
Genome-Wide Association Study of Lint Percentage in Gossypium hirsutum L. Races
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Large Variation in Lint Percentage in G. hirsutum Races
2.2. Genome-Wide Association Study for Lint Percentage
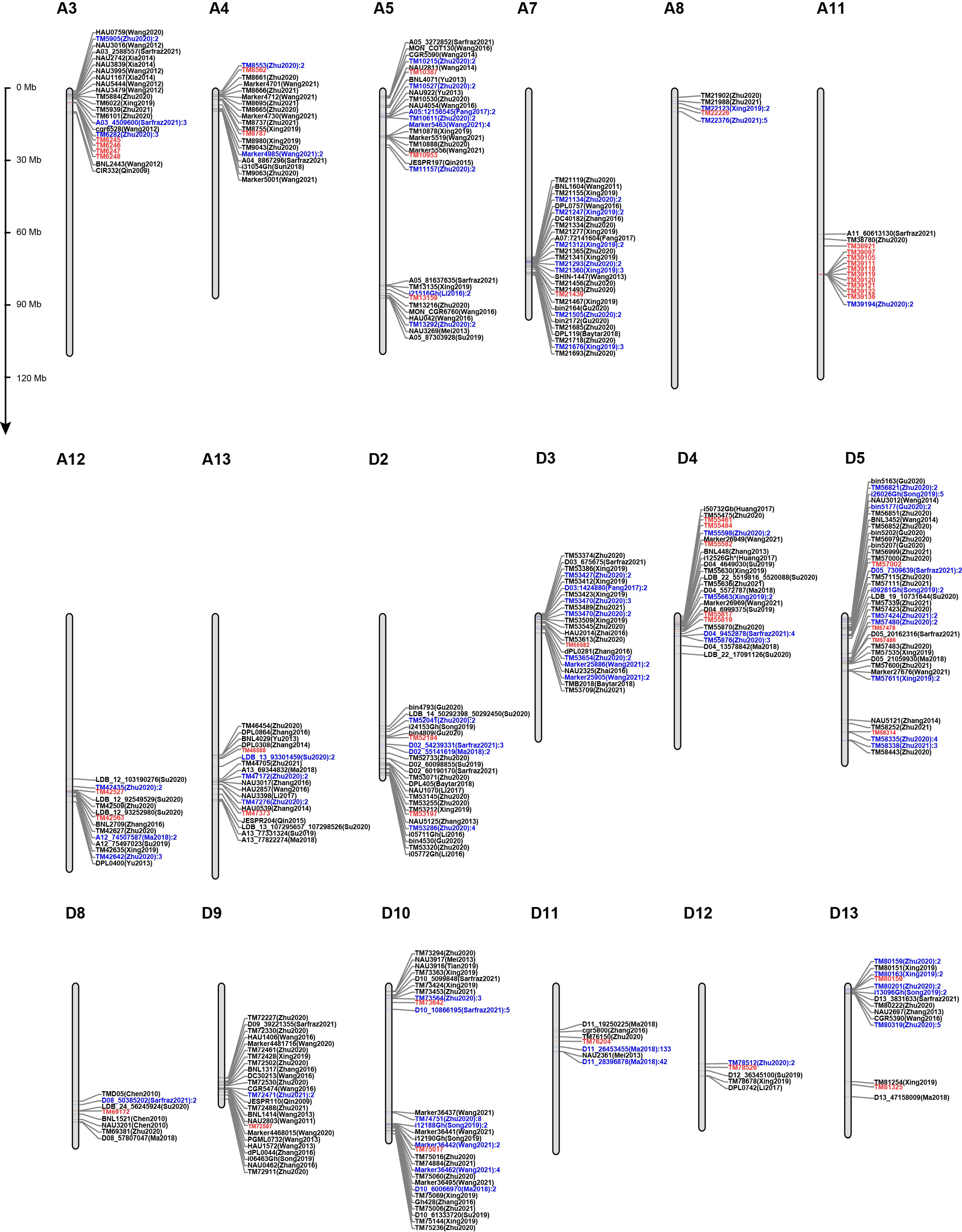
2.3. Validation of the Stability of 45 SNPs Associated with Lint Percentage
2.4. Discovery of Candidate Genes for Lint Percentage
2.5. Gh_A08G0526 and Gh_D12G0934 Were Candidate Genes for Lint Percentage
3. Materials and Methods
3.1. Natural Population Germplasm and Lint Percentage Evaluation
3.2. SNP Genotyping and Population Structure Assessment
3.3. Genome-Wide Association Study of Lint Percentage
3.4. Prediction and Identification of Related Candidate Gene
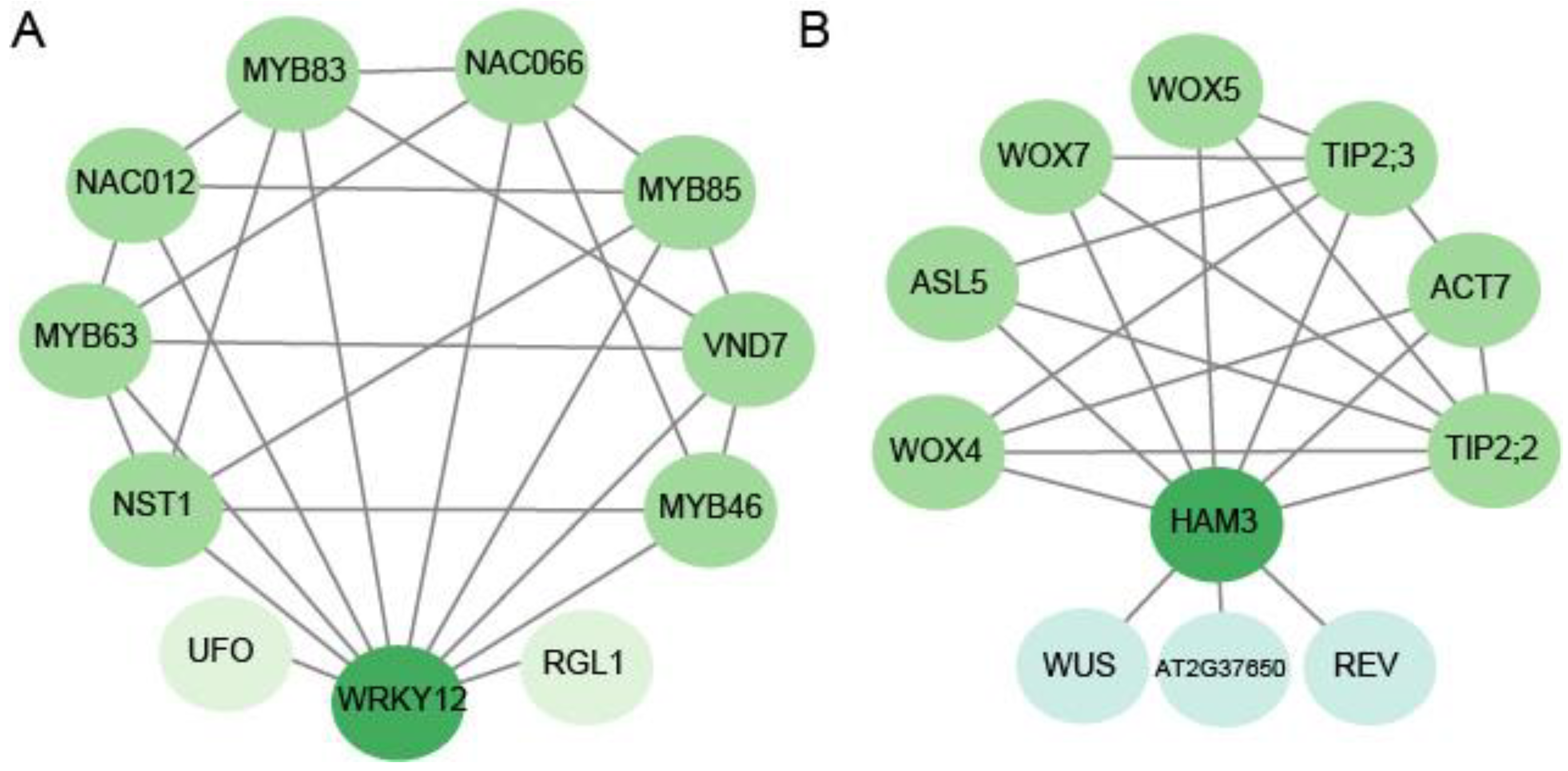
3.5. The Protein–Protein Interaction Analysis, the Cis-Elements, and the Predicted miRNA Targeting the Candidate Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baytar, A.A.; Peynircioğlu, C.; Sezener, V.; Frary, A.; Doğanlar, S. Molecular mapping of QTLs for fiber quality traits in Gossypium hirsutum multi-parent recombinant inbred lines. Euphytica 2021, 217, 181. [Google Scholar] [CrossRef]
- Mehboob-ur-Rahman; Shaheen, T.; Tabbasam, N.; Iqbal, M.A.; Ashraf, M.; Zafar, Y.; Paterson, A.H. Cotton genetic resources. A review. Agron. Sustain. Dev. 2012, 32, 419–432. [Google Scholar]
- Imran, M.; Shakeel, A.; Azhar, F.M.; Farooq, J.; Saleem, M.F.; Saeed, A.; Nazeer, W.; Riaz, M.; Naeem, M.; Javaid, A. Combining ability analysis for within-boll yield components in upland cotton (Gossypium hirsutum L.). Genet. Mol. Res. 2012, 11, 2790–2800. [Google Scholar] [CrossRef]
- Su, J.; Wang, C.; Ma, Q.; Zhang, A.; Shi, C.; Liu, J.; Zhang, X.; Yang, D.; Ma, X. An RTM-GWAS procedure reveals the QTL alleles and candidate genes for three yield-related traits in upland cotton. BMC Plant Biol. 2020, 20, 416. [Google Scholar] [CrossRef]
- Song, C.; Li, W.; Pei, X.; Liu, Y.; Ren, Z.; He, K.; Zhang, F.; Sun, K.; Zhou, X.; Ma, X.; et al. Dissection of the genetic variation and candidate genes of lint percentage by a genome-wide association study in upland cotton. Theor. Appl. Genet. 2019, 132, 1991–2002. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdurakhmonov, I.Y.; Buriev, Z.T.; Saha, S.; Pepper, A.E.; Musaev, J.A.; Almatov, A.; Shermatov, S.E.; Kushanov, F.N.; Mavlonov, G.T.; Reddy, U.K.; et al. Microsatellite markers associated with lint percentage trait in cotton, Gossypium hirsutum. Euphytica 2007, 156, 141–156. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Liu, Z.; Gu, Q.; Zhang, Y.; Li, Z.; Ke, H.; Yang, J.; Wu, J.; Wu, L.; et al. A genome-wide association study uncovers novel genomic regions and candidate genes of yield-related traits in upland cotton. Theor. Appl. Genet. 2018, 131, 2413–2425. [Google Scholar] [CrossRef]
