potential Marker for Oral Cancer. Int. J. Mol. Sci. 2018, 19, 3303. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [Green Version]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.-H.; Chou, C.-H.; Cheng, H.-W.; Chang, K.-W.; Liu, C.-J. Precise Identification of Recurrent Somatic Mutations in Oral Cancer through Whole-Exome Sequencing Using Multiple Mutation Calling Pipelines. Front. Oncol. 2021, 11, 741626. [Google Scholar] [CrossRef]
- Bos, M.K.; Angus, L.; Nasserinejad, K.; Jager, A.; Jansen, M.; Martens, J.W.M.; Sleijfer, S. Whole exome sequencing of cell-free DNA—A systematic review and Bayesian individual patient data meta-analysis. Cancer Treat. Rev. 2020, 83, 101951. [Google Scholar] [CrossRef] [Green Version]
- Tsui, D.W.Y.; Cheng, M.L.; Shady, M.; Yang, J.L.; Stephens, D.; Won, H.; Srinivasan, P.; Huberman, K.; Meng, F.; Jing, X.; et al. Tumor fraction-guided cell-free DNA profiling in metastatic solid tumor patients. Genome Med. 2021, 13, 96. [Google Scholar] [CrossRef]
- Tailor, T.D.; Rao, X.; Campa, M.J.; Wang, J.; Gregory, S.G.; Patz, E.F., Jr. Whole Exome Sequencing of Cell-Free DNA for Early Lung Cancer: A Pilot Study to Differentiate Benign from Malignant CT-Detected Pulmonary Lesions. Front. Oncol. 2019, 9, 317. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.; Kulasinghe, A.; Kenny, L.; Punyadeera, C. The development of a liquid biopsy for head and neck cancers. Oral Oncol. 2016, 61, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Kulasinghe, A.; Allcock, R.J.N.; Tan, L.Y.; Mokany, E.; Kenny, L.; Punyadeera, C. A Pilot Study to Non-Invasively Track PIK3CA Mutation in Head and Neck Cancer. Diagnostics 2018, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagaki, T.; Tamura, M.; Kobashi, K.; Omori, A.; Koyama, R.; Idogawa, M.; Ogi, K.; Hiratsuka, H.; Tokino, T.; Sasaki, Y. Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma. Tumour Biol. 2018, 40, 1010428318800180. [Google Scholar] [CrossRef] [Green Version]
- van Ginkel, J.H.; Huibers, M.M.H.; van Es, R.J.J.; de Bree, R.; Willems, S.M. Droplet digital PCR for detection and quantification of circulating tumor DNA in plasma of head and neck cancer patients. BMC Cancer 2017, 17, 428. [Google Scholar] [CrossRef] [PubMed]
- Braig, F.; Voigtlaender, M.; Schieferdecker, A.; Busch, C.J.; Laban, S.; Grob, T.; Kriegs, M.; Knecht, R.; Bokemeyer, C.; Binder, M. Liquid biopsy monitoring uncovers acquired RAS-mediated resistance to cetuximab in a substantial proportion of patients with head and neck squamous cell carcinoma. Oncotarget 2016, 7, 42988–42995. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.-H.; Cheng, H.-W.; Liu, C.-J. Droplet digital polymerase chain reaction for detection and quantification of cell-free DNA TP53 target somatic mutations in oral cancer. Cancer Biomark. 2022, 33, 29–41. [Google Scholar] [CrossRef]
- Yang, X.; Xu, X.; Zhang, C.; Ji, T.; Wan, T.; Liu, W. The diagnostic value and prospects of gene mutations in circulating tumor DNA for head and neck cancer monitoring. Oral Oncol. 2022, 128, 105846. [Google Scholar] [CrossRef]
- Liebs, S.; Eder, T.; Klauschen, F.; Schutte, M.; Yaspo, M.-L.; Keilholz, U.; Tinhofer, I.; Kidess-Sigal, E.; Braunholz, D. Applicability of liquid biopsies to represent the mutational profile of tumor tissue from different cancer entities. Oncogene 2021, 40, 5204–5212. [Google Scholar] [CrossRef]
