
CRISPR/Cas-Based Approaches to Study Schizophrenia and Other Neurodevelopmental Disorders
 ,
,  , ,
, ,
Abstract
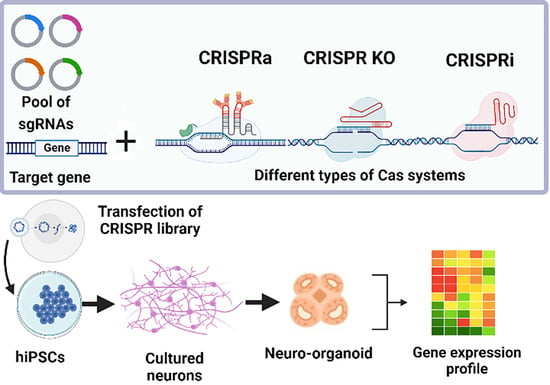
:
1. Introduction
2. Recent Insights into the Genetic Architecture of SZ and Other NDDs
3. Applications of CRISPR-Based Genome Editing Technologies to Study SZ and Other NDDs
4. Epigenetics of SZ and Other NDDs
5. Application of CRISPR-Based Epigenetic Editors to Study SZ and Other NDDs
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garcia-Gutierrez, M.S.; Navarrete, F.; Sala, F.; Gasparyan, A.; Austrich-Olivares, A.; Manzanares, J. Biomarkers in psychiatry: Concept, definition, types and relevance to the clinical reality. Front. Psychiatry 2020, 11, 432. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, C. Genetics in psychiatry: Methods, clinical applications and future perspectives. Psychiatry Clin. Neurosci. Rep. 2022, 1, e6. [Google Scholar] [CrossRef]
- Fuller, T.; Reus, V. Shared genetics of psychiatric disorders. F1000Research 2019, 8, F1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poletti, M.; Raballo, A. Before schizophrenia: Schizophrenic vulnerability in developmental age and its detection. Clin. Neuropsychiatry 2021, 18, 293–295. [Google Scholar]
- Mertens, J.; Wang, Q.W.; Kim, Y.; Yu, D.X.; Pham, S.; Yang, B.; Zheng, Y.; Diffenderfer, K.E.; Zhang, J.; Soltani, S.; et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 2015, 527, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.X.; Di Giorgio, F.P.; Yao, J.; Marchetto, M.C.; Brennand, K.; Wright, R.; Mei, A.; McHenry, L.; Lisuk, D.; Grasmick, J.M.; et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Rep. 2014, 2, 295–310. [Google Scholar] [CrossRef] [Green Version]
- Das, D.; Feuer, K.; Wahbeh, M.; Avramopoulos, D. Modeling psychiatric disorder biology with stem cells. Curr. Psychiatry Rep. 2020, 22, 24. [Google Scholar] [CrossRef]
- Wen, Z.; Nguyen, H.N.; Guo, Z.; Lalli, M.A.; Wang, X.; Su, Y.; Kim, N.S.; Yoon, K.J.; Shin, J.; Zhang, C.; et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014, 515, 414–418. [Google Scholar] [CrossRef] [Green Version]
- Martinez, R.A.; Stein, J.L.; Krostag, A.R.; Nelson, A.M.; Marken, J.S.; Menon, V.; May, R.C.; Yao, Z.; Kaykas, A.; Geschwind, D.H.; et al. Genome engineering of isogenic human ES cells to model autism disorders. Nucleic Acids Res. 2015, 43, e65. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.R.; Gandawijaya, J.; Oguro-Ando, A. A novel method for generating glutamatergic SH-SY5Y neuron-like cells utilizing B-27 supplement. Front. Pharmacol. 2022, 13, 943627. [Google Scholar] [CrossRef]
- Unsicker, C.; Cristian, F.B.; von Hahn, M.; Eckstein, V.; Rappold, G.A.; Berkel, S. SHANK2 mutations impair apoptosis, proliferation and neurite outgrowth during early neuronal differentiation in SH-SY5Y cells. Sci. Rep. 2021, 11, 2128. [Google Scholar] [CrossRef] [PubMed]
- Schrode, N.; Ho, S.M.; Yamamuro, K.; Dobbyn, A.; Huckins, L.; Matos, M.R.; Cheng, E.; Deans, P.J.M.; Flaherty, E.; Barretto, N.; et al. Synergistic effects of common schizophrenia risk variants. Nat. Genet. 2019, 51, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eid, A.; Alshareef, S.; Mahfouz, M.M. CRISPR base editors: Genome editing without double-stranded breaks. Biochem. J. 2018, 475, 1955–1964. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Y.; Birkenshaw, A.; Thomson, T.; Carlaw, T.; Zhang, L.H.; Ross, C.J.D. Increasing the targeting scope of CRISPR base editing system beyond NGG. CRISPR J. 2022, 5, 187–202. [Google Scholar] [CrossRef]
- Hanna, R.E.; Hegde, M.; Fagre, C.R.; DeWeirdt, P.C.; Sangree, A.K.; Szegletes, Z.; Griffith, A.; Feeley, M.N.; Sanson, K.R.; Baidi, Y.; et al. Massively parallel assessment of human variants with base editor screens. Cell 2021, 184, 1064–1080. [Google Scholar] [CrossRef]