- Endrizzi, J.E.; Turcotte, E.L.; Kohel, R.J. Genetics, Cytology, and evolution of Gossypium. In Advances in Genetics; Caspari, E.W., Scandalios, J.G., Eds.; Academic Press: Cambridge, MA, USA, 1985; Volume 23, pp. 271–375. [Google Scholar]
- Said, J.I.; Song, M.; Wang, H.; Lin, Z.; Zhang, X.; Fang, D.D.; Zhang, J. A comparative meta-analysis of QTL between intraspecific Gossypium hirsutum and interspecific G. hirsutum × G. barbadense populations. Mol. Genet. Genom. 2015, 290, 1003–1025. [Google Scholar] [CrossRef]
- Diouf, L.; Magwanga, R.O.; Gong, W.; He, S.; Pan, Z.; Jia, Y.H.; Kirungu, J.N.; Du, X. QTL Mapping of fiber quality and yield-related traits in an intra-specific Upland cotton using genotype by sequencing (GBS). Int. J. Mol. Sci. 2018, 19, 441. [Google Scholar] [CrossRef] [Green Version]
- Abdelraheem, A.; Fang, D.D.; Zhang, J.F. Quantitative trait locus mapping of drought and salt tolerance in an introgressed recombinant inbred line population of Upland cotton under the greenhouse and field conditions. Euphytica 2018, 214, 8. [Google Scholar] [CrossRef]
- Zhu, G.; Gao, W.; Song, X.; Sun, F.; Hou, S.; Liu, N.; Huang, Y.; Zhang, D.; Ni, Z.; Chen, Q.; et al. Genome-wide association reveals genetic variation of lint yield components under salty field conditions in cotton (Gossypium hirsutum L.). BMC Plant Biol. 2020, 20, 23. [Google Scholar] [CrossRef] [Green Version]
- Cavanagh, C.; Morell, M.; Mackay, I.; Powell, W. From mutations to MAGIC: Resources for gene discovery, validation and delivery in crop plants. Curr. Opin. Plant Biol. 2008, 11, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Huang, C.; You, C.; Li, W.; Zhao, W.; Shen, C.; Zhang, B.; Wang, H.; Yan, Z.; Dai, B.; et al. Genome-wide SSR-based association mapping for fiber quality in nation-wide upland cotton inbreed cultivars in China. BMC Genom. 2016, 17, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.; Chen, J.; Liu, B.; Zhang, Z.; Guan, X.; Chen, S.; Zhou, B.; et al. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef]
- Huang, C.; Nie, X.; Shen, C.; You, C.; Li, W.; Zhao, W.; Zhang, X.; Lin, Z. Population structure and genetic basis of the agronomic traits of upland cotton in China revealed by a genome-wide association study using high-density SNPs. Plant Biotechnol. J. 2017, 15, 1374–1386. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Liu, Z.; Gu, Q.; Zhang, Y.; Li, Z.; Ke, H.; Yang, J.; Wu, J.; Wu, L.; et al. Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol. J. 2017, 15, 982–996. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef]
