
Acute Exacerbations of Interstitial Lung Diseases: Focus on Biomarkers

Abstract
:1. Introduction
- -
- A previous or concurrent diagnosis of IPF.
- -
- Deterioration within 30 days.
- -
- New bilateral ground glass opacities and/or consolidation on a background of reticular or honeycomb pattern consistent with usual interstitial pneumonia (UIP).
- -
- No evidence of pulmonary infection by endotracheal aspiration or bronchoalveolar lavage.
- -
- Exclusion of alternative causes, including left heart failure, pulmonary embolism, or an identifiable cause of acute lung injury.
- -
- Previous or concurrent diagnosis of IPF.
- -
- Acute worsening or development of dyspnea, typically less than one-month duration.
- -
- Computed tomography with new bilateral ground-glass opacity and/or consolidation superimposed on a background pattern consistent with the usual interstitial pneumonia pattern.
- -
- Deterioration not fully explained by cardiac failure or fluid overload.
2. Biomarkers for Acute Exacerbations of IPF
2.1. Biomarkers Associated with Alveolar Epithelial Cell Dysfunction
2.2. Biomarkers Associated with Extracellular Matrix Remodeling and Fibroproliferation
2.3. Biomarkers Associated with Immune Dysfunction
2.4. Inflammatory and Anti-Inflammatory Cytokines
2.5. Microbiome
2.6. Non-Coding RNAs
2.7. Mitochondrial DNA
2.8. Coagulation Factors
2.9. Blood Cell Count Derived Inflammation Indexes
2.9.1. Neutrophil-to-Lymphocyte Ratio (NLR)
2.9.2. Platelet-to-Lymphocyte Ratio (PLR)
2.9.3. The Monocyte to Lymphocyte Ratio (MLR)
2.10. Multiple Biomarker Signatures
3. Biomarkers for Acute Exacerbations of NSIP (AE-NSIP)
4. Biomarkers for Acute Exacerbations of Connective Tissue Diseases Interstitial Lung Disease (CTD-ILD)
4.1. Rheumatoid Arthritis
4.2. Systemic Sclerosis
5. Biomarkers for Acute Exacerbations of Hypersensitivity Pneumonitis
6. Biomarkers Associated with AE-ILDs
6.1. KL-6
6.2. Neutrophil-to-Lymphocyte Ratio (NLR)
6.3. D-Dimers
6.4. Interleukin-6 (IL-6)
6.5. Heparin-Binding Protein (HBP)
6.6. BALF Populations
6.7. Proteomic Biomarkers
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wijsenbeek, M.; Cottin, V. Spectrum of Fibrotic Lung Diseases. Reply. N. Engl. J. Med. 2020, 383, 2485–2486. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Moore, B.B.; Flaherty, K.R.; Brown, K.K.; Kaner, R.J.; King, T.E., Jr.; Lasky, J.A.; Loyd, J.E.; Noth, I.; Olman, M.A.; et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Song, J.W.; Hong, S.B.; Lim, C.M.; Koh, Y.; Kim, D.S. Acute exacerbation of idiopathic pulmonary fibrosis: Incidence, risk factors and outcome. Eur. Respir. J. 2011, 37, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Mura, M.; Porretta, M.A.; Bargagli, E.; Sergiacomi, G.; Zompatori, M.; Sverzellati, N.; Taglieri, A.; Mezzasalma, F.; Rottoli, P.; Saltini, C.; et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: A 3-year prospective study. Eur. Respir. J. 2012, 40, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Richeldi, L.; Kim, D.S.; Taniguchi, H.; Tschoepe, I.; Luisetti, M.; Roman, J.; Tino, G.; Schlenker-Herceg, R.; Hallmann, C.; et al. Acute exacerbations in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1601339. [Google Scholar] [CrossRef] [Green Version]
- Moua, T.; Westerly, B.D.; Dulohery, M.M.; Daniels, C.E.; Ryu, J.H.; Lim, K.G. Patients with Fibrotic Interstitial Lung Disease Hospitalized for Acute Respiratory Worsening: A Large Cohort Analysis. Chest 2016, 149, 1205–1214. [Google Scholar] [CrossRef]
- Charokopos, A.; Moua, T.; Ryu, J.H.; Smischney, N.J. Acute exacerbation of interstitial lung disease in the intensive care unit. World J. Crit. Care Med. 2022, 11, 22–32. [Google Scholar] [CrossRef]
- Drakopanagiotakis, F.; Wujak, L.; Wygrecka, M.; Markart, P. Biomarkers in idiopathic pulmonary fibrosis. Matrix Biol. 2018, 68–69, 404–421. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Karampitsakos, T.; Bouros, E.; Tzilas, V.; Liossis, S.N.; Bouros, D. Autoimmune Biomarkers, Antibodies, and Immunologic Evaluation of the Patient with Fibrotic Lung Disease. Clin. Chest Med. 2019, 40, 679–691. [Google Scholar] [CrossRef]
- Zhang, T.; Shen, P.; Duan, C.; Gao, L. KL-6 as an Immunological Biomarker Predicts the Severity, Progression, Acute Exacerbation, and Poor Outcomes of Interstitial Lung Disease: A Systematic Review and Meta-Analysis. Front. Immunol. 2021, 12, 745233. [Google Scholar] [CrossRef]
- Ohshimo, S.; Ishikawa, N.; Horimasu, Y.; Hattori, N.; Hirohashi, N.; Tanigawa, K.; Kohno, N.; Bonella, F.; Guzman, J.; Costabel, U. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir. Med. 2014, 108, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.G.; Choi, S.M.; Lee, J.H.; Yoon, J.K.; Song, J.W. Changes in blood Krebs von den Lungen-6 predict the mortality of patients with acute exacerbation of interstitial lung disease. Sci. Rep. 2022, 12, 4916. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Nagata, N.; Kumazoe, H.; Oda, K.; Ishimoto, H.; Yoshimi, M.; Takata, S.; Hamada, M.; Koreeda, Y.; Takakura, K.; et al. Prognostic value of serial serum KL-6 measurements in patients with idiopathic pulmonary fibrosis. Respir. Investig. 2017, 55, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Kuo, C.W.; Chen, C.W.; Tseng, Y.L.; Wu, C.L.; Lin, S.H. Baseline plasma KL-6 level predicts adverse outcomes in patients with idiopathic pulmonary fibrosis receiving nintedanib: A retrospective real-world cohort study. BMC Pulm. Med. 2021, 21, 165. [Google Scholar] [CrossRef]
- Choi, M.G.; Choi, S.M.; Lee, J.H.; Kim, J.Y.; Song, J.W. Blood Krebs von den Lungen-6 levels predict treatment response to antifibrotic therapy in patients with idiopathic pulmonary fibrosis. Respir. Res. 2022, 23, 334. [Google Scholar] [CrossRef]
- Aloisio, E.; Braga, F.; Puricelli, C.; Panteghini, M. Prognostic role of Krebs von den Lungen-6 (KL-6) measurement in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Clin. Chem. Lab. Med. 2021, 59, 1400–1408. [Google Scholar] [CrossRef]
