
Microglial Activation and Priming in Alzheimer’s Disease: State of the Art and Future Perspectives
 , and
, and
Abstract
:1. Introduction
2. Microglia in Health and Disease
2.1. Microglia Functions
2.1.1. Sensing
2.1.2. Defence
2.1.3. Brain Homeostasis
3. Microglia Phenotypes
4. Microglial Role in AD
4.1. Microglia Activation and Aβ Pathology
4.2. Microglia Activation and Tau Pathology
4.3. Microglia Activation and CX3CL1/CX3CR1 Signalling
5. AD Treatment Strategies Associated with Inflammation and Microglia Activation
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Budd Haeberlein, S.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer disease: An update on pathobiology and treatment strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [Green Version]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [Green Version]
- Mcgeer, P.L.; Itagaki, S.; Tago, H.; Mcgeer, E.G. Reactive microglia in patients with senile dementia of Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright-Jin, E.C.; Gutmann, D.H. Microglia as Dynamic Cellular Mediators of Brain Function. Trends Mol. Med. 2019, 25, 967–979. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, S.E.; Allison, E.K.; Coleman, U.; Kingery-Gallagher, N.D.; El Khoury, J. Heterozygous CX3CR1 Deficiency in Microglia Restores Neuronal β-Amyloid Clearance Pathways and Slows Progression of Alzheimer’s Like-Disease in PS1-APP Mice. Front. Immunol. 2019, 10, 2780. [Google Scholar] [CrossRef] [Green Version]
- Aramideh, J.A.; Vidal-Itriago, A.; Morsch, M.; Graeber, M.B. Cytokine Signalling at the Microglial Penta-Partite Synapse. Int. J. Mol. Sci. 2021, 22, 13186. [Google Scholar] [CrossRef] [PubMed]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional Microglia-Neuron Communication in Health and Disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2017, 101, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef]
- Brennan, E.M.; Martin, L.J.; Johnston, M.V.; Blue, M.E. Ontogeny of non-NMDA glutamate receptors in rat barrel field cortex: II. Alpha-AMPA and kainate receptors. J. Comp. Neurol. 1997, 386, 29–45. [Google Scholar] [CrossRef]
- Bourgognon, J.M.; Cavanagh, J. The role of cytokines in modulating learning and memory and brain plasticity. Brain Neurosci. Adv. 2020, 4, 2398212820979802. [Google Scholar] [CrossRef] [PubMed]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, M.K.; Šimončičová, E.; Bögi, E.; Tremblay, M.E. Shedding Light on the Dark Side of the Microglia. ASN Neuro. 2020, 12, 1759091420925335. [Google Scholar] [CrossRef]
- Cornell, J.; Salinas, S.; Huang, H.Y.; Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural. Regen. Res. 2022, 17, 705–716. [Google Scholar] [PubMed]
- Rohan Walker, F.; Yirmiya, R. Microglia, physiology and behavior: A brief commentary. Brain. Behav. Immun. 2016, 55, 1–5. [Google Scholar] [CrossRef]