- Sarfraz, Z.; Iqbal, M.S.; Geng, X.; Iqbal, M.S.; Nazir, M.F.; Ahmed, H.; He, S.; Jia, Y.; Pan, Z.; Sun, G.; et al. GWAS mediated elucidation of heterosis for metric traits in cotton (Gossypium hirsutum L.) across multiple environments. Front. Plant Sci. 2021, 12, 565552. [Google Scholar] [CrossRef]
- Tyagi, P.; Gore, M.A.; Bowman, D.T.; Campbell, B.T.; Udall, J.A.; Kuraparthy, V. Genetic diversity and population structure in the US Upland cotton (Gossypium hirsutum L.). Theor. Appl. Genet. 2014, 127, 283–295. [Google Scholar] [CrossRef]
- Feng, L.; Zhang, S.; Xing, L.; Yang, B.; Gao, X.; Xie, X.; Zhou, B. QTL analysis for yield and fibre quality traits using three sets of introgression lines developed from three Gossypium hirsutum race stocks. Mol. Genet. Genom. 2019, 294, 789–810. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, T.; Liu, Q.; Gao, X.; Zhu, X.; Zhang, T.; Zhou, B. Quantitative trait locus analysis of boll-related traits in an intraspecific population of Gossypium hirsutum. Euphytica 2015, 203, 121–144. [Google Scholar] [CrossRef]
- Liu, D.; Liu, F.; Shan, X.; Zhang, J.; Tang, S.; Fang, X.; Liu, X.; Wang, W.; Tan, Z.; Teng, Z.; et al. Construction of a high-density genetic map and lint percentage and cottonseed nutrient trait QTL identification in upland cotton (Gossypium hirsutum L.). Mol. Genet. Genom. 2015, 290, 1683–1700. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, X.; Su, Y.; Zhang, X.; Guo, K.; Teng, Z.; Zhang, J.; Liu, D.; Zhang, Z. Genetic mapping and identification of Lgf loci controlling green fuzz in Upland cotton (Gossypium hirsutum L.). Crop J. 2021, 9, 777–784. [Google Scholar] [CrossRef]
- Zhang, S.; Feng, L.; Xing, L.; Yang, B.; Gao, X.; Zhu, X.; Zhang, T.; Zhou, B. New QTLs for lint percentage and boll weight mined in introgression lines from two feral landraces into Gossypium hirsutum acc TM-1. Plant Breed. 2016, 135, 90–101. [Google Scholar] [CrossRef]
- Feng, L.; Zhou, C.; Su, Q.; Xu, M.; Yue, H.; Zhang, S.; Zhou, B. Fine-mapping and candidate gene analysis of qFS-Chr. D02, a QTL for fibre strength introgressed from a semi-wild cotton into Gossypium hirsutum. Plant Sci. 2020, 297, 110524. [Google Scholar] [CrossRef]
- Liu, X.; Yang, L.; Wang, J.; Wang, Y.; Guo, Z.; Li, Q.; Yang, J.; Wu, Y.; Chen, L.; Teng, Z.; et al. Analyzing quantitative trait Loci for fiber quality and yield-related traits from a recombinant inbred line population with Gossypium hirsutum race palmeri as one parent. Front. Plant Sci. 2021, 12, 817748. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhou, Z.L.; Wang, X.X.; Cai, X.Y.; Li, X.N.; Wang, C.Y.; Wang, Y.H.; Fang, L.; Wang, K.B. Genome-wide association mapping of glyphosate-resistance in Gossypium hirsutum races. Euphytica 2016, 209, 209–221. [Google Scholar] [CrossRef]