- Cuella-Martin, R.; Hayward, S.B.; Fan, X.; Chen, X.; Huang, J.W.; Taglialatela, A.; Leuzzi, G.; Zhao, J.; Rabadan, R.; Lu, C.; et al. Functional interrogation of DNA damage response variants with base editing screens. Cell 2021, 184, 1081–1097. [Google Scholar] [CrossRef]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef]
- Hutchinson, A.; Asimit, J.; Wallace, C. Fine-mapping genetic associations. Hum. Mol. Genet. 2020, 29, R81–R88. [Google Scholar] [CrossRef]
- Trubetskoy, V.; Pardinas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef]
- Shimamoto-Mitsuyama, C.; Nakaya, A.; Esaki, K.; Balan, S.; Iwayama, Y.; Ohnishi, T.; Maekawa, M.; Toyota, T.; Dean, B.; Yoshikawa, T. Lipid pathology of the corpus callosum in schizophrenia and the potential role of abnormal gene regulatory networks with reduced microglial marker expression. Cereb. Cortex 2021, 31, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Karmakar, S.; Prasad, M.; Mandal, A.K. Praja1 ubiquitin ligase facilitates degradation of polyglutamine proteins and suppresses polyglutamine-mediated toxicity. Mol. Biol. Cell 2021, 32, 1579–1593. [Google Scholar] [CrossRef] [PubMed]
- Watabe, K.; Kato, Y.; Sakuma, M.; Murata, M.; Niida-Kawaguchi, M.; Takemura, T.; Hanagata, N.; Tada, M.; Kakita, A.; Shibata, N. Praja1 RING-finger E3 ubiquitin ligase suppresses neuronal cytoplasmic TDP-43 aggregate formation. Neuropathology 2020, 40, 570–586. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Suzuki, T.; Raveau, M.; Miyake, N.; Sudo, G.; Tsurusaki, Y.; Watanabe, T.; Sugaya, Y.; Tatsukawa, T.; Mazaki, E.; et al. A recurrent PJA1 variant in trigonocephaly and neurodevelopmental disorders. Ann. Clin. Transl. Neurol. 2020, 7, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.T.; Hinds, D.A.; Tung, J.Y.; Franz, C.; Fan, C.C.; Wang, Y.; Smeland, O.B.; Schork, A.; Holland, D.; Kauppi, K.; et al. Genome-wide analyses for personality traits identify six genomic loci and show correlations with psychiatric disorders. Nat. Genet. 2017, 49, 152–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, T.A.; Akiskal, H.S.; Akiskal, K.K.; Bipolar Genome, S.; Kelsoe, J.R. Genome-wide association study of temperament in bipolar disorder reveals significant associations with three novel Loci. Biol. Psychiatry 2012, 72, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Chauquet, S.; Zhu, Z.; O’Donovan, M.C.; Walters, J.T.R.; Wray, N.R.; Shah, S. Association of Antihypertensive Drug Target Genes with Psychiatric Disorders: A Mendelian Randomization Study. JAMA Psychiatry 2021, 78, 623–631. [Google Scholar] [CrossRef]
- Cosarderelioglu, C.; Nidadavolu, L.S.; George, C.J.; Oh, E.S.; Bennett, D.A.; Walston, J.D.; Abadir, P.M. Brain Renin-Angiotensin System at the Intersect of Physical and Cognitive Frailty. Front. Neurosci. 2020, 14, 586314. [Google Scholar] [CrossRef]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362, eaat8127. [Google Scholar] [CrossRef] [Green Version]
- Maritzen, T.; Koo, S.J.; Haucke, V. Turning CALM into excitement: AP180 and CALM in endocytosis and disease. Biol. Cell. 2012, 104, 588–602. [Google Scholar] [CrossRef]
- Galvan, L.; Francelle, L.; Gaillard, M.C.; de Longprez, L.; Carrillo-de Sauvage, M.A.; Liot, G.; Cambon, K.; Stimmer, L.; Luccantoni, S.; Flament, J.; et al. The striatal kinase DCLK3 produces neuroprotection against mutant huntingtin. Brain 2018, 141, 1434–1454. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Bray, N.J. Schizophrenia Genomics: Convergence on Synaptic Development, Adult Synaptic Plasticity, or Both? Biol. Psychiatry 2022, 91, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.F.; Daly, M.J.; O’Donovan, M. Genetic architectures of psychiatric disorders: The emerging picture and its implications. Nat. Rev. Genet. 2012, 13, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genomics, C.; Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984–994. [Google Scholar]