- Glennon, K.I.; Maralani, M.; Abdian, N.; Paccard, A.; Montermini, L.; Nam, A.J.; Arseneault, M.; Staffa, A.; Jandaghi, P.; Meehan, B.; et al. Rational Development of Liquid Biopsy Analysis in Renal Cell Carcinoma. Cancers 2021, 13, 5825. [Google Scholar] [CrossRef]
- Hata, T.; Mizuma, M.; Motoi, F.; Omori, Y.; Ishida, M.; Nakagawa, K.; Hayashi, H.; Morikawa, T.; Kamei, T.; Furukawa, T.; et al. GNAS mutation detection in circulating cell-free DNA is a specific predictor for intraductal papillary mucinous neoplasms of the pancreas, especially for intestinal subtype. Sci. Rep. 2020, 10, 17761. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraj Shaglouf, L.H.; Ranjpour, M.; Wajid, S.; Jain, S.K. Elevated expression of cellular SYNE1, MMP10, and GTPase1 and their regulatory role in hepatocellular carcinoma progression. Protoplasma 2020, 257, 157–167. [Google Scholar] [CrossRef]
- Abudureheman, A.; Ainiwaer, J.; Hou, Z.; Niyaz, M.; Turghun, A.; Hasim, A.; Zhang, H.; Lu, X.; Sheyhidin, I. High MLL2 expression predicts poor prognosis and promotes tumor progression by inducing EMT in esophageal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Ni, S.; Hu, J.; Duan, Y.; Shi, S.; Li, R.; Wu, H.; Qu, Y.; Li, Y. Down expression of LRP1B promotes cell migration via RhoA/Cdc42 pathway and actin cytoskeleton remodeling in renal cell cancer. Cancer Sci. 2013, 104, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Song, F.; Zheng, H.; Hu, L.; Lu, H.; Liu, P.; Hao, X.; Zhang, W.; Chen, K. Functional SNP in the microRNA-367 binding site in the 3’UTR of the calcium channel ryanodine receptor gene 3 (RYR3) affects breast cancer risk and calcification. Proc. Natl. Acad. Sci. USA 2011, 108, 13653–13658. [Google Scholar] [CrossRef] [Green Version]
- Wan, T.; Wang, H.; Gou, M.; Si, H.; Wang, Z.; Yan, H.; Liu, T.; Chen, S.; Fan, R.; Qian, N.; et al. LncRNA HEIH promotes cell proliferation, migration and invasion in cholangiocarcinoma by modulating miR-98-5p/HECTD4. Biomed. Pharmacother. 2020, 125, 109916. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Chen, J.; Wang, J.; Xu, J.; Liu, Y. TTN mutations predict a poor prognosis in patients with thyroid cancer. Biosci. Rep. 2022, 42, BSR20221168. [Google Scholar] [CrossRef] [PubMed]
- Ardeshir-Larijani, F.; Bhateja, P.; Lipka, M.B.; Sharma, N.; Fu, P.; Dowlati, A. KMT2D Mutation Is Associated with Poor Prognosis in Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2018, 19, e489–e501. [Google Scholar] [CrossRef]
- Rao, R.C.; Dou, Y. Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Yu, W.; Zhang, C.; Zhao, S.; Yu, Z.; Xiao, X.; Tang, R.; Xuan, Y.; Yang, W.; Hao, J.; et al. CREB-binding protein regulates lung cancer growth by targeting MAPK and CPSF4 signaling pathway. Mol. Oncol. 2016, 10, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaira, V.; Faversani, A.; Martin, N.M.; Garlick, D.S.; Ferrero, S.; Nosotti, M.; Kissil, J.L.; Bosari, S.; Altieri, D.C. Regulation of lung cancer metastasis by Klf4-Numb-like signaling. Cancer Res. 2013, 73, 2695–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.; Qi, Z.; Pang, Y.; Li, H.; Xie, H.; Wu, J.; Huang, Y.; Zhu, Y.; Shen, Y.; Zhu, Y.; et al. Retinoic Acid-Related Orphan Receptor C Regulates Proliferation, Glycolysis, and Chemoresistance via the PD-L1/ITGB6/STAT3 Signaling Axis in Bladder Cancer. Cancer Res. 2019, 79, 2604–2618. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, W.; Li, K.; Guo, Y.; Liu, D.; Yao, Z.; Lin, X.; Li, S.; Jiang, Z.; Liu, Q.; et al. The oncogenic roles of nuclear receptor coactivator 1 in human esophageal carcinoma. Cancer Med. 2018, 7, 5205–5216. [Google Scholar] [CrossRef]