- Guo, X.; Wang, Y.; Hou, Y.; Zhou, Z.; Sun, R.; Qin, T.; Wang, K.; Liu, F.; Wang, Y.; Huang, Z.; et al. Genome-wide dissection of the genetic basis for drought tolerance in Gossypium hirsutum L. races. Front. Plant Sci. 2022, 13, 876095. [Google Scholar] [CrossRef]
- Xu, Y.; Magwanga, R.O.; Yang, X.; Jin, D.; Cai, X.; Hou, Y.; Wei, Y.; Zhou, Z.; Wang, K.; Liu, F. Genetic regulatory networks for salt-alkali stress in Gossypium hirsutum with differing morphological characteristics. BMC Genom. 2020, 21, 15. [Google Scholar] [CrossRef] [Green Version]
- Xing, H.; Yuan, Y.; Zhang, H.; Wang, L.; Mao, L.; Tao, J.; Wang, X.; Feng, W.; Wang, H.; Wang, Q.; et al. Multi-environments and multi-models association mapping identified candidate genes of lint percentage and seed index in Gossypium hirsutum L. Mol. Breed. 2019, 39, 149. [Google Scholar] [CrossRef]
- Qin, H.; Chen, M.; Yi, X.; Bie, S.; Zhang, C.; Zhang, Y.; Lan, J.; Meng, Y.; Yuan, Y.; Jiao, C. Identification of associated SSR markers for yield component and fiber quality traits based on frame map and Upland cotton collections. PLoS ONE 2015, 10, e0118073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, H.; Ge, Q.; Shang, H.; Yuan, Y. Inheritance, QTLs, and candidate genes of lint percentage in Upland cotton. Front. Genet. 2022, 13, 855574. [Google Scholar] [CrossRef] [PubMed]
- Said, J.I.; Lin, Z.; Zhang, X.; Song, M.; Zhang, J. A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom. 2013, 14, 776. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, M.D.; Chen-Plotkin, A.S. The post-GWAS era: From association to function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, T.; Guo, W. The im mutant gene negatively affects many aspects of fiber quality traits and lint percentage in cotton. Crop Sci. 2013, 53, 27–37. [Google Scholar] [CrossRef]
- Yang, P.; Sun, X.; Liu, X.; Wang, W.; Hao, Y.; Chen, L.; Liu, J.; He, H.; Zhang, T.; Bao, W.; et al. Identification of candidate genes for lint percentage and fiber quality through QTL mapping and transcriptome analysis in an allotetraploid interspecific cotton CSSLs population. Front. Plant Sci. 2022, 13, 882051. [Google Scholar] [CrossRef]
- Wang, H.; Jia, X.; Kang, M.; Li, W.; Fu, X.; Ma, L.; Lu, J.; Wei, H.; Yu, S. QTL mapping and candidate gene identification of lint percentage based on a recombinant inbred line population of upland cotton. Euphytica 2021, 217, 102. [Google Scholar] [CrossRef]