- Polke, M.; Kondoh, Y.; Wijsenbeek, M.; Cottin, V.; Walsh, S.L.F.; Collard, H.R.; Chaudhuri, N.; Avdeev, S.; Behr, J.; Calligaro, G.; et al. Management of Acute Exacerbation of Idiopathic Pulmonary Fibrosis in Specialised and Non-specialised ILD Centres Around the World. Front. Med. 2021, 8, 699644. [Google Scholar] [CrossRef]
- Collard, H.R.; Calfee, C.S.; Wolters, P.J.; Song, J.W.; Hong, S.B.; Brady, S.; Ishizaka, A.; Jones, K.D.; King, T.E., Jr.; Matthay, M.A.; et al. Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L3–L7. [Google Scholar] [CrossRef] [Green Version]
- White, E.S.; Xia, M.; Murray, S.; Dyal, R.; Flaherty, C.M.; Flaherty, K.R.; Moore, B.B.; Cheng, L.; Doyle, T.J.; Villalba, J.; et al. Plasma Surfactant Protein-D, Matrix Metalloproteinase-7, and Osteopontin Index Distinguishes Idiopathic Pulmonary Fibrosis from Other Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2016, 194, 1242–1251. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Ju, Q.; Cao, J.; Tang, W.; Zhang, J. Impact of serum SP-A and SP-D levels on comparison and prognosis of idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Medicine 2017, 96, e7083. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Akira, M.; Sugimoto, C.; Tachibana, K.; Inoue, Y.; Shintani, S.; Okuma, T.; Kasai, T.; Hayashi, S.; Inoue, Y. Seroradiologic prognostic evaluation of acute exacerbation in patients with idiopathic interstitial pneumonia: A retrospective observational study. J. Thorac. Dis. 2020, 12, 4132–4147. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.E.; Armanios, M. Short telomeres: A repeat offender in IPF. Lancet Respir. Med. 2014, 2, 513–514. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [Green Version]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [Green Version]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A., 3rd; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Tomos, I.; Karakatsani, A.; Manali, E.D.; Kottaridi, C.; Spathis, A.; Argentos, S.; Papiris, S.A. Telomere length across different UIP fibrotic-Interstitial Lung Diseases: A prospective Greek case-control study. Pulmonology 2022, 28, 254–261. [Google Scholar] [CrossRef]
- Cao, M.; Gu, L.; Guo, L.; Liu, M.; Wang, T.; Zhang, J.; Zhang, H.; Zhang, Y.; Shi, Y.; Zhao, Y.; et al. Elevated Expression of Growth Differentiation Factor-15 Is Associated with Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Front. Immunol. 2022, 13, 891448. [Google Scholar] [CrossRef]
- Henry, M.T.; McMahon, K.; Mackarel, A.J.; Prikk, K.; Sorsa, T.; Maisi, P.; Sepper, R.; Fitzgerald, M.X.; O’Connor, C.M. Matrix metalloproteinases and tissue inhibitor of metalloproteinase-1 in sarcoidosis and IPF. Eur. Respir. J. 2002, 20, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- Suga, M.; Iyonaga, K.; Okamoto, T.; Gushima, Y.; Miyakawa, H.; Akaike, T.; Ando, M. Characteristic elevation of matrix metalloproteinase activity in idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2000, 162, 1949–1956. [Google Scholar] [CrossRef] [Green Version]
- Craig, V.J.; Zhang, L.; Hagood, J.S.; Owen, C.A. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am. J. Respir. Cell. Mol. Biol. 2015, 53, 585–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, A.; Beltrao, M.; Sokhatska, O.; Costa, D.; Melo, N.; Mota, P.; Marques, A.; Delgado, L. Serum metalloproteinases 1 and 7 in the diagnosis of idiopathic pulmonary fibrosis and other interstitial pneumonias. Respir. Med. 2015, 109, 1063–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, Y.; White, E.S.; de Bernard, S.; Cornelisse, P.; Leconte, I.; Morganti, A.; Roux, S.; Nayler, O. MMP-7 is a predictive biomarker of disease progression in patients with idiopathic pulmonary fibrosis. ERJ Open. Res. 2017, 3, 00074-2016. [Google Scholar] [CrossRef] [Green Version]
- Richards, T.J.; Kaminski, N.; Baribaud, F.; Flavin, S.; Brodmerkel, C.; Horowitz, D.; Li, K.; Choi, J.; Vuga, L.J.; Lindell, K.O.; et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 185, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.F.; Garcia, J.G.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.E.; et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef]
- Sand, J.M.B.; Tanino, Y.; Karsdal, M.A.; Nikaido, T.; Misa, K.; Sato, Y.; Togawa, R.; Wang, X.; Leeming, D.J.; Munakata, M. A Serological Biomarker of Versican Degradation is Associated with Mortality Following Acute Exacerbations of Idiopathic Interstitial Pneumonia. Respir. Res. 2018, 19, 82. [Google Scholar] [CrossRef] [Green Version]
- Ge, J.; Tang, L.; Mu, P.; Zhu, F.; Xie, L.; Tang, Y. Association of ADAM17 Expression Levels in Patients with Interstitial Lung Disease. Immunol. Investig. 2020, 49, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Meng, K.; Gao, Y.; Zhang, J.; Xie, M.; Tian, Y.; Liu, X.; Ma, M.; Cai, Y.; Wu, H.; et al. Elevated serum human epididymis protein 4 is associated with disease severity and worse survival in idiopathic pulmonary fibrosis: A cohort study. Ann. Transl. Med. 2022, 10, 992. [Google Scholar] [CrossRef]
- Chien, J.W.; Richards, T.J.; Gibson, K.F.; Zhang, Y.; Lindell, K.O.; Shao, L.; Lyman, S.K.; Adamkewicz, J.I.; Smith, V.; Kaminski, N.; et al. Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur. Respir. J. 2014, 43, 1430–1438. [Google Scholar] [CrossRef] [Green Version]
- Alhamad, E.H.; Shakoor, Z.; Al-Kassimi, F.A.; Almogren, A.; ElRab, M.O.G.; Maharaj, S.; Kolb, M. Rapid detection of circulating fibrocytes by flowcytometry in idiopathic pulmonary fibrosis. Ann. Thorac. Med. 2015, 10, 279–283. [Google Scholar] [CrossRef]
- Moeller, A.; Gilpin, S.E.; Ask, K.; Cox, G.; Cook, D.; Gauldie, J.; Margetts, P.J.; Farkas, L.; Dobranowski, J.; Boylan, C.; et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 588–594. [Google Scholar] [CrossRef]