- Kang, J.M.; Park, S.; Kim, S.J.; Hong, H.Y.; Jeong, J.; Kim, H.S.; Kim, S.J. CBL enhances breast tumor formation by inhibiting tumor suppressive activity of TGF-beta signaling. Oncogene 2012, 31, 5123–5131. [Google Scholar] [CrossRef] [Green Version]
- Baek, J.H.; Yun, H.S.; Kwon, G.T.; Kim, J.Y.; Lee, C.W.; Song, J.Y.; Um, H.D.; Kang, C.M.; Park, J.K.; Kim, J.S.; et al. PLOD3 promotes lung metastasis via regulation of STAT3. Cell Death Dis. 2018, 9, 1138. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Yu, S.; Wang, G.; Ma, Z.; Fu, X.; Cao, Y.; Li, Q.; Xu, Z. Elevation of EIF4G1 promotes non-small cell lung cancer progression by activating mTOR signalling. J. Cell. Mol. Med. 2021, 25, 2994–3005. [Google Scholar] [CrossRef]
- Glatzel-Plucinska, N.; Piotrowska, A.; Dziegiel, P.; Podhorska-Okolow, M. The Role of SATB1 in Tumour Progression and Metastasis. Int. J. Mol. Sci. 2019, 20, 4156. [Google Scholar] [CrossRef] [Green Version]
- Haesen, D.; Abbasi Asbagh, L.; Derua, R.; Hubert, A.; Schrauwen, S.; Hoorne, Y.; Amant, F.; Waelkens, E.; Sablina, A.; Janssens, V. Recurrent PPP2R1A Mutations in Uterine Cancer Act through a Dominant-Negative Mechanism to Promote Malignant Cell Growth. Cancer Res. 2016, 76, 5719–5731. [Google Scholar] [CrossRef] [Green Version]
- Okamura, S.; Yoshino, H.; Kuroshima, K.; Tsuruda, M.; Osako, Y.; Sakaguchi, T.; Yonemori, M.; Yamada, Y.; Tatarano, S.; Nakagawa, M.; et al. EHHADH contributes to cisplatin resistance through regulation by tumor-suppressive microRNAs in bladder cancer. BMC Cancer 2021, 21, 48. [Google Scholar] [CrossRef]
- Ayyagari, V.N.; Wang, X.; Diaz-Sylvester, P.L.; Groesch, K.; Brard, L. Assessment of acyl-CoA cholesterol acyltransferase (ACAT-1) role in ovarian cancer progression-An in vitro study. PLoS ONE 2020, 15, e0228024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Klevebring, D.; Neiman, M.; Sundling, S.; Eriksson, L.; Darai Ramqvist, E.; Celebioglu, F.; Czene, K.; Hall, P.; Egevad, L.; Gronberg, H.; et al. Evaluation of exome sequencing to estimate tumor burden in plasma. PLoS ONE 2014, 9, e104417. [Google Scholar] [CrossRef] [Green Version]
- Dietz, S.; Schirmer, U.; Merce, C.; von Bubnoff, N.; Dahl, E.; Meister, M.; Muley, T.; Thomas, M.; Sultmann, H. Low Input Whole-Exome Sequencing to Determine the Representation of the Tumor Exome in Circulating DNA of Non-Small Cell Lung Cancer Patients. PLoS ONE 2016, 11, e0161012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Trigg, R.M.; Martinson, L.J.; Parpart-Li, S.; Shaw, J.A. Factors that influence quality and yield of circulating-free DNA: A systematic review of the methodology literature. Heliyon 2018, 4, e00699. [Google Scholar] [CrossRef] [Green Version]
- Mauger, F.; Horgues, C.; Pierre-Jean, M.; Oussada, N.; Mesrob, L.; Deleuze, J.F. Comparison of commercially available whole-genome sequencing kits for variant detection in circulating cell-free DNA. Sci. Rep. 2020, 10, 6190. [Google Scholar] [CrossRef] [Green Version]