- Luo, P.; Chu, S.F.; Zhang, Z.; Xia, C.Y.; Chen, N.H. Fractalkine/CX3CR1 is involved in the cross-talk between neuron and glia in neurological diseases. Brain Res. Bull. 2019, 146, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, J.; Gao, J.; Wu, Y.; Yin, M.; Zhao, W. CD200-, CX3CL1-, and TREM2-mediated neuron-microglia interactions and their involvements in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, G.K.; Murphy, K.J. Neuron-glia crosstalk in health and disease: Fractalkine and CX3CR1 take centre stage. Open Biol. 2013, 3, 130181. [Google Scholar] [CrossRef] [Green Version]
- Rogers, J.T.; Morganti, J.M.; Bachstetter, A.D.; Hudson, C.E.; Peters, M.M.; Grimmig, B.A.; Weeber, E.J.; Bickford, P.C.; Gemma, C. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 2011, 31, 16241–16250. [Google Scholar] [CrossRef]
- Williams, J.L.; Holman, D.W.; Klein, R.S. Chemokines in the balance: Maintenance of homeostasis and protection at CNS barriers. Front. Cell Neurosci. 2014, 8, 154. [Google Scholar] [CrossRef] [Green Version]
- Bachstetter, A.D.; Morganti, J.M.; Jernberg, J.; Schlunk, A.; Mitchell, S.H.; Brewster, K.W.; Hudson, C.E.; Cole, M.J.; Harrison, J.K.; Bickford, P.C.; et al. Fractalkine and CX3CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol. Aging 2011, 32, 2030–2044. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef]
- Savage, J.C.; Carrier, M.; Tremblay, M.E. Morphology of Microglia Across Contexts of Health and Disease. Methods Mol. Biol. 2019, 2034, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sánchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [Green Version]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.J.; Au, N.P.B.; Ma, C.H.E. Functional and Phenotypic Diversity of Microglia: Implication for Microglia-Based Therapies for Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 896852. [Google Scholar] [CrossRef]
- Streit, W.J.; Braak, H.; Xue, Q.S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Bittner, T.; Jung, C.K.E.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2010, 13, 411–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Chinnathambi, S. Microglial priming of antigen presentation and adaptive stimulation in Alzheimer’s disease. Cell Mol. Life Sci. 2019, 76, 3681–3694. [Google Scholar] [CrossRef]
- Preeti, K.; Sood, A.; Fernandes, V. Metabolic Regulation of Glia and Their Neuroinflammatory Role in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2022, 42, 2527–2551. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.N.; Barbosa-Silva, M.C.; Maron-Gutierrez, T. Microglial priming in infections and its risk to neurodegenerative diseases. Front. Cell. Neurosci. 2022, 16, 878987. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Spittau, B. Aging Microglia-Phenotypes, Functions and Implications for Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 194. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.K.; Bhattacharya, M.; Ferguson, S.S.; Rylett, R.J. Identification of a novelnuclear localization signal common to 69- and 82-kDa human choline acetyltrans-ferase. J. Biol. Chem. 2003, 278, 20217–20224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baazaoui, N.; Iqbal, K. Alzheimer’s Disease: Challenges and a Therapeutic Opportunity to Treat It with a Neurotrophic Compound. Biomolecules 2022, 12, 1409. [Google Scholar] [CrossRef] [PubMed]
- Filiou, M.D.; Arefin, A.S.; Moscato, P.; Graeber, M.B. ‘Neuroinflammation’ differs categorically from inflammation: Transcriptomes of Alzheimer’s disease, Parkinson’s disease, schizophrenia and inflammatory diseases compared. Neurogenetics 2014, 15, 201–212. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Haure-Mirande, J.V.; Audrain, M.; Ehrlich, M.E.; Gandy, S. Microglial TYROBP/DAP12 in Alzheimer’s disease: Transduction of physiological and pathological signals across TREM2. Mol. Neurodegener. 2022, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, M.I.; Chu, S.; Pierce, A.L.; Brubaker, W.D.; Hauhart, R.E.; Mastroeni, D.; Clarke, E.V.; Rogers, J.; Atkinson, J.P.; Tenner, A.J. Analysis of the putative role of CR1 in Alzheimer’s disease: Genetic association, expression and function. PLoS ONE 2016, 11, e0149792. [Google Scholar] [CrossRef] [Green Version]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Paresce, D.M.; Chung, H.; Maxfield, F.R. Slow degradation of aggregates of the Alzheimer’s disease amyloid beta-protein by microglial cells. J. Biol. Chem. 1997, 272, 29390–29397. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [Green Version]
- White, C.S.; Lawrence, C.B.; Brough, D.; Rivers-Auty, J. Inflammasomes as therapeutic targets for Alzheimer’s disease. Brain Pathol. 2017, 27, 223–234. [Google Scholar] [CrossRef]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-kB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Morales, I.; Jiménez, J.M.; Mancilla, M.; Maccioni, R.B. Tau oligomers and fibrils induce activation of microglial cells. J. Alzheimers Dis. 2013, 37, 849–856. [Google Scholar] [CrossRef]
- Sekiya, M.; Wang, M.; Fujisaki, N.; Sakakibara, Y.; Quan, X.; Ehrlich, M.E.; De Jager, P.L.; Bennett, D.A.; Schadt, E.E.; Gandy, S.; et al. Integrated biology approach reveals molecular and pathological interactions among Alzheimer’s Aβ42, Tau, TREM2, and TYROBP in Drosophila models. Genome Med. 2018, 10, 26. [Google Scholar] [CrossRef]
- Takahashi, H.; Klein, Z.A.; Bhagat, S.M.; Kaufman, A.C.; Kostylev, M.A.; Ikezu, T.; Strittmatter, S.M. Alzheimer’s Disease Neuroimaging Initiative. Opposing effects of progranulin deficiency on amyloid and tau pathologies via microglial TYROBP network. Acta Neuropathol. 2017, 133, 785–807. [Google Scholar] [CrossRef] [Green Version]