- Shimizu, H.; Sakamoto, S.; Okamoto, M.; Isshiki, T.; Ono, J.; Shimizu, S.; Hoshino, T.; Izuhara, K.; Homma, S. Association of serum monomeric periostin level with outcomes of acute exacerbation of idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumonia. Ann. Transl. Med. 2021, 9, 739. [Google Scholar] [CrossRef]
- Murata, K.; Koga, Y.; Kasahara, N.; Hachisu, Y.; Nunomura, S.; Nakajima, N.; Yokoo, H.; Kaira, K.; Maeno, T.; Dobashi, K.; et al. Accumulation of periostin in acute exacerbation of familial idiopathic pulmonary fibrosis. J. Thorac. Dis. 2018, 10, E587–E591. [Google Scholar] [CrossRef]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M.; et al. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005, 2, e251. [Google Scholar] [CrossRef] [Green Version]
- Gui, X.; Qiu, X.; Xie, M.; Tian, Y.; Min, C.; Huang, M.; Hongyan, W.; Chen, T.; Zhang, X.; Chen, J.; et al. Prognostic Value of Serum Osteopontin in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Biomed. Res. Int. 2020, 2020, 3424208. [Google Scholar] [CrossRef] [Green Version]
- O’Dwyer, D.N.; Armstrong, M.E.; Kooblall, M.; Donnelly, S.C. Targeting defective Toll-like receptor-3 function and idiopathic pulmonary fibrosis. Expert. Opin. Ther. Targets 2015, 19, 507–514. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Armstrong, M.E.; Trujillo, G.; Cooke, G.; Keane, M.P.; Fallon, P.G.; Simpson, A.J.; Millar, A.B.; McGrath, E.E.; Whyte, M.K.; et al. The Toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- McElroy, A.N.; Invernizzi, R.; Laskowska, J.W.; O’Neill, A.; Doroudian, M.; Moghoofei, M.; Mostafaei, S.; Li, F.; Przybylski, A.A.; O’Dwyer, D.N.; et al. Candidate Role for Toll-like Receptor 3 L412F Polymorphism and Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2022, 205, 550–562. [Google Scholar] [CrossRef]
- Oldham, J.M.; Ma, S.F.; Martinez, F.J.; Anstrom, K.J.; Raghu, G.; Schwartz, D.A.; Valenzi, E.; Witt, L.; Lee, C.; Vij, R.; et al. TOLLIP, MUC5B, and the Response to N-Acetylcysteine among Individuals with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1475–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonella, F.; Campo, I.; Zorzetto, M.; Boerner, E.; Ohshimo, S.; Theegarten, D.; Taube, C.; Costabel, U. Potential clinical utility of MUC5B und TOLLIP single nucleotide polymorphisms (SNPs) in the management of patients with IPF. Orphanet J. Rare Dis. 2021, 16, 111. [Google Scholar] [CrossRef]
- Cole, A.M.; Waring, A.J. The role of defensins in lung biology and therapy. Am. J. Respir. Med. 2002, 1, 249–259. [Google Scholar] [CrossRef]
- Sakamoto, N.; Ishimatsu, Y.; Kakugawa, T.; Yura, H.; Tomonaga, M.; Harada, T.; Nakashima, S.; Hara, S.; Hara, A.; Ishimoto, H.; et al. Elevated plasma alpha-defensins in patients with acute exacerbation of fibrotic interstitial pneumonia. Respir. Med. 2015, 109, 265–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuga, L.J.; Tedrow, J.R.; Pandit, K.V.; Tan, J.; Kass, D.J.; Xue, J.; Chandra, D.; Leader, J.K.; Gibson, K.F.; Kaminski, N.; et al. C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 189, 966–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Enomoto, N.; Hozumi, H.; Isayama, T.; Naoi, H.; Aono, Y.; Katsumata, M.; Yasui, H.; Karayama, M.; Suzuki, Y.; et al. Serum S100A8 and S100A9 as prognostic biomarkers in acute exacerbation of idiopathic pulmonary fibrosis. Respir. Investig. 2021, 59, 827–836. [Google Scholar] [CrossRef]
- Kagimoto, A.; Tsutani, Y.; Kushitani, K.; Kambara, T.; Mimae, T.; Miyata, Y.; Takeshima, Y.; Okada, M. Serum S100 calcium-binding protein A4 as a novel predictive marker of acute exacerbation of interstitial pneumonia after surgery for lung cancer. BMC Pulm. Med. 2021, 21, 186. [Google Scholar] [CrossRef] [PubMed]
- Feghali-Bostwick, C.A.; Tsai, C.G.; Valentine, V.G.; Kantrow, S.; Stoner, M.W.; Pilewski, J.M.; Gadgil, A.; George, M.P.; Gibson, K.F.; Choi, A.M.; et al. Cellular and humoral autoreactivity in idiopathic pulmonary fibrosis. J. Immunol. 2007, 179, 2592–2599. [Google Scholar] [CrossRef] [Green Version]
- Gilani, S.R.; Vuga, L.J.; Lindell, K.O.; Gibson, K.F.; Xue, J.; Kaminski, N.; Valentine, V.G.; Lindsay, E.K.; George, M.P.; Steele, C.; et al. CD28 down-regulation on circulating CD4 T-cells is associated with poor prognoses of patients with idiopathic pulmonary fibrosis. PLoS ONE 2010, 5, e8959. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y.; Dobashi, K.; Endou, K.; Ono, A.; Yanagitani, N.; Utsugi, M.; Sano, T.; Ishizuka, T.; Shimizu, K.; Tanaka, S.; et al. Decreased interstitial FOXP3(+) lymphocytes in usual interstitial pneumonia with discrepancy of CXCL12/CXCR4 axis. Int. J. Immunopathol. Pharmacol. 2010, 23, 449–461. [Google Scholar] [CrossRef]
- Hata, K.; Yanagihara, T.; Matsubara, K.; Kunimura, K.; Suzuki, K.; Tsubouchi, K.; Eto, D.; Ando, H.; Uehara, M.; Ikegame, S.; et al. Mass cytometry identifies characteristic immune cell subsets in bronchoalveolar lavage fluid from interstitial lung diseases. Front. Immunol. 2023, 14, 1145814. [Google Scholar] [CrossRef]
- Xaubet, A.; Agusti, C.; Luburich, P.; Barbera, J.A.; Carrion, M.; Ayuso, M.C.; Roca, J.; Rodriguez-Roisin, R. Interleukin-8 expression in bronchoalveolar lavage cells in the evaluation of alveolitis in idiopathic pulmonary fibrosis. Respir. Med. 1998, 92, 338–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegenhagen, M.W.; Zabel, P.; Zissel, G.; Schlaak, M.; Muller-Quernheim, J. Serum level of interleukin 8 is elevated in idiopathic pulmonary fibrosis and indicates disease activity. Am. J. Respir. Crit. Care Med. 1998, 157, 762–768. [Google Scholar] [CrossRef]
- Papiris, S.A.; Tomos, I.P.; Karakatsani, A.; Spathis, A.; Korbila, I.; Analitis, A.; Kolilekas, L.; Kagouridis, K.; Loukides, S.; Karakitsos, P.; et al. High levels of IL-6 and IL-8 characterize early-on idiopathic pulmonary fibrosis acute exacerbations. Cytokine 2018, 102, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Tomos, I.; Dimakopoulou, K.; Manali, E.D.; Papiris, S.A.; Karakatsani, A. Long-term personal air pollution exposure and risk for acute exacerbation of idiopathic pulmonary fibrosis. Environ. Health 2021, 20, 99. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Y.; Zou, R.; Chen, H.; Liu, X.; Qiu, X.; Xiao, Y.; Cai, H.; Dai, J. Associations of Serological Biomarkers of sICAM-1, IL-1beta, MIF, and su-PAR with 3-Month Mortality in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Mediat. Inflamm. 2020, 2020, 4534272. [Google Scholar] [CrossRef]