- Woo, H.J.; Yu, C.; Kumar, K.; Reifman, J. Large-scale interaction effects reveal missing heritability in schizophrenia, bipolar disorder and posttraumatic stress disorder. Transl. Psychiatry 2017, 7, e1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, T.; Poterba, T.; Curtis, D.; Akil, H.; Al Eissa, M.; Barchas, J.D.; Bass, N.; Bigdeli, T.B.; Breen, G.; Bromet, E.J.; et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 2022, 604, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Hsu, S.H.; Tsai, H.Y.; Cheng, F.Y.; Cheng, M.C. Transcriptomic and Proteomic Analysis of CRISPR/Cas9-Mediated ARC-Knockout HEK293 Cells. Int. J. Mol. Sci. 2022, 23, 4498. [Google Scholar] [CrossRef]
- Wang, J.; Li, S.; Li, X.; Liu, J.; Yang, J.; Li, Y.; Li, W.; Yang, Y.; Li, J.; Chen, R.; et al. Functional variant rs2270363 on 16p13.3 confers schizophrenia risk by regulating NMRAL1. Brain 2020, 145, 2569–2585. [Google Scholar] [CrossRef]
- Li, S.; Li, J.; Liu, J.; Wang, J.; Li, X.; Huo, Y.; Li, Y.; Liu, Y.; Li, M.; Xiao, X.; et al. Regulatory variants at 2q33.1 confer schizophrenia risk by modulating distal gene TYW5 expression. Brain 2022, 145, 770–786. [Google Scholar] [CrossRef]
- Torres-Ruiz, R.; Benitez-Burraco, A.; Martinez-Lage, M.; Rodriguez-Perales, S.; Garcia-Bellido, P. Functional characterization of two enhancers located downstream FOXP2. BMC Med. Genet. 2019, 20, 65. [Google Scholar] [CrossRef]
- Sanders, B.; D’Andrea, D.; Collins, M.O.; Rees, E.; Steward, T.G.J.; Zhu, Y.; Chapman, G.; Legge, S.E.; Pardinas, A.F.; Harwood, A.J. Transcriptional programs regulating neuronal differentiation are disrupted in DLG2 knockout human embryonic stem cells and enriched for schizophrenia and related disorders risk variants. Nat. Commun. 2022, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Ortolano, N.A.; Romero-Morales, A.I.; Rasmussen, M.L.; Bodnya, C.; Kline, L.A.; Joshi, P.; Connelly, J.P.; Rose, K.L.; Pruett-Miller, S.M.; Gama, V. A proteomics approach for the identification of cullin-9 (CUL9) related signaling pathways in induced pluripotent stem cell models. PLoS ONE 2021, 16, e0248000. [Google Scholar] [CrossRef] [PubMed]
- Amador, A.; Bostick, C.D.; Olson, H.; Peters, J.; Camp, C.R.; Krizay, D.; Chen, W.; Han, W.; Tang, W.; Kanber, A.; et al. Modelling and treating GRIN2A developmental and epileptic encephalopathy in mice. Brain 2020, 143, 2039–2057. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Brown, L.A.; Cais, O.; Watson, J.; Clayton, A.J.; Chang, V.T.; Biggs, D.; Preece, C.; Hernandez-Pliego, P.; Krohn, J.; et al. A point mutation in the ion conduction pore of AMPA receptor GRIA3 causes dramatically perturbed sleep patterns as well as intellectual disability. Hum. Mol. Genet. 2017, 26, 3869–3882. [Google Scholar] [CrossRef] [Green Version]
- Linda, K.; Lewerissa, E.I.; Verboven, A.H.A.; Gabriele, M.; Frega, M.; Klein Gunnewiek, T.M.; Devilee, L.; Ulferts, E.; Hommersom, M.; Oudakker, A.; et al. Imbalanced autophagy causes synaptic deficits in a human model for neurodevelopmental disorders. Autophagy 2022, 18, 423–442. [Google Scholar] [CrossRef]
- Wang, S.; Rhijn, J.V.; Akkouh, I.; Kogo, N.; Maas, N.; Bleeck, A.; Ortiz, I.S.; Lewerissa, E.; Wu, K.M.; Schoenmaker, C.; et al. Loss-of-function variants in the schizophrenia risk gene SETD1A alter neuronal network activity in human neurons through the cAMP/PKA pathway. Cell Rep. 2022, 39, 110790. [Google Scholar] [CrossRef]
- Arruda, N.L.; Carico, Z.M.; Justice, M.; Liu, Y.F.; Zhou, J.; Stefan, H.C.; Dowen, J.M. Distinct and overlapping roles of STAG1 and STAG2 in cohesin localization and gene expression in embryonic stem cells. Epigenetics Chromatin. 2020, 13, 32. [Google Scholar] [CrossRef]
- Tuvikene, J.; Esvald, E.E.; Rahni, A.; Uustalu, K.; Zhuravskaya, A.; Avarlaid, A.; Makeyev, E.V.; Timmusk, T. Intronic enhancer region governs transcript-specific Bdnf expression in rodent neurons. Elife 2021, 10, e65161. [Google Scholar] [CrossRef]