- Finneran, D.J.; Morgan, D.; Gordon, M.N.; Nash, K.R. 3 CNS-Wide over Expression of Fractalkine Improves Cognitive Functioning in a Tauopathy Model. J. Neuroimmune Pharmacol. 2019, 14, 312–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhao, J.; Fan, C.; Zhu, L.; Huang, C.; Li, Q.; Gan, D.; Wen, C.; Chen, M.; Lu, D. Delivery of exogenous proteins by mesenchymal stem cells attenuates early memory deficits in a murine model of Alzheimer’s disease. Neurobiol. Aging 2020, 86, 81–91. [Google Scholar] [CrossRef]
- Bemiller, S.M.; Maphis, N.M.; Formica, S.V.; Wilson, G.N.; Miller, C.M.; Xu, G.; Kokiko-Cochran, O.N.; Kim, K.-W.; Jung, S.; Cannon, J.L.; et al. Genetically enhancing the expression of chemokine domain of CX3CL1 fails to prevent tau pathology in mouse models of tauopathy. J. Neuroinflammation 2018, 15, 278. [Google Scholar] [CrossRef] [Green Version]
- Bolos, M.; Llorens-Martin, M.; Perea, J.R.; Jurado-Arjona, J.; Rabano, A.; Hernandez, F.; Avila, J. Absence of CX3CR1 impairs the internalization of tau by microglia. Mol. Neurodegener 2017, 12, 59. [Google Scholar] [CrossRef] [Green Version]
- Perea, J.R.; Lleo, A.; Alcolea, D.; Fortea, J.; Avila, J.; Bolos, M. Decreased CX3CL1 levels in the cerebrospinal fluid of patients with Alzheimer’s disease. Front. Neurosci. 2018, 12, 609. [Google Scholar] [CrossRef] [Green Version]
- Guedes, J.R.; Lao, T.; Cardoso, A.L.; El Khoury, J. Roles of Microglial and Monocyte Chemokines and Their Receptors in Regulating Alzheimer’s Disease-Associated Amyloid-β and Tau Pathologies. Front Neurol. 2018, 9, 549. [Google Scholar] [CrossRef] [Green Version]
- Bivona, G.; Iemmolo, M.; Piccoli, T.; Agnello, L.; Lo Sasso, B.; Ciaccio, M.; Ghersi, G. High Cerebrospinal Fluid CX3CL1 Levels in Alzheimer’s Disease Patients but Not in Non-Alzheimer’s Disease Dementia. J. Clin. Med. 2022, 11, 5498. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Xie, H.; Zhang, C.; Wang, T.; Tian, H.; Lu, L.; Xu, J.Y.; Xu, G.T.; Liu, L.; Zhang, J. Enhancing fractalkine/CX3CR1 signalling pathway can reduce neuroinflammation by attenuating microglia activation in experimental diabetic retinopathy. J. Cell. Mol. Med. 2022, 26, 1229–1244. [Google Scholar] [CrossRef] [PubMed]
- Miguel-Álvarez, M.; Santos-Lozano, A.; Sanchis-Gomar, F.; Fiuza-Luces, C.; Pareja-Galeano, H.; Garatachea, N.; Lucia, A. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: A systematic review and meta-analysis of treatment effect. Drugs Aging 2015, 32, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zhou, X.W.; Wang, J.Z. The dual roles of cytokines in Alzheimer’s disease: Update on interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener 2016, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, L.; Ammit, A.J. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 2010, 58, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Shen, X.; Shen, X.; Li, X.; Jin, Z.; Ouyang, X.; Lu, J.; Zhu, D.; Wang, J.; Shen, X. Miltefosine as a PPM1A activator improves AD-like pathology in mice by alleviating tauopathy via microglia/neurons crosstalk. Brain Behav. Immun. Health. 2022, 26, 100546. [Google Scholar] [CrossRef] [PubMed]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.; Sampathkumar, N.K.; Carre, I.; Mondal, M.; Chennell, G.; Vernon, A.C.; Ruepp, M.D.; Mitchell, J.C. Heterozygous expression of the Alzheimer’s disease-protective PLCγ2 P522R variant enhances Aβ clearance while preserving synapses. Cell. Mol. Life Sci. 2022, 79, 453. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Almouhanna, F.; Chausse, B. Interferon γ: A master cytokine in microglia-mediated neural network dysfunction and neurodegeneration. Trends Neurosci. 2022, 45, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Subbarayan, M.S.; Joly-Amado, A.; Bickford, P.C.; Nash, K.R. CX3CL1/CX3CR1 signaling targets for the treatment of neurodegenerative diseases. Pharmacol. Ther. 2022, 231, 107989. [Google Scholar] [CrossRef]
- Cho, S.H.; Sun, B.; Zhou, Y.; Kauppinen, T.M.; Halabisky, B.; Wes, P.; Ransohoff, R.M.; Gan, L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 32713–32722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Xu, G.; Jay, T.R.; Bhatta, S.; Kim, K.W.; Jung, S.; Landreth, G.E.; Ransohoff, R.M.; Lamb, B.T. Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. J. Neurosci. 2014, 34, 12538–12546. [Google Scholar] [CrossRef] [PubMed]


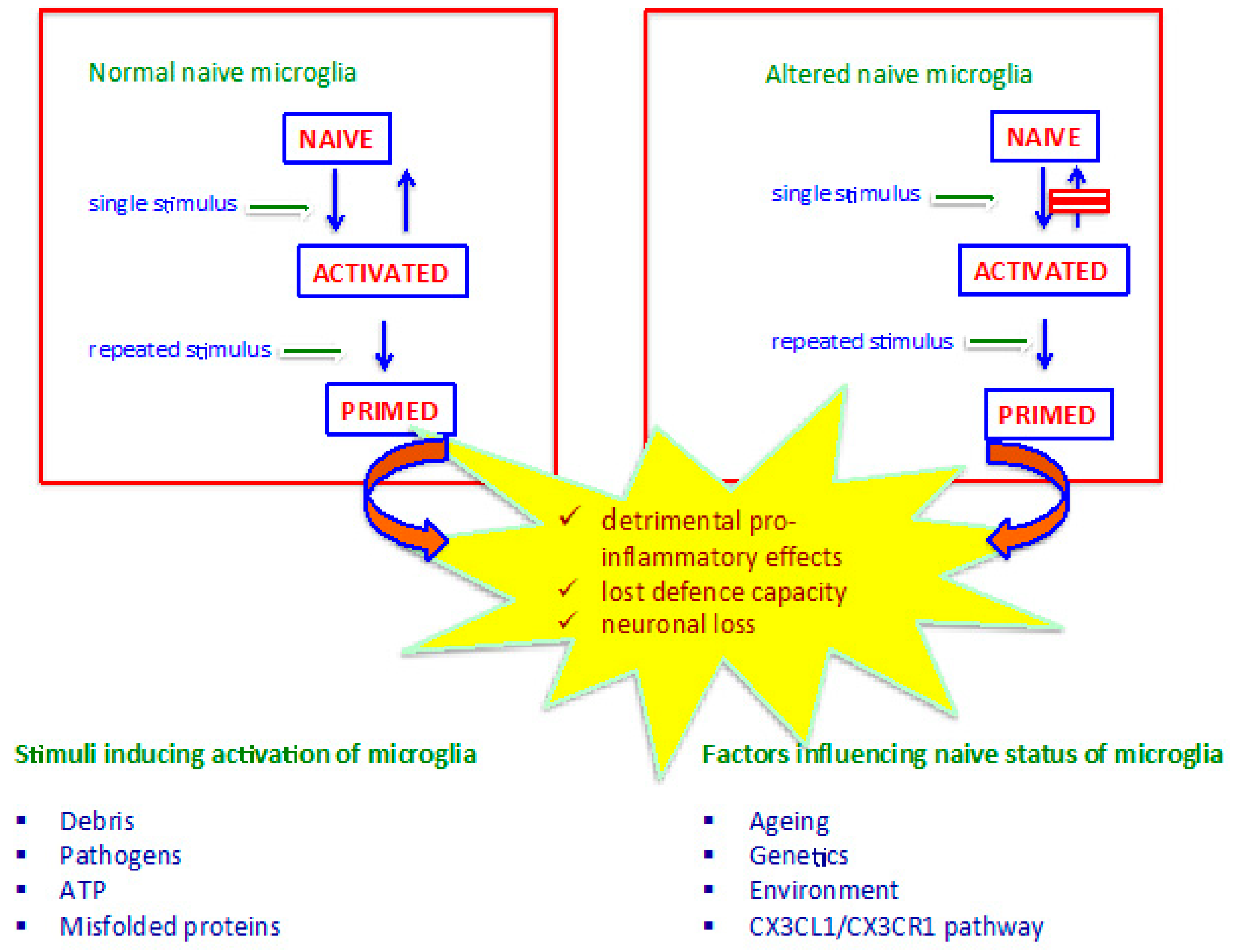{kind=link}
| Phenotype | Main Features |
|---|---|
|
|
|
|
|
|
|
|
|
|
| Activation | Priming |
|---|---|
|
|
|
|
|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bivona, G.; Iemmolo, M.; Agnello, L.; Lo Sasso, B.; Gambino, C.M.; Giglio, R.V.; Scazzone, C.; Ghersi, G.; Ciaccio, M. Microglial Activation and Priming in Alzheimer’s Disease: State of the Art and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 884. https://doi.org/10.3390/ijms24010884
Bivona G, Iemmolo M, Agnello L, Lo Sasso B, Gambino CM, Giglio RV, Scazzone C, Ghersi G, Ciaccio M. Microglial Activation and Priming in Alzheimer’s Disease: State of the Art and Future Perspectives. International Journal of Molecular Sciences. 2023; 24(1):884. https://doi.org/10.3390/ijms24010884
Chicago/Turabian StyleBivona, Giulia, Matilda Iemmolo, Luisa Agnello, Bruna Lo Sasso, Caterina Maria Gambino, Rosaria Vincenza Giglio, Concetta Scazzone, Giulio Ghersi, and Marcello Ciaccio. 2023. "Microglial Activation and Priming in Alzheimer’s Disease: State of the Art and Future Perspectives" International Journal of Molecular Sciences 24, no. 1: 884. https://doi.org/10.3390/ijms24010884