- Oishi, K.; Mimura-Kimura, Y.; Miyasho, T.; Aoe, K.; Ogata, Y.; Katayama, H.; Murata, Y.; Ueoka, H.; Matsumoto, T.; Mimura, Y. Association between cytokine removal by polymyxin B hemoperfusion and improved pulmonary oxygenation in patients with acute exacerbation of idiopathic pulmonary fibrosis. Cytokine 2013, 61, 84–89. [Google Scholar] [CrossRef]
- Lee, J.H.; Jang, J.H.; Park, J.H.; Jang, H.J.; Park, C.S.; Lee, S.; Kim, S.H.; Kim, J.Y.; Kim, H.K. The role of interleukin-6 as a prognostic biomarker for predicting acute exacerbation in interstitial lung diseases. PLoS ONE 2021, 16, e0255365. [Google Scholar] [CrossRef]
- Osuna-Gomez, R.; Barril, S.; Mulet, M.; Atenza, C.Z.; Millan-Billi, P.; Pardessus, A.; Brough, D.E.; Sabzevari, H.; Semnani, R.T.; Castillo, D.; et al. The immunoregulatory role of IL-35 in patients with interstitial lung disease. Immunology 2023, 168, 610–621. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.J.; Wells, A.U.; Kim, H.C.; Ji, W.; Cookson, W.O.; Moffatt, M.F.; Kim, D.S.; Maher, T.M. Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 29. [Google Scholar] [CrossRef] [Green Version]
- Weng, D.; Chen, X.Q.; Qiu, H.; Zhang, Y.; Li, Q.H.; Zhao, M.M.; Wu, Q.; Chen, T.; Hu, Y.; Wang, L.S.; et al. The Role of Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Mediat. Inflamm. 2019, 2019, 5160694. [Google Scholar] [CrossRef]
- Invernizzi, R.; Molyneaux, P.L. The contribution of infection and the respiratory microbiome in acute exacerbations of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2019, 28, 190045. [Google Scholar] [CrossRef]
- Ntolios, P.; Tzilas, V.; Bouros, E.; Avdoula, E.; Karakasiliotis, I.; Bouros, D.; Steiropoulos, P. The Role of Microbiome and Virome in Idiopathic Pulmonary Fibrosis. Biomedicines 2021, 9, 442. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Kaminski, N. Epigenetics in idiopathic pulmonary fibrosis. Biochem. Cell Biol. 2015, 93, 159–170. [Google Scholar] [CrossRef]
- Min, H.; Fan, S.; Song, S.; Zhuang, Y.; Li, H.; Wu, Y.; Cai, H.; Yi, L.; Dai, J.; Gao, Q. Plasma microRNAs are associated with acute exacerbation in idiopathic pulmonary fibrosis. Diagn. Pathol. 2016, 11, 135. [Google Scholar] [CrossRef] [Green Version]
- Oak, S.R.; Murray, L.; Herath, A.; Sleeman, M.; Anderson, I.; Joshi, A.D.; Coelho, A.L.; Flaherty, K.R.; Toews, G.B.; Knight, D.; et al. A micro RNA processing defect in rapidly progressing idiopathic pulmonary fibrosis. PLoS ONE 2011, 6, e21253. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Cheresh, P.; Jablonski, R.P.; Williams, D.B.; Kamp, D.W. The Role of Mitochondrial DNA in Mediating Alveolar Epithelial Cell Apoptosis and Pulmonary Fibrosis. Int. J. Mol. Sci. 2015, 16, 21486–21519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, C.; Sun, H.; Gulati, M.; Herazo-Maya, J.D.; Chen, Y.; Osafo-Addo, A.; Brandsdorfer, C.; Winkler, J.; Blaul, C.; Faunce, J.; et al. Extracellular Mitochondrial DNA Is Generated by Fibroblasts and Predicts Death in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Ishimoto, H.; Yamada, S.; Kushima, H.; Ishii, H.; Imanaga, T.; Harada, T.; Ishimatsu, Y.; Matsumoto, N.; Naito, K.; et al. Autopsy analyses in acute exacerbation of idiopathic pulmonary fibrosis. Respir. Res. 2014, 15, 109. [Google Scholar] [CrossRef] [Green Version]
- Kotani, I.; Sato, A.; Hayakawa, H.; Urano, T.; Takada, Y.; Takada, A. Increased procoagulant and antifibrinolytic activities in the lungs with idiopathic pulmonary fibrosis. Thromb. Res. 1995, 77, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Kondoh, Y.; Azuma, A.; Inoue, Y.; Ogura, T.; Sakamoto, S.; Tsushima, K.; Johkoh, T.; Fujimoto, K.; Ichikado, K.; Matsuzawa, Y.; et al. Thrombomodulin Alfa for Acute Exacerbation of Idiopathic Pulmonary Fibrosis. A Randomized, Double-Blind Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2020, 201, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Mikolasch, T.A.; George, P.M.; Sahota, J.; Nancarrow, T.; Barratt, S.L.; Woodhead, F.A.; Kouranos, V.; Cope, V.S.A.; Creamer, A.W.; Fidan, S.; et al. Multi-center evaluation of baseline neutrophil-to-lymphocyte (NLR) ratio as an independent predictor of mortality and clinical risk stratifier in idiopathic pulmonary fibrosis. EClinicalMedicine 2023, 55, 101758. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Mehta, J.; Stauffer, J.; Morgenthien, E.; Yang, M.; Limb, S.L.; Bhorade, S. Changes in Neutrophil-Lymphocyte or Platelet-Lymphocyte Ratios and Their Associations with Clinical Outcomes in Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2021, 10, 1427. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cai, J.; Zhang, M.; Yan, X. Prognostic Role of NLR, PLR and MHR in Patients with Idiopathic Pulmonary Fibrosis. Front. Immunol. 2022, 13, 882217. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Willis-Owen, S.A.G.; Cox, M.J.; James, P.; Cowman, S.; Loebinger, M.; Blanchard, A.; Edwards, L.M.; Stock, C.; Daccord, C.; et al. Host-Microbial Interactions in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 195, 1640–1650. [Google Scholar] [CrossRef]
- Zinellu, A.; Paliogiannis, P.; Sotgiu, E.; Mellino, S.; Mangoni, A.A.; Zinellu, E.; Negri, S.; Collu, C.; Pintus, G.; Serra, A.; et al. Blood Cell Count Derived Inflammation Indexes in Patients with Idiopathic Pulmonary Fibrosis. Lung 2020, 198, 821–827. [Google Scholar] [CrossRef]
- Zinellu, A.; Collu, C.; Nasser, M.; Paliogiannis, P.; Mellino, S.; Zinellu, E.; Traclet, J.; Ahmad, K.; Mangoni, A.A.; Carru, C.; et al. The Aggregate Index of Systemic Inflammation (AISI): A Novel Prognostic Biomarker in Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2021, 10, 4134. [Google Scholar] [CrossRef]