- Brighi, C.; Salaris, F.; Soloperto, A.; Cordella, F.; Ghirga, S.; de Turris, V.; Rosito, M.; Porceddu, P.F.; D’Antoni, C.; Reggiani, A.; et al. Novel fragile X syndrome 2D and 3D brain models based on human isogenic FMRP-KO iPSCs. Cell Death Dis. 2021, 12, 498. [Google Scholar] [CrossRef]
- Fair, S.R.; Schwind, W.; Julian, D.; Biel, A.; Guo, G.; Rutherford, R.; Ramadesikan, S.; Westfall, J.; Miller, K.E.; Kararoudi, M.N.; et al. Cerebral organoids containing an AUTS2 missense variant model microcephaly. Brain 2021, awac244. [Google Scholar] [CrossRef]
- Feron, F.; Perry, C.; Hirning, M.H.; McGrath, J.; Mackay-Sim, A. Altered adhesion, proliferation and death in neural cultures from adults with schizophrenia. Schizophr. Res. 1999, 40, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.A.; O’Connell, K.S.; Lekva, T.; Szabo, A.; Akkouh, I.A.; Osete, J.R.; Agartz, I.; Engh, J.A.; Andreou, D.; Boye, B.; et al. Systemic cell adhesion molecules in severe mental illness: Potential role of intercellular CAM-1 in linking peripheral and neuroinflammation. Biol. Psychiatry 2023, 93, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.Q.; Weickert, T.W.; Catts, V.S.; Balzan, R.; Galletly, C.; Liu, D.; O’Donnell, M.; Shannon Weickert, C. Altered levels of immune cell adhesion molecules are associated with memory impairment in schizophrenia and healthy controls. Brain Behav. Immun. 2020, 89, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Komada, M.; Nishimura, Y. Epigenetics and Neuroinflammation Associated with Neurodevelopmental Disorders: A Microglial Perspective. Front. Cell Dev. Biol. 2022, 10, 852752. [Google Scholar] [CrossRef]
- Harper, J.W.; Schulman, B.A. Cullin-RING ubiquitin ligase regulatory circuits: A quarter century beyond the F-Box hypothesis. Annu. Rev. Biochem. 2021, 90, 403–429. [Google Scholar] [CrossRef]
- Pei, X.H.; Bai, F.; Li, Z.; Smith, M.D.; Whitewolf, G.; Jin, R.; Xiong, Y. Cytoplasmic CUL9/PARC ubiquitin ligase is a tumor suppressor and promotes p53-dependent apoptosis. Cancer Res. 2011, 71, 2969–2977. [Google Scholar] [CrossRef] [Green Version]
- Fazia, T.; Marzanati, D.; Carotenuto, A.L.; Beecham, A.; Hadjixenofontos, A.; McCauley, J.L.; Saddi, V.; Piras, M.; Bernardinelli, L.; Gentilini, D. Homozygosity Haplotype and Whole-Exome Sequencing Analysis to Identify Potentially Functional Rare Variants Involved in Multiple Sclerosis among Sardinian Families. Curr. Issues Mol. Biol. 2021, 43, 1778–1793. [Google Scholar] [CrossRef]
- Cuadrado, A.; Losada, A. Specialized functions of cohesins STAG1 and STAG2 in 3D genome architecture. Curr. Opin. Genet. Dev. 2020, 61, 9–16. [Google Scholar] [CrossRef]
- Di Muro, E.; Palumbo, P.; Benvenuto, M.; Accadia, M.; Di Giacomo, M.C.; Manieri, S.; Abate, R.; Tagliente, M.; Castellana, S.; Mazza, T.; et al. Novel STAG1 Frameshift mutation in a patient affected by a syndromic form of neurodevelopmental disorder. Genes 2021, 12, 1116. [Google Scholar] [CrossRef]
- Soardi, F.C.; Machado-Silva, A.; Linhares, N.D.; Zheng, G.; Qu, Q.; Pena, H.B.; Martins, T.M.M.; Vieira, H.G.S.; Pereira, N.B.; Melo-Minardi, R.C.; et al. Familial STAG2 germline mutation defines a new human cohesinopathy. NPJ Genom. Med. 2017, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Zang, W.; Zheng, X. Structure and functions of cellular redox sensor HSCARG/NMRAL1, a linkage among redox status, innate immunity, DNA damage response, and cancer. Free. Radic Biol. Med. 2020, 160, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Zhou, J.; Wong, A.K.; Chen, K.M.; Theesfeld, C.L.; Darnell, R.B.; Troyanskaya, O.G. Genome-wide landscape of RNA-binding protein target site dysregulation reveals a major impact on psychiatric disorder risk. Nat. Genet. 2021, 53, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, X.; Zhao, L.; Liang, R.; Deng, W.; Guo, W.; Wang, Q.; Hu, X.; Du, X.; Sham, P.C.; et al. Comprehensive and integrative analyses identify TYW5 as a schizophrenia risk gene. BMC Med. 2022, 20, 169. [Google Scholar] [CrossRef] [PubMed]