- Su, J.; Fan, S.; Li, L.; Wei, H.; Wang, C.; Wang, H.; Song, M.; Zhang, C.; Gu, L.; Zhao, S.; et al. Detection of favorable QTL alleles and candidate genes for lint percentage by GWAS in Chinese Upland cotton. Front. Plant Sci. 2016, 7, 1576. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Wang, C.; Hao, F.; Ma, Q.; Wang, J.; Li, J.; Ning, X. Genetic detection of lint percentage applying single-locus and multi-locus genome-wide association studies in Chinese early-maturity Upland cotton. Front. Plant Sci. 2019, 10, 964. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Andres, R.J.; Zhang, K.; Kuraparthy, V. High-density linkage map construction and QTL analysis of fiber quality and lint percentage in tetraploid cotton. Crop Sci. 2021, 61, 3340–3360. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, Q.; Chen, F.; Wang, M.; Dixon, R.A. NAC domain function and transcriptional control of a secondary cell wall master switch. Plant J. 2011, 68, 1104–1114. [Google Scholar] [CrossRef]
- Sun, W.; Gao, Z.; Wang, J.; Huang, Y.; Chen, Y.; Li, J.; Lv, M.; Wang, J.; Luo, M.; Zuo, K. Cotton fiber elongation requires the transcription factor GhMYB212 to regulate sucrose transportation into expanding fibers. New Phytol. 2019, 222, 864–881. [Google Scholar] [CrossRef]
- Walford, S.A.; Wu, Y.; Llewellyn, D.J.; Dennis, E.S. GhMYB25-like: A key factor in early cotton fibre development. Plant J. 2011, 65, 785–797. [Google Scholar] [CrossRef]
- Machado, A.; Wu, Y.; Yang, Y.; Llewellyn, D.J.; Dennis, E.S. The MYB transcription factor GhMYB25 regulates early fibre and trichome development. Plant J. 2009, 59, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, F.; Guo, Y.; Gan, X.; Yang, M.; Zeng, W.; Persson, S.; Li, J.; Xu, W. GhMYB7 promotes secondary wall cellulose deposition in cotton fibres by regulating GhCesA gene expression through three distinct cis-elements. New Phytol. 2021, 232, 1718–1737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Luo, F.; Zhong, Y.; He, J.; Li, L. Modulation of NAC transcription factor NST1 activity by XYLEM NAC DOMAIN1 regulates secondary cell wall formation in Arabidopsis. J. Exp. Bot. 2020, 71, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, G.Q.; Zou, D.; Yan, J.Q.; Li, Y.; Hu, S.; Li, X.B. The cotton (Gossypium hirsutum) NAC transcription factor (FSN1) as a positive regulator participates in controlling secondary cell wall biosynthesis and modification of fibers. New Phytol. 2018, 217, 625–640. [Google Scholar] [CrossRef] [Green Version]
- Zhong, R.; Lee, C.; Ye, Z.H. Global analysis of direct targets of secondary wall NAC master switches in Arabidopsis. Mol. Plant 2010, 3, 1087–1103. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Mitsuda, N.; Ohtani, M.; Ohme-Takagi, M.; Kato, K.; Demura, T. VASCULAR-RELATED NAC-DOMAIN7 directly regulates the expression of a broad range of genes for xylem vessel formation. Plant J. 2011, 66, 579–590. [Google Scholar] [CrossRef]