- Rosas, I.O.; Richards, T.J.; Konishi, K.; Zhang, Y.; Gibson, K.; Lokshin, A.E.; Lindell, K.O.; Cisneros, J.; Macdonald, S.D.; Pardo, A.; et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008, 5, e93. [Google Scholar] [CrossRef] [Green Version]
- Herazo-Maya, J.D.; Sun, J.; Molyneaux, P.L.; Li, Q.; Villalba, J.A.; Tzouvelekis, A.; Lynn, H.; Juan-Guardela, B.M.; Risquez, C.; Osorio, J.C.; et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: An international, multicentre, cohort study. Lancet Respir. Med. 2017, 5, 857–868. [Google Scholar] [CrossRef]
- Clynick, B.; Corte, T.J.; Jo, H.E.; Stewart, I.; Glaspole, I.N.; Grainge, C.; Maher, T.M.; Navaratnam, V.; Hubbard, R.; Hopkins, P.M.A.; et al. Biomarker signatures for progressive idiopathic pulmonary fibrosis. Eur. Respir. J. 2022, 59, 2101181. [Google Scholar] [CrossRef]
- Fraioli, F.; Lyasheva, M.; Porter, J.C.; Bomanji, J.; Shortman, R.I.; Endozo, R.; Wan, S.; Bertoletti, L.; Machado, M.; Ganeshan, B.; et al. Synergistic application of pulmonary (18)F-FDG PET/HRCT and computer-based CT analysis with conventional severity measures to refine current risk stratification in idiopathic pulmonary fibrosis (IPF). Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2023–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, K.; Gibson, K.F.; Lindell, K.O.; Richards, T.J.; Zhang, Y.; Dhir, R.; Bisceglia, M.; Gilbert, S.; Yousem, S.A.; Song, J.W.; et al. Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, S.; Shimizu, H.; Isshiki, T.; Nakamura, Y.; Usui, Y.; Kurosaki, A.; Isobe, K.; Takai, Y.; Homma, S. New risk scoring system for predicting 3-month mortality after acute exacerbation of idiopathic pulmonary fibrosis. Sci. Rep. 2022, 12, 1134. [Google Scholar] [CrossRef] [PubMed]
- Carleo, A.; Landi, C.; Prasse, A.; Bergantini, L.; D’Alessandro, M.; Cameli, P.; Janciauskiene, S.; Rottoli, P.; Bini, L.; Bargagli, E. Proteomic characterization of idiopathic pulmonary fibrosis patients: Stable versus acute exacerbation. Monaldi Arch. Chest Dis. 2020, 90. [Google Scholar] [CrossRef] [PubMed]
- Corrin, B. Classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2014, 189, 1008–1009. [Google Scholar] [CrossRef]
- Papanikolaou, I.C.; Drakopanagiotakis, F.; Polychronopoulos, V.S. Acute exacerbations of interstitial lung diseases. Curr. Opin. Pulm. Med. 2010, 16, 480–486. [Google Scholar] [CrossRef]
- Yamakawa, H.; Hagiwara, E.; Ikeda, S.; Iwasawa, T.; Otoshi, R.; Tabata, E.; Okuda, R.; Sekine, A.; Baba, T.; Iso, S.; et al. Evaluation of changes in the serum levels of Krebs von den Lungen-6 and surfactant protein-D over time as important biomarkers in idiopathic fibrotic nonspecific interstitial pneumonia. Respir. Investig. 2019, 57, 422–429. [Google Scholar] [CrossRef]
- Ichiyasu, H.; Ichikado, K.; Yamashita, A.; Iyonaga, K.; Sakamoto, O.; Suga, M.; Kohrogi, H. Pneumocyte biomarkers KL-6 and surfactant protein D reflect the distinct findings of high-resolution computed tomography in nonspecific interstitial pneumonia. Respiration 2012, 83, 190–197. [Google Scholar] [CrossRef]
- Bennett, D.; Salvini, M.; Fui, A.; Cillis, G.; Cameli, P.; Mazzei, M.A.; Fossi, A.; Refini, R.M.; Rottoli, P. Calgranulin B and KL-6 in Bronchoalveolar Lavage of Patients with IPF and NSIP. Inflammation 2019, 42, 463–470. [Google Scholar] [CrossRef]
- Ohara, I.; Aida, S.; Shimazaki, H.; Kobayashi, H.; Tsuda, H.; Toda, T.; Nakanishi, K.; Tamai, S. Proteomic analysis in usual and nonspecific interstitial pneumonia. Histol. Histopathol. 2014, 29, 377–386. [Google Scholar] [CrossRef]
- Yoo, H.; Hino, T.; Hwang, J.; Franks, T.J.; Han, J.; Im, Y.; Lee, H.Y.; Chung, M.P.; Hatabu, H.; Lee, K.S. Connective tissue disease-related interstitial lung disease (CTD-ILD) and interstitial lung abnormality (ILA): Evolving concept of CT findings, pathology and management. Eur. J. Radiol. Open. 2022, 9, 100419. [Google Scholar] [CrossRef]
- Faverio, P.; Stainer, A.; Conti, S.; Madotto, F.; De Giacomi, F.; Della Zoppa, M.; Vancheri, A.; Pellegrino, M.R.; Tonelli, R.; Cerri, S.; et al. Differences between Acute Exacerbations of Idiopathic Pulmonary Fibrosis and Other Interstitial Lung Diseases. Diagnostics 2021, 11, 1623. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, R.; Afeltra, A.; Alunno, A.; Bartoloni-Bocci, E.; Berardicurti, O.; Bombardieri, M.; Bortoluzzi, A.; Caporali, R.; Caso, F.; Cervera, R.; et al. Guidelines for biomarkers in autoimmune rheumatic diseases—Evidence based analysis. Autoimmun. Rev. 2019, 18, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Natalini, J.G.; Baker, J.F.; Singh, N.; Mahajan, T.D.; Roul, P.; Thiele, G.M.; Sauer, B.C.; Johnson, C.R.; Kawut, S.M.; Mikuls, T.R.; et al. Autoantibody Seropositivity and Risk for Interstitial Lung Disease in a Prospective Male-Predominant Rheumatoid Arthritis Cohort of U.S. Veterans. Ann. Am. Thorac. Soc. 2021, 18, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Harlow, L.; Rosas, I.O.; Gochuico, B.R.; Mikuls, T.R.; Dellaripa, P.F.; Oddis, C.V.; Ascherman, D.P. Identification of citrullinated hsp90 isoforms as novel autoantigens in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheum. 2013, 65, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Juge, P.A.; Lee, J.S.; Ebstein, E.; Furukawa, H.; Dobrinskikh, E.; Gazal, S.; Kannengiesser, C.; Ottaviani, S.; Oka, S.; Tohma, S.; et al. MUC5B Promoter Variant and Rheumatoid Arthritis with Interstitial Lung Disease. N. Engl. J. Med. 2018, 379, 2209–2219. [Google Scholar] [CrossRef]
- Palomaki, A.; FinnGen Rheumatology Clinical Expert Group; Palotie, A.; Koskela, J.; Eklund, K.K.; Pirinen, M.; FinnGen; Ripatti, S.; Laitinen, T.; Mars, N. Lifetime risk of rheumatoid arthritis-associated interstitial lung disease in MUC5B mutation carriers. Ann. Rheum. Dis. 2021, 80, 1530–1536. [Google Scholar] [CrossRef]