- Kunadirek, P.; Chuaypen, N.; Jenjaroenpun, P.; Wongsurawat, T.; Pinjaroen, N.; Sirichindakul, P.; Nookaew, I.; Tangkijvanich, P. Cell-Free DNA Analysis by Whole-Exome Sequencing for Hepatocellular Carcinoma: A Pilot Study in Thailand. Cancers 2021, 13, 2229. [Google Scholar] [CrossRef]
- Ma, X.; Shao, Y.; Tian, L.; Flasch, D.A.; Mulder, H.L.; Edmonson, M.N.; Liu, Y.; Chen, X.; Newman, S.; Nakitandwe, J.; et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biol. 2019, 20, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijg, J. Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev. 2014, 26, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Springer, S.; Mulvey, C.L.; Silliman, N.; Schaefer, J.; Sausen, M.; James, N.; Rettig, E.M.; Guo, T.; Pickering, C.R.; et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Sci. Transl. Med. 2015, 7, 293ra104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galot, R.; van Marcke, C.; Helaers, R.; Mendola, A.; Goebbels, R.M.; Caignet, X.; Ambroise, J.; Wittouck, K.; Vikkula, M.; Limaye, N.; et al. Liquid biopsy for mutational profiling of locoregional recurrent and/or metastatic head and neck squamous cell carcinoma. Oral Oncol. 2020, 104, 104631. [Google Scholar] [CrossRef]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018, 6, 271–281.e7. [Google Scholar] [CrossRef] [Green Version]
- Karimnezhad, A.; Palidwor, G.A.; Thavorn, K.; Stewart, D.J.; Campbell, P.A.; Lo, B.; Perkins, T.J. Accuracy and reproducibility of somatic point mutation calling in clinical-type targeted sequencing data. BMC Med. Genom. 2020, 13, 156. [Google Scholar] [CrossRef]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM Disrupts Cancer Progression. Front. Oncol. 2015, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- D’Aniello, C.; Patriarca, E.J.; Phang, J.M.; Minchiotti, G. Proline Metabolism in Tumor Growth and Metastatic Progression. Front. Oncol. 2020, 10, 776. [Google Scholar] [CrossRef]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Sivanand, S.; Vander Heiden, M.G. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell 2020, 37, 147–156. [Google Scholar] [CrossRef]
- Burgener, J.M.; Zou, J.; Zhao, Z.; Zheng, Y.; Shen, S.Y.; Huang, S.H.; Keshavarzi, S.; Xu, W.; Liu, F.-F.; Liu, G.; et al. Tumor-Naive Multimodal Profiling of Circulating Tumor DNA in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2021, 27, 4230–4244. [Google Scholar] [CrossRef]
- Wilson, H.L.; D’Agostino, R.B., Jr.; Meegalla, N.; Petro, R.; Commander, S.; Topaloglu, U.; Zhang, W.; Porosnicu, M. The Prognostic and Therapeutic Value of the Mutational Profile of Blood and Tumor Tissue in Head and Neck Squamous Cell Carcinoma. Oncologist 2021, 26, e279–e289. [Google Scholar] [CrossRef]
- Wu, P.; Xie, C.; Yang, L.; Liu, Y.; Zeng, J.; Li, X.; Fang, X.; Fan, Y.; Zhao, S.; Kuang, N.; et al. The genomic architectures of tumour-adjacent tissues, plasma and saliva reveal evolutionary underpinnings of relapse in head and neck squamous cell carcinoma. Br. J. Cancer 2021, 125, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Kim, H.-S.; Cho, E.S.; Han, D.; Park, J.A.; Park, J.Y.; Nam, W.; Kim, H.J.; Cha, I.-H.; Cha, Y.H. Longitudinal detection of somatic mutations in saliva and plasma for the surveillance of oral squamous cell carcinomas. PLoS ONE 2021, 16, e0256979. [Google Scholar] [CrossRef]