- Ohashi-Ito, K.; Iwamoto, K.; Fukuda, H. LOB DOMAIN-CONTAINING PROTEIN 15 positively regulates expression of VND7, a master regulator of tracheary elements. Plant Cell Physiol. 2018, 59, 989–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, R.L.; Zhong, R.; Ye, Z.H. MYB83 is a direct target of SND1 and acts redundantly with MYB46 in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell Physiol. 2009, 50, 1950–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.N.; Li, Y.; Chen, Y.H.; Lu, R.; Zhou, L.; Wang, Y.; Zheng, Y.; Li, X.B. Phosphorylation of WRKY16 by MPK3-1 is essential for its transcriptional activity during fiber initiation and elongation in cotton (Gossypium hirsutum). Plant Cell 2021, 33, 2736–2752. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, M.K.; McKinney, E.C.; Meagher, R.B. A single vegetative actin isovariant overexpressed under the control of multiple regulatory sequences is sufficient for normal Arabidopsis development. Plant Cell 2009, 21, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Gilliland, L.U.; Pawloski, L.C.; Kandasamy, M.K.; Meagher, R.B. Arabidopsis actin gene ACT7 plays an essential role in germination and root growth. Plant J. 2003, 33, 319–328. [Google Scholar] [CrossRef]
- Ji, J.; Strable, J.; Shimizu, R.; Koenig, D.; Sinha, N.; Scanlon, M.J. WOX4 promotes procambial development. Plant Physiol. 2010, 152, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Xu, L. Transcription factors WOX11/12 directly activate WOX5/7 to promote root primordia initiation and organogenesis. Plant Physiol. 2016, 172, 2363–2373. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Feng, Z.; Meng, L. Ectopic expression of the Arabidopsis ASYMMETRIC LEAVES2-LIKE5 (ASL5) gene in cockscomb (Celosia cristata) generates vascular-pattern modifications in lateral organs. Plant Cell Tissue Organ Cult. 2012, 110, 163–169. [Google Scholar] [CrossRef]
- Engstrom, E.M.; Andersen, C.M.; Gumulak-Smith, J.; Hu, J.; Orlova, E.; Sozzani, R.; Bowman, J.L. Arabidopsis homologs of the petunia hairy meristem gene are required for maintenance of shoot and root indeterminacy. Plant Physiol. 2011, 155, 735–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.; Li, C.; Zhang, J.; Li, F.; Ma, L.; Tan, Y.; Wang, Q.; Zhang, B. Differential expression of microRNAs during fiber development between fuzzless-lintless mutant and its wild-type allotetraploid cotton. Sci. Rep. 