- Rehbach, K.; Fernando, M.B.; Brennand, K.J. Integrating CRISPR Engineering and hiPSC-Derived 2D Disease Modeling Systems. J. Neurosci. 2020, 40, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Sidhaye, J.; Knoblich, J.A. Brain organoids: An ensemble of bioassays to investigate human neurodevelopment and disease. Cell Death Differ. 2021, 28, 52–67. [Google Scholar] [CrossRef]
- Richter, J.D.; Zhao, X. The molecular biology of FMRP: New insights into fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef]
- Sunamura, N.; Iwashita, S.; Enomoto, K.; Kadoshima, T.; Isono, F. Loss of the fragile X mental retardation protein causes aberrant differentiation in human neural progenitor cells. Sci. Rep. 2018, 8, 11585. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.H.F.; Lai, T.K.Y.; Su, P.; Liu, F. Altered cortical Cytoarchitecture in the Fmr1 knockout mouse. Mol. Brain 2019, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Zhou, Y.; Li, Y.; Han, Y.; Xu, J.; Niu, W.; Li, Z.; Liu, S.; Feng, H.; Huang, W.; et al. A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat. Neurosci. 2021, 24, 1377–1391. [Google Scholar] [CrossRef]
- Kim, Y.J.; Khoshkhoo, S.; Frankowski, J.C.; Zhu, B.; Abbasi, S.; Lee, S.; Wu, Y.E.; Hunt, R.F. Chd2 Is Necessary for Neural Circuit Development and Long-Term Memory. Neuron 2018, 100, 1180–1193. [Google Scholar] [CrossRef] [Green Version]
- De Maria, B.; Balestrini, S.; Mei, D.; Melani, F.; Pellacani, S.; Pisano, T.; Rosati, A.; Scaturro, G.M.; Giordano, L.; Cantalupo, G.; et al. Expanding the genetic and phenotypic spectrum of CHD2-related disease: From early neurodevelopmental disorders to adult-onset epilepsy. Am. J. Med. Genet. A 2022, 188, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.Y.; Ju, X.C.; Li, Y.; Zeng, P.M.; Wu, J.; Zhou, Y.Y.; Shen, L.B.; Dong, J.; Chen, Y.J.; Luo, Z.G. Generation of vascularized brain organoids to study neurovascular interactions. Elife 2022, 11, e76707. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.; An, J.H.; Yang, H.J.; Lee, D.G.; Kim, J.; Koh, H.; Park, Y.H.; Song, B.S.; Sim, B.W.; Lee, H.J.; et al. Human Blood Vessel Organoids Penetrate Human Cerebral Organoids and Form a Vessel-Like System. Cells 2021, 10, 2036. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N. Assembloids. Nat. Methods 2021, 18, 27. [Google Scholar] [CrossRef]
- Miura, Y.; Li, M.Y.; Revah, O.; Yoon, S.J.; Narazaki, G.; Pasca, S.P. Engineering brain assembloids to interrogate human neural circuits. Nat. Protoc. 2022, 17, 15–35. [Google Scholar] [CrossRef]
- Abbott, A.E.; Linke, A.C.; Nair, A.; Jahedi, A.; Alba, L.A.; Keown, C.L.; Fishman, I.; Muller, R.A. Repetitive behaviors in autism are linked to imbalance of corticostriatal connectivity: A functional connectivity MRI study. Soc. Cogn. Affect. Neurosci. 2018, 13, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Blazer, A.; Chengappa, K.N.R.; Foran, W.; Parr, A.C.; Kahn, C.E.; Luna, B.; Sarpal, D.K. hanges in corticostriatal connectivity and striatal tissue iron associated with efficacy of clozapine for treatmentresistant schizophrenia. Psychopharmacology 2022, 239, 2503–2514. [Google Scholar] [CrossRef]
- Guo, W.; Wang, H.; Kumar, T.A.; Couthouis, J.; Braems, E.; Masrori, P.; van Schoor, E.; Fan, Y.; Ahuja, K.; Moisse, M.; et al. CRISPR/Cas9 screen in human iPSC-derived cortical neurons identifies NEK6 as a novel disease modifier of C9orf72 poly(PR) toxicity. Alzheimers Dement. 2022. [Google Scholar]
- Meng, X.; Yao, D.; Chen, X.; Kelley, K.W.; Reis, N.; Thete, M.V.; Kulkarni, S.; Bassik, M.C.; Pașca, S.P. CRISPR screens in 3D assembloids reveal disease genes associated with human interneuron development. bioRxiv 2022, 506845. [Google Scholar] [CrossRef]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Burmeister, M.; McInnis, M.G.; Zollner, S. Psychiatric genetics: Progress amid controversy. Nat. Rev. Genet. 2008, 9, 527–540. [Google Scholar] [CrossRef] [PubMed]