- Porter, A.; Natsuhara, M.; Daniels, G.A.; Patel, S.P.; Sacco, A.G.; Bykowski, J.; Banks, K.C.; Cohen, E.E.W. Next generation sequencing of cell free circulating tumor DNA in blood samples of recurrent and metastatic head and neck cancer patients. Transl. Cancer Res. 2020, 9, 203–209. [Google Scholar] [CrossRef]
- Angeles, A.K.; Janke, F.; Bauer, S.; Christopoulos, P.; Riediger, A.L.; Sultmann, H. Liquid Biopsies beyond Mutation Calling: Genomic and Epigenomic Features of Cell-Free DNA in Cancer. Cancers 2021, 13, 5615. [Google Scholar] [CrossRef]
- Chao, J.; Lee, J.; Kim, K.; Kang, S.Y.; Lee, T.; Kim, K.M.; Kim, S.T.; Klempner, S.J.; Lee, H. A Pilot Study of Baseline Spatial Genomic Heterogeneity in Primary Gastric Cancers Using Multi-Region Endoscopic Sampling. Front. Oncol. 2020, 10, 225. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, R.; Yuan, M.; Wang, D.; Fu, C.; Chen, R.; Lei, C.; Zhou, Q. One-stop molecular classification of endometrial carcinoma using comprehensive next-generation sequencing. Int. J. Cancer 2022, 151, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Avanzini, S.; Kurtz, D.M.; Chabon, J.J.; Moding, E.J.; Hori, S.S.; Gambhir, S.S.; Alizadeh, A.A.; Diehn, M.; Reiter, J.G. A mathematical model of ctDNA shedding predicts tumor detection size. Sci. Adv. 2020, 6, eabc4308. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.; Casadevall, D.; Bellosillo, B.; Pericay, C.; Garcia-Carbonero, R.; Losa, F.; Layos, L.; Alonso, V.; Capdevila, J.; Gallego, J.; et al. Clinical Impact of Presurgery Circulating Tumor DNA after Total Neoadjuvant Treatment in Locally Advanced Rectal Cancer: A Biomarker Study from the GEMCAD 1402 Trial. Clin. Cancer Res. 2021, 27, 2890–2898. [Google Scholar] [CrossRef]
- Bachet, J.B.; Bouche, O.; Taieb, J.; Dubreuil, O.; Garcia, M.L.; Meurisse, A.; Normand, C.; Gornet, J.M.; Artru, P.; Louafi, S.; et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: The AGEO RASANC prospective multicenter study. Ann. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodriguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Gundem, G.; Van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.C.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.L.; Hognas, G.; Annala, M.; et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015, 520, 353–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef] [Green Version]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Brandwein-Gensler, M.; Smith, R.V. Prognostic indicators in head and neck oncology including the new 7th edition of the AJCC staging system. Head Neck Pathol. 2010, 4, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Larson, D.E.; Harris, C.C.; Chen, K.; Koboldt, D.C.; Abbott, T.E.; Dooling, D.J.; Ley, T.J.; Mardis, E.R.; Wilson, R.K.; Ding, L. SomaticSniper: Identification of somatic point mutations in whole genome sequencing data. Bioinformatics 2012, 28, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Ewing, A.D.; Houlahan, K.E.; Hu, Y.; Ellrott, K.; Caloian, C.; Yamaguchi, T.N.; Bare, J.C.; P’ng, C.; Waggott, D.; Sabelnykova, V.Y.; et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat. Methods 2015, 12, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, J.; Nilsson, L.M.; Mitra, S.; Alsen, S.; Shelke, G.V.; Sah, V.R.; Forsberg, E.M.V.; Stierner, U.; All-Eriksson, C.; Einarsdottir, B.; et al. Molecular profiling of driver events in metastatic uveal melanoma. Nat. Commun. 2020, 11, 1894. [Google Scholar] [CrossRef] [Green Version]