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Tu, L.; Tang, W.; Gao, W.; Lindsey, K.; Zhang, X. Small RNA and degradome profiling reveals a role for miRNAs and their targets in the developing fibers of Gossypium barbadense. Plant J. 2014, 80, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Spring: New York, NY, USA, 2018. [Google Scholar]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Artico, S.; Nardeli, S.M.; Brilhante, O.; Grossi-de-Sa, M.F.; Alves-Ferreira, M. Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC Plant Biol. 2010, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Saeed, A.I.; Bhagabati, N.K.; Braisted, J.C.; Liang, W.; Sharov, V.; Howe, E.A.; Li, J.; Thiagarajan, M.; White, J.A.; Quackenbush, J. TM4 microarray software suite. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2006; Volume 411, pp. 134–193. [Google Scholar]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Dehais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouze, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, J.; Li, D.; Zhang, Z.; Liu, F.; Zhou, X.; Wang, T.; Ling, Y.; Su, Z. PMRD: Plant microRNA database. Nucleic Acids Res. 2010, 38, D806–D813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Env. | Max | Min | Mean | Std. | CV | Skewness | Kurtosis |
|---|---|---|---|---|---|---|---|
| E1 | 47.55 | 12.03 | 30.31 | 5.86 | 19.34 | 0.14 | −0.15 |
| E2 | 54.77 | 8.29 | 28 | 6.55 | 23.39 | 0.43 | 0.85 |
| E3 | 43.8 | 14.47 | 28.04 | 6.21 | 22.14 | 0.48 | −0.24 |
| E4 | 43.86 | 12.35 | 26.61 | 6.21 | 23.34 | 0.39 | −0.39 |
| E5 | 42.43 | 14.87 | 26.37 | 5.76 | 21.83 | 0.51 | −0.03 |
| SNP | Chr | Pos. (bp) | GAPIT | Tassel | ||||
|---|---|---|---|---|---|---|---|---|
| FaST-LMM | FarmCPU | BLINK | MLMM | MLM | TMLM | |||
| TM6245 | A3 | 6,000,457 | BLUP (4.90) | BLUP (4.90) | ||||
| TM6246 | A3 | 6,008,064 | BLUP (5.10) | BLUP (6.99) | BLUP (5.10) | |||
| TM6247 | A3 | 6,015,363 | BLUP (5.10) | BLUP (5.10) | ||||
| TM6248 | A3 | 6,020,227 | BLUP (4.90) | BLUP (4.90) | ||||
| TM8562 | A4 | 1,704,160 | E5 (6.37) | E5 (6.23) | ||||
| TM8787 | A4 | 5,125,340 | E1 (7.94) | E1 (10.43) | ||||
| TM10387 | A5 | 7,465,603 | E5 (4.71) | E5 (4.71) | ||||
| TM10953 | A5 | 21,122,062 | E3 (4.97) | E3 (9.45) | ||||
| TM13159 | A5 | 83,646,730 | E3 (6.75); E4 (6.30); E5 (6.66) | E3 (17.39); E4 (6.90); E5 (9.84) | ||||
| TM21439 | A7 | 73,820,001 | E2 (5.42) | E2 (5.42) | ||||
| TM22226 | A8 | 8,180,911 | E4 (15.51) | E4 (11.19); E5 (6.70) | ||||
| TM38921 | A11 | 65,498,934 | E2 (6.45) | E2 (7.92) | ||||
| TM39097 | A11 | 77,128,215 | E2 (5.41) | E2 (5.01) | ||||
| TM39105 | A11 | 77,162,385 | E2 (4.94) | E2 (5.23) | ||||
| TM39111 | A11 | 77,201,721 | E2 (4.85) | E2 (5.06) | ||||
| TM39118 | A11 | 77,245,152 | E2 (5.41) | E2 (4.73) | ||||
| TM39119 | A11 | 77,250,629 | E2 (6.57) | E2 (7.05) | E2 (4.99) | E2 (6.57) | E2 (5.48) | E2 (4.80) |
| TM39120 | A11 | 77,254,990 | E2 (5.35) | E2 (5.42) | ||||