- Furukawa, H.; Oka, S.; Higuchi, T.; Shimada, K.; Hashimoto, A.; Matsui, T.; Tohma, S. Biomarkers for interstitial lung disease and acute-onset diffuse interstitial lung disease in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X211022506. [Google Scholar] [CrossRef]
- Pulito-Cueto, V.; Remuzgo-Martinez, S.; Genre, F.; Atienza-Mateo, B.; Mora-Cuesta, V.M.; Iturbe-Fernandez, D.; Lera-Gomez, L.; Mora-Gil, M.S.; Prieto-Pena, D.; Portilla, V.; et al. Elevated VCAM-1, MCP-1 and ADMA serum levels related to pulmonary fibrosis of interstitial lung disease associated with rheumatoid arthritis. Front. Mol. Biosci. 2022, 9, 1056121. [Google Scholar] [CrossRef]
- Pulito-Cueto, V.; Genre, F.; Lopez-Mejias, R.; Mora-Cuesta, V.M.; Iturbe-Fernandez, D.; Portilla, V.; Mora-Gil, M.S.; Ocejo-Vinyals, J.G.; Gualillo, O.; Blanco, R.; et al. Endothelin-1 as a Biomarker of Idiopathic Pulmonary Fibrosis and Interstitial Lung Disease Associated with Autoimmune Diseases. Int. J. Mol. Sci. 2023, 24, 1275. [Google Scholar] [CrossRef]
- Zheng, W.; Hu, X.; Zou, M.; Hu, N.; Song, W.; Wang, R.; Liu, Y.; Hou, Q.; Liu, Y.; Chen, X.; et al. Plasma IL-36alpha and IL-36gamma as Potential Biomarkers in Interstitial Lung Disease Associated with Rheumatoid Arthritis: A Pilot Study in the Chinese Population. Inflammation 2022, 46, 285–296. [Google Scholar] [CrossRef]
- Lin, T.; Xu, S.; Wang, Y.; Nian, X.; Shan, X.; Jiang, T.; Qiu, M. Human epididymis protein 4 as a new diagnostic biomarker for rheumatoid arthritis-associated interstitial lung disease. Clin. Exp. Rheumatol. 2022, 40, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.A.; Oldham, J.M.; Ley, B.; Anand, V.; Adegunsoye, A.; Liu, G.; Batra, K.; Torrealba, J.; Kozlitina, J.; Glazer, C.; et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur. Respir. J. 2019, 53, 1801641. [Google Scholar] [CrossRef]
- Doyle, T.J.; Patel, A.S.; Hatabu, H.; Nishino, M.; Wu, G.; Osorio, J.C.; Golzarri, M.F.; Traslosheros, A.; Chu, S.G.; Frits, M.L.; et al. Detection of Rheumatoid Arthritis-Interstitial Lung Disease Is Enhanced by Serum Biomarkers. Am. J. Respir. Crit. Care Med. 2015, 191, 1403–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, H.; Oka, S.; Shimada, K.; Hashimoto, A.; Komiya, A.; Matsui, T.; Fukui, N.; Tohma, S. Serum Metabolomic Profiles of Rheumatoid Arthritis Patients with Acute-Onset Diffuse Interstitial Lung Disease. Biomark. Insights 2019, 14, 1177271919870472. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Hirata, S.; Nagasawa, H.; Kameda, H.; Seto, Y.; Atsumi, T.; Dohi, M.; Koike, T.; Miyasaka, N.; Harigai, M. A retrospective study of serum KL-6 levels during treatment with biological disease-modifying antirheumatic drugs in rheumatoid arthritis patients: A report from the Ad Hoc Committee for Safety of Biological DMARDs of the Japan College of Rheumatology. Mod. Rheumatol. 2013, 23, 297–303. [Google Scholar] [CrossRef]
- Tanaka, N.; Nishimura, K.; Waki, D.; Kadoba, K.; Murabe, H.; Yokota, T. Annual variation rate of KL-6 for predicting acute exacerbation in patients with rheumatoid arthritis-associated interstitial lung disease. Mod. Rheumatol. 2021, 31, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wang, Y.; Meng, F.; Feng, M.; Zhao, X.; Gao, C.; Luo, J. Identification of biomarkers by machine learning classifiers to assist diagnose rheumatoid arthritis-associated interstitial lung disease. Arthritis Res. Ther. 2022, 24, 115. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.; Zheng, S.; Lin, J.; Hu, S.; Zhuang, J.; Lin, Q.; Xie, X.; Zheng, K.; Zhang, W.; et al. The role of lung ultrasound B-lines and serum KL-6 in the screening and follow-up of rheumatoid arthritis patients for an identification of interstitial lung disease: Review of the literature, proposal for a preliminary algorithm, and clinical application to cases. Arthritis Res. Ther. 2021, 23, 212. [Google Scholar] [CrossRef]
- Venerito, V.; Manfredi, A.; Lopalco, G.; Lavista, M.; Cassone, G.; Scardapane, A.; Sebastiani, M.; Iannone, F. Radiomics to predict the mortality of patients with rheumatoid arthritis-associated interstitial lung disease: A proof-of-concept study. Front. Med. 2022, 9, 1069486. [Google Scholar] [CrossRef]
- Bonhomme, O.; Andre, B.; Gester, F.; de Seny, D.; Moermans, C.; Struman, I.; Louis, R.; Malaise, M.; Guiot, J. Biomarkers in systemic sclerosis-associated interstitial lung disease: Review of the literature. Rheumatology 2019, 58, 1534–1546. [Google Scholar] [CrossRef] [Green Version]
- Fertig, N.; Domsic, R.T.; Rodriguez-Reyna, T.; Kuwana, M.; Lucas, M.; Medsger, T.A., Jr.; Feghali-Bostwick, C.A. Anti-U11/U12 RNP antibodies in systemic sclerosis: A new serologic marker associated with pulmonary fibrosis. Arthritis Rheum. 2009, 61, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Steen, V.D. Autoantibodies in systemic sclerosis. Semin. Arthritis Rheum. 2005, 35, 35–42. [Google Scholar] [CrossRef]
- Fields, A.; Potel, K.N.; Cabuhal, R.; Aziri, B.; Stewart, I.D.; Schock, B.C. Mediators of systemic sclerosis-associated interstitial lung disease (SSc-ILD): Systematic review and meta-analyses. Thorax 2022. online ahead of print. [Google Scholar] [CrossRef]
- Hant, F.N.; Ludwicka-Bradley, A.; Wang, H.J.; Li, N.; Elashoff, R.; Tashkin, D.P.; Silver, R.M.; Scleroderma Lung Study Research Group. Surfactant protein D and KL-6 as serum biomarkers of interstitial lung disease in patients with scleroderma. J. Rheumatol. 2009, 36, 773–780. [Google Scholar] [CrossRef]
- Elhai, M.; Hoffmann-Vold, A.M.; Avouac, J.; Pezet, S.; Cauvet, A.; Leblond, A.; Fretheim, H.; Garen, T.; Kuwana, M.; Molberg, O.; et al. Performance of Candidate Serum Biomarkers for Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2019, 71, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Schupp, J.; Becker, M.; Gunther, J.; Muller-Quernheim, J.; Riemekasten, G.; Prasse, A. Serum CCL18 is predictive for lung disease progression and mortality in systemic sclerosis. Eur. Respir. J. 2014, 43, 1530–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiev, K.P.; Hua-Huy, T.; Kettaneh, A.; Gain, M.; Duong-Quy, S.; Toledano, C.; Cabane, J.; Dinh-Xuan, A.T. Serum CC chemokine ligand-18 predicts lung disease worsening in systemic sclerosis. Eur. Respir. J. 2011, 38, 1355–1360. [Google Scholar] [CrossRef]