- van Calker, D.; Serchov, T. The “missing heritability”-Problem in psychiatry: Is the interaction of genetics, epigenetics and transposable elements a potential solution? Neurosci. Biobehav. Rev. 2021, 126, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Griffin, A.; Mahesh, A.; Tiwari, V.K. Disruption of the gene regulatory programme in neurodevelopmental disorders. Biochim. Biophys Acta. Gene Regul. Mech. 2022, 1865, 194860. [Google Scholar] [CrossRef] [PubMed]
- Gopinathan, G.; Diekwisch, T.G.H. Epigenetics and Early Development. J. Dev. Biol. 2022, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Li, D.; Jin, W.; Shi, Y.; Li, Z.; Ma, P.; Sun, J.; Chen, S.; Li, P.; Lin, P. Research Progress on the Correlation between Epigenetics and Schizophrenia. Front. Neurosci. 2021, 15, 688727. [Google Scholar] [CrossRef]
- Davies, M.N.; Volta, M.; Pidsley, R.; Lunnon, K.; Dixit, A.; Lovestone, S.; Coarfa, C.; Harris, R.A.; Milosavljevic, A.; Troakes, C.; et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012, 13, R43. [Google Scholar] [CrossRef] [Green Version]
- Olova, N.; Krueger, F.; Andrews, S.; Oxley, D.; Berrens, R.V.; Branco, M.R.; Reik, W. Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome Biol. 2018, 19, 33. [Google Scholar] [CrossRef] [Green Version]
- Perzel Mandell, K.A.; Eagles, N.J.; Wilton, R.; Price, A.J.; Semick, S.A.; Collado-Torres, L.; Ulrich, W.S.; Tao, R.; Han, S.; Szalay, A.S.; et al. Genome-wide sequencing-based identification of methylation quantitative trait loci and their role in schizophrenia risk. Nat. Commun. 2021, 12, 5251. [Google Scholar] [CrossRef]
- Yu, H.; Cheng, W.; Zhang, X.; Wang, X.; Yue, W. Integration analysis of methylation quantitative trait loci and GWAS identify three schizophrenia risk variants. Neuropsychopharmacology 2020, 45, 1179–1187. [Google Scholar] [CrossRef]
- Deneris, E.S.; Wyler, S.C. Serotonergic transcriptional networks and potential importance to mental health. Nat. Neurosci. 2012, 15, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.L.; Spencer, W.C.; Tabuchi, N.; Kitt, M.M.; Deneris, E.S. Reorganization of postmitotic neuronal chromatin accessibility for maturation of serotonergic identity. Elife 2022, 11, e75970. [Google Scholar] [CrossRef] [PubMed]
- Heavner, W.E.; Smith, S.E.P. Resolving the Synaptic versus Developmental Dichotomy of Autism Risk Genes. Trends Neurosci. 2020, 43, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Mazille, M.; Scheiffele, P.; Mauger, O. Cue-specific remodeling of the neuronal transcriptome through intron retention programs. bioRxiv 2021, 463312. [Google Scholar] [CrossRef]
- Mews, P.; Calipari, E.S.; Day, J.; Lobo, M.K.; Bredy, T.; Abel, T. From circuits to chromatin: The emerging role of epigenetics in mental health. J. Neurosci. 2021, 41, 873–882. [Google Scholar] [CrossRef]
- Lepack, A.E.; Werner, C.T.; Stewart, A.F.; Fulton, S.L.; Zhong, P.; Farrelly, L.A.; Smith, A.C.W.; Ramakrishnan, A.; Lyu, Y.; Bastle, R.M.; et al. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 2020, 368, 197–201. [Google Scholar] [CrossRef]
- Jansen, R.; Penninx, B.W.; Madar, V.; Xia, K.; Milaneschi, Y.; Hottenga, J.J.; Hammerschlag, A.R.; Beekman, A.; van der Wee, N.; Smit, J.H.; et al. Gene expression in major depressive disorder. Mol. Psychiatry 2016, 21, 339–347. [Google Scholar] [CrossRef]
- Jaffe, A.E.; Straub, R.E.; Shin, J.H.; Tao, R.; Gao, Y.; Collado-Torres, L.; Kam-Thong, T.; Xi, H.S.; Quan, J.; Chen, Q.; et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat. Neurosci. 2018, 21, 1117–1125. [Google Scholar] [CrossRef]