- Steensma, D.P. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2018, 2, 3404–3410. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lin, L.H.; Yu, S.Y.; Kao, S.Y.; Chang, K.W.; Cheng, H.W.; Liu, C.J. FAT1 somatic mutations in head and neck carcinoma are associated with tumor progression and survival. Carcinogenesis 2018, 39, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, F.; Muinos, F.; Sentis, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, A.; Nouspikel, T.; Christinat, Y.; Ho, L.; McKee, T.; Addeo, A. Tissue-Plasma TMB Comparison and Plasma TMB Monitoring in Patients with Metastatic Non-small Cell Lung Cancer Receiving Immune Checkpoint Inhibitors. Front. Oncol. 2020, 10, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, I.; Rashid, M.; Tomas, I.M.; Williams, M.J.; Graham, T.A.; Adams, D.J.; Vigilante, A.; Watt, F.M. Genomic landscape and clonal architecture of mouse oral squamous cell carcinomas dictate tumour ecology. Nat. Commun. 2020, 11, 5671. [Google Scholar] [CrossRef]
- Vettore, A.L.; Ramnarayanan, K.; Poore, G.; Lim, K.; Ong, C.K.; Huang, K.K.; Leong, H.S.; Chong, F.T.; Lim, T.K.; Lim, W.K.; et al. Mutational landscapes of tongue carcinoma reveal recurrent mutations in genes of therapeutic and prognostic relevance. Genome Med. 2015, 7, 98. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | N | Mean of PMB ± SD | p-Value | |
|---|---|---|---|---|
| Age | ||||
| ≤60 | 23 | 16.41 ± 26.43 | 0.661 | |
| >60 | 27 | 20.19 ± 31.78 | ||
| Gender | ||||
| Male | 48 | 19.05 ± 29.67 | 0.766 | |
| Female | 2 | 4.13 ± 1.68 | ||
| T stage | ||||
| T1–3 | 13 | 20.68 ± 36.41 | 0.196 | |
| T4 | 37 | 17.67 ± 26.78 | ||
| N stage | ||||
| N0 | 28 | 18.03 ± 32.42 | 0.358 | |
| N+ | 22 | 19.0 ± 25.29 | ||
| Clinical stage | ||||
| I–III | 11 | 9.89 ± 27.75 | 0.019 | |
| IV | 39 | 20.87 ± 29.47 | ||
| Metastasis | ||||
| No | 41 | 14.23 ± 33.22 | 0.002 | |
| Yes | 9 | 37.7 ± 19.32 | ||
| Perineural invasion | ||||
| No | 33 | 20.79 ± 28.25 | 0.616 | |
| Yes | 17 | 13.92 ± 33.68 | ||
| Lymphovascular invasion | ||||
| No | 39 | 17.56 ± 4.52 | 0.991 | |
| Yes | 11 | 21.63 ± 10.15 | ||
| HPV status | ||||
| p16 negative | 46 | 19.82 ± 30.08 | 0.391 | |
| p16 positive | 4 | 2.67 ± 1.00 | ||
| Gene | Total Mutations | IGV PASS | IGV Non-PASS | ||
|---|---|---|---|---|---|
| Mutations (%) | Mutations (%) | ||||
| MUC19 | 57 | 29 | (50.9%) | 28 | (49.1%) |
| OBSCN | 78 | 48 | (61.5%) | 30 | (38.5%) |
| KMT2D | 62 | 40 | (64.5%) | 22 | (35.5%) |
| RYR1 | 51 | 39 | (76.5%) | 12 | (23.5%) |
| MUC16 | 111 | 86 | (77.5%) | 25 | (22.5%) |
| DNAH1 | 50 | 39 | (78.0%) | 11 | (22.0%) |
| PLEC | 51 | 40 | (78.4%) | 11 | (21.6%) |
| DNHD1 | 39 | 31 | (79.5%) | 8 | (20.5%) |
| MACF1 | 56 | 45 | (80.4%) | 11 | (19.6%) |
| USH2A | 37 | 30 | (81.1%) | 7 | (18.9%) |
| UBR4 | 57 | 47 | (82.5%) | 10 | (17.5%) |
| FAT4 | 35 | 29 | (82.9%) | 6 | (17.1%) |
| SYNE2 | 53 | 44 | (83.0%) | 9 | (17.0%) |
| LRP1 | 43 | 36 | (83.7%) | 7 | (16.3%) |