- Omatsu, J.; Saigusa, R.; Miyagawa, T.; Fukui, Y.; Toyama, S.; Awaji, K.; Ikawa, T.; Norimatsu, Y.; Yoshizaki, A.; Sato, S.; et al. Serum S100A12 levels: Possible association with skin sclerosis and interstitial lung disease in systemic sclerosis. Exp. Dermatol. 2021, 30, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, L.; E, L.; Xu, K.; Wang, X.F.; Zhang, B.; Su, J.; Meng, Z. Increased levels of HE4 (WFDC2) in systemic sclerosis: A novel biomarker reflecting interstitial lung disease severity? Ther. Adv. Chronic Dis. 2020, 11, 2040622320956420. [Google Scholar] [CrossRef]
- Atilla, N.; Cetin, G.Y.; Balkarli, A. Association of neutrophil/lymphocyte ratio with the degree ofinterstitial lung disease in systemic sclerosis. Turk. J. Med. Sci. 2016, 46, 1871–1874. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef] [PubMed]
- Tzilas, V.; Tzouvelekis, A.; Bouros, E.; Karampitsakos, T.; Ntasiou, M.; Katsaras, M.; Costabel, U.; Wells, A.; Bouros, D. Diagnostic value of BAL lymphocytosis in patients with indeterminate for usual interstitial pneumonia imaging pattern. Eur. Respir. J. 2019, 54, 1901144. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Diez, S.; Munoz, X.; Ojanguren, I.; Romero-Mesones, C.; Espejo, D.; Villar, A.; Gomez-Olles, S.; Cruz, M.J. YKL-40 and KL-6 Levels in Serum and Sputum of Patients Diagnosed With Hypersensitivity Pneumonitis. J. Allergy Clin. Immunol. Pract. 2022, 10, 2414–2423. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhang, J.; Ren, X. Krebs von den lungen-6 as a clinical marker for hypersensitivity pneumonitis: A meta-analysis and bioinformatics analysis. Front. Immunol. 2022, 13, 1041098. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Fujii, M.; Furusawa, H.; Tsuchiya, K.; Miyazaki, Y.; Inase, N. The usefulness of KL-6 and SP-D for the diagnosis and management of chronic hypersensitivity pneumonitis. Respir. Med. 2015, 109, 1576–1581. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.; Grutters, J.C.; Sato, H.; van Velzen-Blad, H.; Zanen, P.; Kohno, N.; Welsh, K.I.; du Bois, R.M.; van den Bosch, J.M. Analysis of KL-6 and SP-D as disease markers in bird fancier’s lung. Sarcoidosis Vasc. Diffuse Lung Dis. 2005, 22, 51–57. [Google Scholar]
- Onishi, Y.; Kawamura, T.; Higashino, T.; Kagami, R.; Hirata, N.; Miyake, K. Clinical features of chronic summer-type hypersensitivity pneumonitis and proposition of diagnostic criteria. Respir. Investig. 2020, 58, 59–67. [Google Scholar] [CrossRef]
- Ejima, M.; Okamoto, T.; Suzuki, T.; Miyazaki, Y. Role of serum surfactant protein-D as a prognostic predictor in fibrotic hypersensitivity pneumonitis. Respir. Investig. 2022, 60, 369–378. [Google Scholar] [CrossRef]
- Watanabe, M.; Horimasu, Y.; Iwamoto, H.; Yamaguchi, K.; Sakamoto, S.; Masuda, T.; Nakashima, T.; Miyamoto, S.; Ohshimo, S.; Fujitaka, K.; et al. C-C Motif Chemokine Ligand 15 May Be a Useful Biomarker for Predicting the Prognosis of Patients with Chronic Hypersensitivity Pneumonitis. Respiration 2019, 98, 212–220. [Google Scholar] [CrossRef] [Green Version]
- d’Alessandro, M.; Bergantini, L.; Cameli, P.; Lanzarone, N.; Perillo, F.; Perrone, A.; Bargagli, E. BAL and serum multiplex lipid profiling in idiopathic pulmonary fibrosis and fibrotic hypersensitivity pneumonitis. Life Sci. 2020, 256, 117995. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Unoura, K.; Tateishi, T.; Akashi, T.; Takemura, T.; Tomita, M.; Inase, N.; Yoshizawa, Y. Higher serum CCL17 may be a promising predictor of acute exacerbations in chronic hypersensitivity pneumonitis. Respir. Res. 2013, 14, 57. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; He, X.; Ohshimo, S.; Griese, M.; Sarria, R.; Guzman, J.; Costabel, U.; Bonella, F. Serum YKL-40 as predictor of outcome in hypersensitivity pneumonitis. Eur. Respir. J. 2017, 49, 1501924. [Google Scholar] [CrossRef] [Green Version]
- Vietri, L.; d’Alessandro, M.; Bergantini, L.; Carleo, A.; Cameli, P.; Mazzei, M.A.; Sestini, P.; Bargagli, E. Specificity of serum amyloid A as a biomarker of idiopathic pulmonary fibrosis. Intern. Med. J. 2020, 50, 1571–1574. [Google Scholar] [CrossRef] [PubMed]
- Bergantini, L.; d’Alessandro, M.; Vietri, L.; Rana, G.D.; Cameli, P.; Acerra, S.; Sestini, P.; Bargagli, E. Utility of serological biomarker’ panels for diagnostic accuracy of interstitial lung diseases. Immunol. Res. 2020, 68, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Sterclova, M.; Matej, R.; Mandakova, P.; Skibova, J.; Vasakova, M. Role of interleukin 4 and its receptor in clinical presentation of chronic extrinsic allergic alveolitis: A pilot study. Multidiscip. Respir. Med. 2013, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, Y.; Kasahara, T.; Mukaida, N.; Matsushima, K.; Kitamura, S. Chemokines in bronchoalveolar lavage fluid in summer-type hypersensitivity pneumonitis. Eur. Respir. J. 1995, 8, 1084–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nukui, Y.; Miyazaki, Y.; Masuo, M.; Okamoto, T.; Furusawa, H.; Tateishi, T.; Kishino, M.; Tateishi, U.; Ono, J.; Ohta, S.; et al. Periostin as a predictor of prognosis in chronic bird-related hypersensitivity pneumonitis. Allergol. Int. 2019, 68, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.J.; Hur, J.; Go, D.J.; Kang, E.H.; Park, J.K.; Lee, E.Y.; Shin, K.; Lee, E.B.; Song, Y.W.; Lee, Y.J. Baseline peripheral blood neutrophil-to-lymphocyte ratio could predict survival in patients with adult polymyositis and dermatomyositis: A retrospective observational study. PLoS ONE 2018, 13, e0190411. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, G.; Acquah, S.O.; Salvatore, M.; Padilla, M.L. Elevated serum D-dimer level is associated with an increased risk of acute exacerbation in interstitial lung disease. Respir. Med. 2017, 128, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Zhang, T.; Lin, R.; Zeng, Y.; Cheng, Z.J.; Li, N.; Zheng, P.; Huang, H.; Zhang, X.D.; Wang, H.; et al. Clinical utility of heparin-binding protein as an acute-phase inflammatory marker in interstitial lung disease. J. Leukoc. Biol. 2022, 112, 861–873. [Google Scholar] [CrossRef]