- Boks, M.P.; Houtepen, L.C.; Xu, Z.; He, Y.; Ursini, G.; Maihofer, A.X.; Rajarajan, P.; Yu, Q.; Xu, H.; Wu, Y.; et al. Genetic vulnerability to DUSP22 promoter hypermethylation is involved in the relation between in utero famine exposure and schizophrenia. NPJ Schizophr. 2018, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- McKinney, B.C.; McClain, L.L.; Hensler, C.M.; Wei, Y.; Klei, L.; Lewis, D.A.; Devlin, B.; Wang, J.; Ding, Y.; Sweet, R.A. Schizophrenia-associated differential DNA methylation in brain is distributed across the genome and annotated to MAD1L1, a locus at which DNA methylation and transcription phenotypes share genetic variation with schizophrenia risk. Transl. Psychiatry 2022, 12, 340. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Imbeault, M.; Helleboid, P.Y.; Trono, D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017, 543, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Alerasool, N.; Segal, D.; Lee, H.; Taipale, M. An efficient KRAB domain for CRISPRi applications in human cells. Nat Methods. 2020, 17, 1093–1096. [Google Scholar] [PubMed]
- Yeo, N.C.; Chavez, A.; Lance-Byrne, A.; Chan, Y.; Menn, D.; Milanova, D.; Kuo, C.C.; Guo, X.; Sharma, S.; Tung, A.; et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 2018, 15, 611–616. [Google Scholar] [CrossRef]
- Ding, L.; Schmitt, L.T.; Brux, M.; Surun, D.; Augsburg, M.; Lansing, F.; Mircetic, J.; Theis, M.; Buchholz, F. DNA methylation-independent long-term epigenetic silencing with dCRISPR/Cas9 fusion proteins. Life Sci. Alliance 2022, 5, e202101321. [Google Scholar] [CrossRef] [PubMed]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- O’Geen, H.; Bates, S.L.; Carter, S.S.; Nisson, K.A.; Halmai, J.; Fink, K.D.; Rhie, S.K.; Farnham, P.J.; Segal, D.J. Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenetics Chromatin. 2019, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- O’Geen, H.; Tomkova, M.; Combs, J.A.; Tilley, E.K.; Segal, D.J. Determinants of heritable gene silencing for KRAB-dCas9 + DNMT3 and Ezh2-dCas9 + DNMT3 hit-and-run epigenome editing. Nucleic Acids Res. 2022, 50, 3239–3253. [Google Scholar] [CrossRef]
- Nakamura, M.; Ivec, A.E.; Gao, Y.; Qi, L.S. Durable CRISPR-Based Epigenetic Silencing. BioDesign Res. 2021, 2021, 9815820. [Google Scholar] [CrossRef]
- Moses, C.; Hodgetts, S.I.; Nugent, F.; Ben-Ary, G.; Park, K.K.; Blancafort, P.; Harvey, A.R. Transcriptional repression of PTEN in neural cells using CRISPR/dCas9 epigenetic editing. Sci. Rep. 2020, 10, 11393. [Google Scholar] [CrossRef]
- Carullo, N.V.N.; Hinds, J.E.; Revanna, J.S.; Tuscher, J.J.; Bauman, A.J.; Day, J.J. A Cre-Dependent CRISPR/dCas9 System for Gene Expression Regulation in Neurons. eNeuro 2021, 8. [Google Scholar] [CrossRef]
- Duke, C.G.; Bach, S.V.; Revanna, J.S.; Sultan, F.A.; Southern, N.T.; Davis, M.N.; Carullo, N.V.N.; Bauman, A.J.; Phillips, R.A., 3rd; Day, J.J. An Improved CRISPR/dCas9 Interference Tool for Neuronal Gene Suppression. Front. Genome Ed. 2020, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Colasante, G.; Lignani, G.; Brusco, S.; Di Berardino, C.; Carpenter, J.; Giannelli, S.; Valassina, N.; Bido, S.; Ricci, R.; Castoldi, V.; et al. dCas9-Based Scn1a Gene Activation Restores Inhibitory Interneuron Excitability and Attenuates Seizures in Dravet Syndrome Mice. Mol. Ther. 2020, 28, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Ortega, J.A.; Quinlan, K.A.; Santos, D.P.; Gu, H.; Martin, E.J.; Galonska, C.; Pop, R.; Maidl, S.; Di Pardo, A.; et al. Dissecting the Functional Consequences of De Novo DNA Methylation Dynamics in Human Motor Neuron Differentiation and Physiology. Cell Stem Cell. 2018, 22, 559–574. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Li, A.; Sekiya, M.; Beckmann, N.D.; Quan, X.; Schrode, N.; Fernando, M.B.; Yu, A.; Zhu, L.; Cao, J.; et al. Transformative Network Modeling of Multi-omics Data Reveals Detailed Circuits, Key Regulators, and Potential Therapeutics for Alzheimer’s Disease. Neuron 2021, 109, 257–272. [Google Scholar] [CrossRef]