| TM39121 | A11 | 77,259,287 | E2 (5.39) | E2 (4.72) | ||||
| TM39122 | A11 | 77,267,198 | E2 (5.33) | E2 (5.42) | ||||
| TM39136 | A11 | 77,356,582 | E3 (9.26); E5 (12.70) | E1 (5.59); E2 (4.84); E3 (14.49); E4 (5.33); E5 (13.96) | ||||
| TM42527 | A12 | 73,222,779 | E1 (5.91) | E1 (13.91) | ||||
| TM42563 | A12 | 73,856,986 | E2 (5.34) | E2 (7.14) | ||||
| TM46588 | A13 | 60,134,206 | E1 (6.86) | E1 (14.22) | ||||
| TM47373 | A13 | 72,835,782 | E3 (8.73) | E3 (7.06) | ||||
| TM52184 | D2 | 51,543,776 | E1 (4.89); E4 (10.43) | E4 (6.60) | ||||
| TM53197 | D2 | 64,206,361 | E1 (4.77) | E3 (6.30) | ||||
| TM53588 | D3 | 4,016,585 | E4 (15.33) | E1 (13.39); E4 (14.39); E5 (10.63) | ||||
| TM55461 | D4 | 2,087,134 | E2 (6.19) | E2 (9.29) | E2 (6.19) | E2 (5.23) | ||
| TM55484 | D4 | 2,902,882 | E5 (9.59) | E5 (13.00) | ||||
| TM55582 | D4 | 3,824,554 | E2 (6.80) | E2 (6.86) | ||||
| TM55817 | D4 | 7,622,861 | E2 (5.35) | E2 (5.35) | ||||
| TM55819 | D4 | 7,631,258 | E2 (5.35) | E2 (5.35) | ||||
| TM57002 | D5 | 6,810,289 | E1 (4.85); E3 (5.82) | |||||
| TM57478 | D5 | 19,820,229 | E4 (4.95) | E4 (22.33); E5 (4.79) | E4 (12.47) | E4 (4.95) | E4 (4.76) | |
| TM57486 | D5 | 20,068,395 | E2 (5.45) | E3 (4.87) | ||||
| TM58314 | D5 | 49,232,321 | E1 (4.90); E3 (4.77) | |||||
| TM69172 | D8 | 53,049,470 | E3 (13.74) | E3 (8.17) | ||||
| TM72587 | D9 | 43,752,600 | E3 (7.45) | E3 (7.83) | ||||
| TM73642 | D10 | 8,030,303 | E3 (5.42) | E3 (6.93) | ||||
| TM75017 | D10 | 58,300,092 | E4 (4.72) | E4 (13.12) | ||||
| TM76204 | D11 | 24,111,532 | E3 (8.00) | E3 (14.52) | ||||
| TM78526 | D12 | 34,893,826 | E2 (4.84) | E3 (5.16) | E2 (4.84) | |||
| TM80159 | D13 | 2,590,468 | E5 (5.39) | E5 (9.39) | ||||
| TM81325 | D13 | 42,144,468 | E1 (7.99); E2 (9.96) | E1 (6.54) | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Guo, X.; Cai, X.; Xu, Y.; Sun, R.; Umer, M.J.; Wang, K.; Qin, T.; Hou, Y.; Wang, Y.; et al. Genome-Wide Association Study of Lint Percentage in Gossypium hirsutum L. Races. Int. J. Mol. Sci. 2023, 24, 10404. https://doi.org/10.3390/ijms241210404
Wang Y, Guo X, Cai X, Xu Y, Sun R, Umer MJ, Wang K, Qin T, Hou Y, Wang Y, et al. Genome-Wide Association Study of Lint Percentage in Gossypium hirsutum L. Races. International Journal of Molecular Sciences. 2023; 24(12):10404. https://doi.org/10.3390/ijms241210404
Chicago/Turabian StyleWang, Yuanyuan, Xinlei Guo, Xiaoyan Cai, Yanchao Xu, Runrun Sun, Muhammad Jawad Umer, Kunbo Wang, Tengfei Qin, Yuqing Hou, Yuhong Wang, and et al. 2023. "Genome-Wide Association Study of Lint Percentage in Gossypium hirsutum L. Races" International Journal of Molecular Sciences 24, no. 12: 10404. https://doi.org/10.3390/ijms241210404