- Kono, M.; Miyashita, K.; Hirama, R.; Oshima, Y.; Takeda, K.; Mochizuka, Y.; Tsutsumi, A.; Miwa, H.; Miki, Y.; Hashimoto, D.; et al. Prognostic significance of bronchoalveolar lavage cellular analysis in patients with acute exacerbation of interstitial lung disease. Respir. Med. 2021, 186, 106534. [Google Scholar] [CrossRef] [PubMed]
- Bowman, W.S.; Newton, C.A.; Linderholm, A.L.; Neely, M.L.; Pugashetti, J.V.; Kaul, B.; Vo, V.; Echt, G.A.; Leon, W.; Shah, R.J.; et al. Proteomic biomarkers of progressive fibrosing interstitial lung disease: A multicentre cohort analysis. Lancet Respir. Med. 2022, 10, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug. Discov. 2017, 16, 755–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiot, J.; Moermans, C.; Henket, M.; Corhay, J.L.; Louis, R. Blood Biomarkers in Idiopathic Pulmonary Fibrosis. Lung 2017, 195, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzouvelekis, A.; Herazo-Maya, J.D.; Slade, M.; Chu, J.H.; Deiuliis, G.; Ryu, C.; Li, Q.; Sakamoto, K.; Ibarra, G.; Pan, H.; et al. Validation of the prognostic value of MMP-7 in idiopathic pulmonary fibrosis. Respirology 2017, 22, 486–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, T.M.; Oballa, E.; Simpson, J.K.; Porte, J.; Habgood, A.; Fahy, W.A.; Flynn, A.; Molyneaux, P.L.; Braybrooke, R.; Divyateja, H.; et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: An analysis from the multicentre PROFILE cohort study. Lancet Respir. Med. 2017, 5, 946–955. [Google Scholar] [CrossRef] [Green Version]


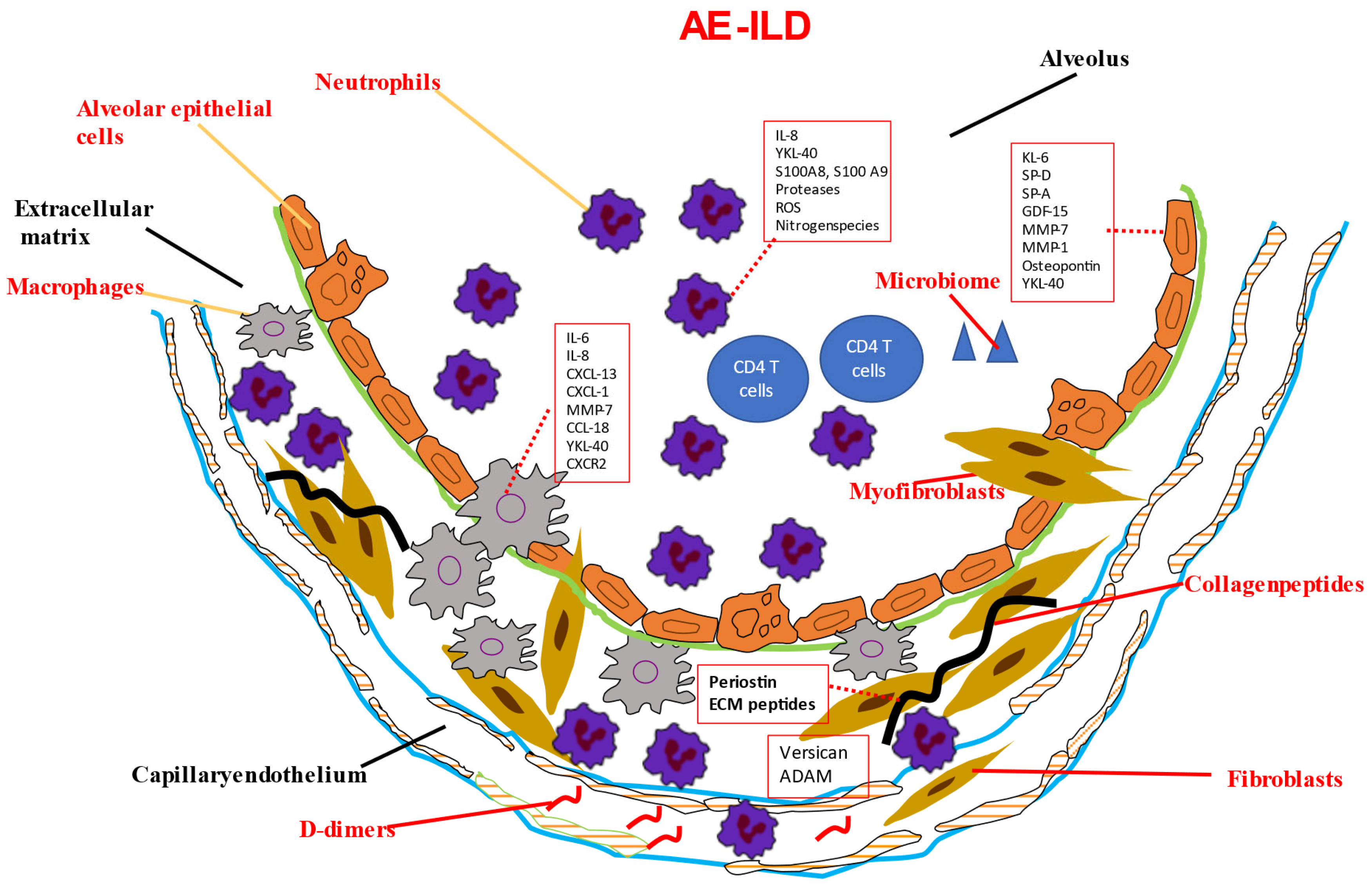{kind=link}
| ILD Subtype | Biomarker | Compartment | Function | Reference |
|---|---|---|---|---|
| IPF | KL-6 | Serum | Predictive of development of AE-IPF | [12,13,15,16,17] |
| Serum | Descriptive of AE-IPF | [14] | ||
| Serum | Predictive of mortality of AE-IPF | [14] | ||
| SP-D | Serum | Descriptive of AE-IPF | [20,22] | |
| Serum | Predictive of AE-IPF | [22] | ||
| Serum | Predictive of mortality of AE-IPF | [23] | ||
| SP-A | Serum | Descriptive of AE-IPF | [20,22] | |
| Serum | Predictive of AE-IPF | [22] | ||
| Telomere length | Serum circulating leukocytes | Descriptive of AE-IPF | [28] | |
| GDF-15 | Serum, lung | Descriptive of AE-IPF | [29] | |
| Serum | Predictive of AE-IPF | [29] | ||
| Versican | Serum | Descriptive of AE-IPF | [37] | |
| Serum | Predictive of mortality of AE-IPF | [37] | ||
| ADAM-17 | Serum | Descriptive of AE-IPF | [38] | |
| Human epididymis protein 4 | Serum | Descriptive of AE-IPF | [39] | |
| Circulating fibrocytes | Blood | Descriptive of AE-IPF | [42] | |
| Periostin | Serum | Descriptive of AE-IPF | [43] | |
| Serum | Predictive of mortality of AE-IPF | [43] | ||
| Osteopontin | Serum | Descriptive of AE-IPF | [46] | |
| Serum | Predictive of AE-IPF | [46] | ||
| L412-F | Blood fibroblasts | Predictive of AE-IPF | [49] | |
| TOLLIP rs5743890 | Blood leukocytes | Predictive of AE-IPF | [51] | |
| CXCL13 | Serum | Descriptive of AE-IPF | [54] | |
| Serum | Predictive of AE-IPF | [54] | ||
| S100A8 and S100A9 | Serum | Predictive of AE-IPF | [55,56] | |
| IL-8 | Serum | Descriptive of AE-IPF | [63] | |
| Serum | Predictive of mortality of AE-IPF | [63] | ||
| IL-1b | Serum | Descriptive of AE-IPF | [64,65,66] | |
| Serum | Predictive of mortality of AE-IPF | [65] | ||
| IL-6 | Serum | Predictive of AE-IPF | [63,67] | |
| Lung microbiome changes (increased Proteobacteria, Campylobacter spp. and Stenotrophomona spp., decreased Veillonella spp. and Campylobacter spp.) | BAL | Descriptive of AE-IPF | [69] | |
| miR-25-3p | Serum | Descriptive of AE-IPF | [74] | |
| let-7d-5p | Serum | Descriptive of AE-IPF | [74] | |
| D-dimers | Serum | Descriptive of AE-IPF | [20] | |
| Neutrophil-to-lymphocyte ratio | Blood | Predictive of AE-IPF | [81,82] | |
| Blood | Predictive of mortality of AE-IPF | [83] | ||
| CCNA2 | Lung | Descriptive of AE-IPF | [91] | |
| sICAM-1, MIF, IL-1b, and su-PAR | Serum | Descriptive of AE-IPF | [65] | |
| Serum | Predictive of mortality of AE-IPF | [65] | ||
| Proteomic analysis (IGKC, S100A9, PEDF, IGHG1, ALDOA, A1AT, HPT, CO3, and PIGR) | BALF | Descriptive of AE-IPF | [93] | |
| BALF neutrophilia and lymphopenia | BALF | Predictive of mortality of AE-IPF | [151] | |
| CTD-ILD | D-dimers | Serum | Predictive of AE-ILD | [149] |
| IL-6 | Serum | Predictive of AE-ILD | [67] | |
| BALF neutrophilia and lymphopenia | BALF | Predictive of mortality of AE-ILD | [151] | |
| RA-ILD | KL-6 | Serum | Prognostic of AE-ILD | [116] |
| Mannosamine, Alliin, Kynurenine, 2-hydroxybutyric acid | Serum | Diagnostic of AE-ILD | [114] | |
| HP | KL-6 | Serum | Diagnostic of AE-ILD | [135] |
| Periostin | Serum | Diagnostic of AE-ILD | [147] | |
| CCL-17 | Serum | Diagnostic of AE-ILD | [141] | |
| Serum | Predictive of AE-ILD | [141] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drakopanagiotakis, F.; Markart, P.; Steiropoulos, P. Acute Exacerbations of Interstitial Lung Diseases: Focus on Biomarkers. Int. J. Mol. Sci. 2023, 24, 10196. https://doi.org/10.3390/ijms241210196
Drakopanagiotakis F, Markart P, Steiropoulos P. Acute Exacerbations of Interstitial Lung Diseases: Focus on Biomarkers. International Journal of Molecular Sciences. 2023; 24(12):10196. https://doi.org/10.3390/ijms241210196
Chicago/Turabian StyleDrakopanagiotakis, Fotios, Philipp Markart, and Paschalis Steiropoulos. 2023. "Acute Exacerbations of Interstitial Lung Diseases: Focus on Biomarkers" International Journal of Molecular Sciences 24, no. 12: 10196. https://doi.org/10.3390/ijms241210196