| LRP1B | 44 | 37 | (84.1%) | 7 | (15.9%) |
| HECTD4 | 47 | 40 | (85.1%) | 7 | (14.9%) |
| INTS1 | 30 | 26 | (86.7%) | 4 | (13.3%) |
| SYNE1 | 87 | 78 | (89.7%) | 9 | (10.3%) |
| RYR3 | 62 | 56 | (90.3%) | 6 | (9.7%) |
| TTN | 116 | 105 | (90.5%) | 11 | (9.5%) |
| DMD | 48 | 44 | (91.7%) | 4 | (8.3%) |
| ATM | 36 | 33 | (91.7%) | 3 | (8.3%) |
| DNAH11 | 39 | 36 | (92.3%) | 3 | (7.7%) |
| NBEAL1 | 26 | 24 | (92.3%) | 2 | (7.7%) |
| MYCBP2 | 40 | 37 | (92.5%) | 3 | (7.5%) |
| Clinical Stage | Metastasis | |||||
|---|---|---|---|---|---|---|
| Mutation Types | I–III | IV | p Value | No | Yes | p Value |
| Synonymous mutations | 234.7 ± 174.7 | 508.0 ± 115.4 | 0.089 | 350.1 ± 105.0 | 893.3 ± 219.6 | 0.003 |
| Missense mutations | 572.9 ± 449.7 | 1231.0 ± 279.6 | 0.013 | 832.4 ± 249.0 | 2242.4 ± 594.1 | 0.002 |
| Inserted stop mutations | 80.5 ± 71.6 | 130.7 ± 37.9 | 0.083 | 92.3 ± 36.2 | 244.2 ± 74.4 | 0.003 |
| Splicing site mutations | 10.0 ± 8.6 | 26.3 ± 6.0 | 0.006 | 17.5 ± 5.5 | 46.3 ± 10.6 | 0.001 |
| Stop loss mutations | 1.0 ± 0.8 | 2.0 ± 0.8 | 0.264 | 1.7 ± 0.7 | 2.3 ± 0.9 | 0.023 |
| Lost start mutations | 1.5 ± 1.5 | 2.1 ± 0.6 | 0.055 | 1.6 ± 0.6 | 3.6 ± 0.8 | 0.001 |
| Retained stop mutations | 0.0 ± 0.0 | 0.2 ± 0.1 | 0.170 | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.911 |
| In-frame mutations | 0.2 ± 0.1 | 0.5 ± 0.1 | 0.327 | 0.4 ± 0.1 | 0.8 ± 0.4 | 0.200 |
| Frameshift mutations | 0.0 ± 0.0 | 0.2 ± 0.1 | 0.170 | 0.1 ± 0.1 | 0.2 ± 0.1 | 0.324 |
| KEGG Pathway | FDR p-Value | Molecules |
|---|---|---|
| All Patients | ||
| ECM-receptor interaction | <0.001 | FRAS1, COL4A4, COL4A6, COL6A3, COL6A5, HSPG2, LAMA2, LAMA3, TNXB, VWF |
| Calcium signaling pathway | 0.002 | ATP2B3, CACNA1B, CACNA1D, CACNA1G, ERBB4, ITPR1, ITPR2, PHKB, RYR1, RYR2, RYR3 |
| Notch signaling pathway | 0.017 | CREBBP, NOTCH1, NOTCH2, NOTCH4, NCOR2 |
| Patients with metastatic OSCC | ||
| Choline metabolism in cancer | 0.018 | RAF1, SP1, WAS, DGKK, DGKQ, DGKZ, PIK3CB, SLC22A3, SLC44A1, SLC44A2 |
| O-glycan biosynthesis | 0.019 | RFNG, ST6GAL1, GALNT14, GALNT18, GALNT5, GALNT8 |
| Protein processing in endoplasmic reticulum | 0.044 | DNAJC10, NPLOC4, SEC23B, SEL1L, SEL1L2, STT3A, TRAF2, UGGT2, EIF2AK1, HSPA4L, P4HB, PDIA4, VCP |
| Expired patients | ||
| BCAA catabolism | 0.022 | HMGCS2, AUH, ACAT1, ACADSB, EHHADH |
| ECM-receptor interaction | 0.036 | AGRN, COL2A1, COL9A1, ITGA9, ITGB5, LAMC1 |
| Hypoxia-related | 0.042 | ARNT, EPAS1, PAK3, RAF1, TFE3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, L.-H.; Chang, K.-W.; Cheng, H.-W.; Liu, C.-J. Identification of Somatic Mutations in Plasma Cell-Free DNA from Patients with Metastatic Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 10408. https://doi.org/10.3390/ijms241210408
Lin L-H, Chang K-W, Cheng H-W, Liu C-J. Identification of Somatic Mutations in Plasma Cell-Free DNA from Patients with Metastatic Oral Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2023; 24(12):10408. https://doi.org/10.3390/ijms241210408
Chicago/Turabian StyleLin, Li-Han, Kuo-Wei Chang, Hui-Wen Cheng, and Chung-Ji Liu. 2023. "Identification of Somatic Mutations in Plasma Cell-Free DNA from Patients with Metastatic Oral Squamous Cell Carcinoma" International Journal of Molecular Sciences 24, no. 12: 10408. https://doi.org/10.3390/ijms241210408