- Ho, S.M.; Hartley, B.J.; Flaherty, E.; Rajarajan, P.; Abdelaal, R.; Obiorah, I.; Barretto, N.; Muhammad, H.; Phatnani, H.P.; Akbarian, S.; et al. Evaluating Synthetic Activation and Repression of Neuropsychiatric-Related Genes in hiPSC-Derived NPCs, Neurons, and Astrocytes. Stem Cell Rep. 2017, 9, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Shen, W.; Zhang, J.; Yang, B.; Liu, Y.N.; Qi, H.; Yu, X.; Lu, S.Y.; Chen, Y.; Xu, Y.Z.; et al. CRISPR interference-based specific and efficient gene inactivation in the brain. Nat. Neurosci. 2018, 21, 447–454. [Google Scholar] [CrossRef]
- Papes, F.; Camargo, A.P.; de Souza, J.S.; Carvalho, V.M.A.; Szeto, R.A.; LaMontagne, E.; Teixeira, J.R.; Avansini, S.H.; Sanchez-Sanchez, S.M.; Nakahara, T.S.; et al. Transcription Factor 4 loss-of-function is associated with deficits in progenitor proliferation and cortical neuron content. Nat. Commun. 2022, 13, 2387. [Google Scholar] [CrossRef]
- Casas-Mollano, J.A.; Zinselmeier, M.H.; Erickson, S.E.; Smanski, M.J. CRISPR-Cas Activators for Engineering Gene Expression in Higher Eukaryotes. CRISPR J. 2020, 3, 350–364. [Google Scholar] [CrossRef]
- Chakravarti, R.; Lenka, S.K.; Gautam, A.; Singh, R.; Ravichandiran, V.; Roy, S.; Ghosh, D. A Review on CRISPR-mediated Epigenome Editing: A Future Directive for Therapeutic Management of Cancer. Curr. Drug Targets 2022, 23, 836–853. [Google Scholar]
- Pacesa, M.; Lin, C.H.; Clery, A.; Saha, A.; Arantes, P.R.; Bargsten, K.; Irby, M.J.; Allain, F.H.; Palermo, G.; Cameron, P.; et al. Structural basis for Cas9 off-target activity. Cell 2022, 185, 4067–4081. [Google Scholar] [CrossRef]
- Teixeira, J.R.; Szeto, R.A.; Carvalho, V.M.A.; Muotri, A.R.; Papes, F. Transcription factor 4 and its association with psychiatric disorders. Transl. Psychiatry 2021, 11, 19. [Google Scholar] [CrossRef]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Gachechiladze, M.A.; Ludwig, C.H.; Laurie, M.T.; Hong, J.Y.; Nathaniel, D.; Prabhu, A.V.; Fernandopulle, M.S.; Patel, R.; Abshari, M.; et al. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron 2019, 104, 239–255. [Google Scholar] [CrossRef]
- Kim, Y.S.; Choi, J.; Yoon, B.E. Neuron-Glia Interactions in Neurodevelopmental Disorders. Cells 2020, 9, 2176. [Google Scholar] [CrossRef]
- Vallee, A. Neuroinflammation in Schizophrenia: The Key Role of the WNT/beta-Catenin Pathway. Int. J. Mol. Sci. 2022, 23, 2810. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Leng, K.; Rose, I.V.L.; Kim, H.; Xia, W.; Romero-Fernandez, W.; Rooney, B.; Koontz, M.; Li, E.; Ao, Y.; Wang, S.; et al. CRISPRi screens in human iPSC-derived astrocytes elucidate regulators of distinct inflammatory reactive states. Nat. Neurosci. 2022, 25, 1528–1542. [Google Scholar] [CrossRef]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Townsley, K.G.; Li, A.; Deans, P.M.; Fullard, J.F.; Yu, A.; Cartwright, S.; Zhang, W.; Wang, M.; Voloudakis, G.; Girdhar, K.; et al. Convergent impact of schizophrenia risk genes. bioRxiv 2022, 486286. [Google Scholar] [CrossRef]


{kind=link}
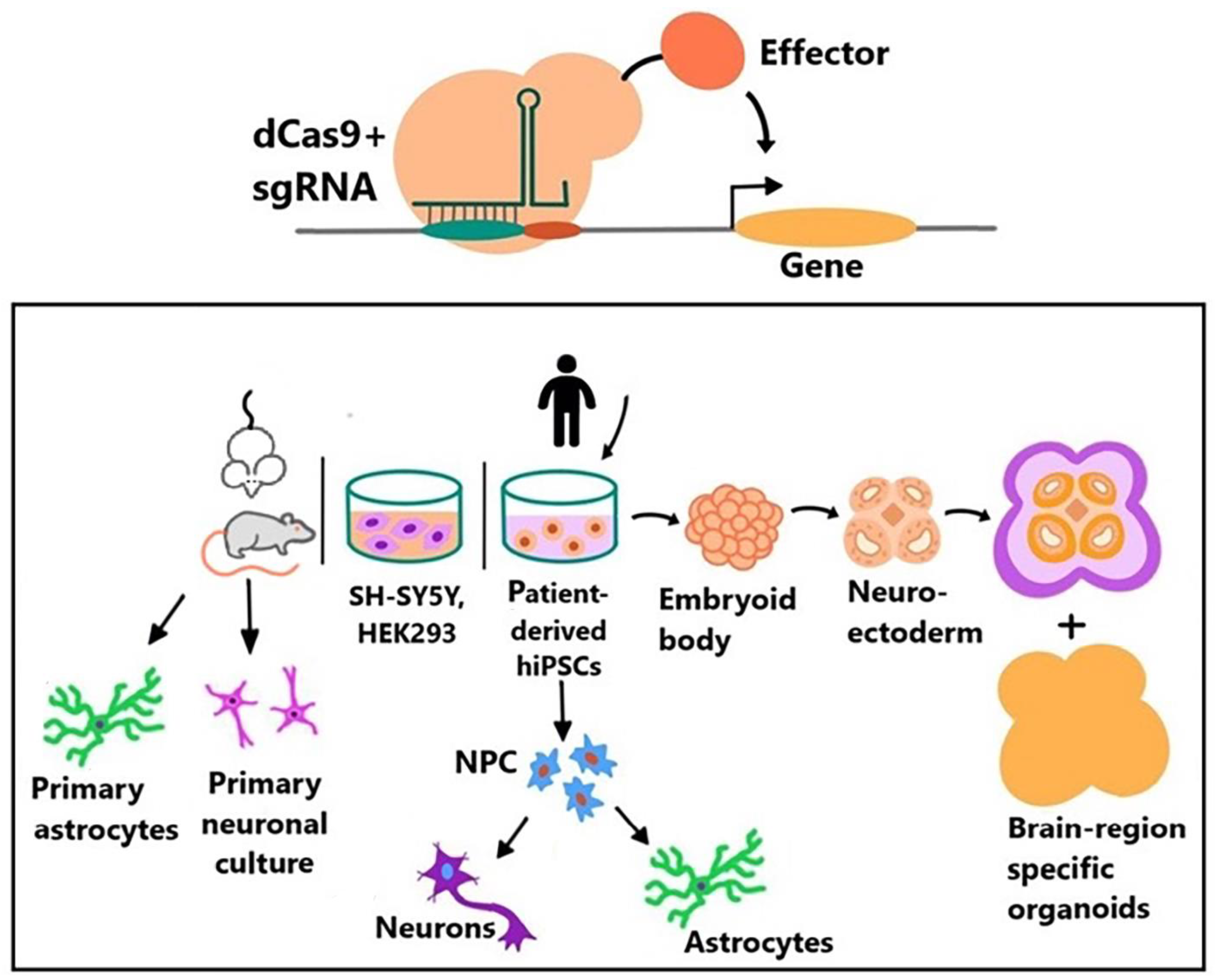{kind=link}
| Gene/SNP | Genome Editor/Genetic Modification | Molecular/Phenotypic Changes | References |
|---|---|---|---|
| ARC | CRISPR/Cas9/gene knockout | Differentially expressed genes and proteins are linked to the extracellular matrix and synapse function | [37] |
| rs2270363 at 16p13.3 | CRISPR/Cas9/deletion in HEK-293T, SH-SY5Y, SK-N-SH, mouse NSCs and rat primary cortical neurons | SNP decreases the expression of NMRAL1, which leads to impaired proliferation and differentiation of NSCs and significantly reduces the density of dendritic spikes on neurons. | [38] |
| rs796364 and rs281759 at 2q33.1 | CRISPR/Cas9/deletion in HEK-293T, SH-SY5Y, SK-N-SH, mouse NSCs and rat primary cortical neurons | SNPs involved in the regulation of TYW5. Deregulation of TYW5 leads to defects in NSC proliferation and differentiation as well as dendritic spin density in neurons | [39] |
| FOXP2 | CRISPR/Cas9/deletion two downstream enhancers in SK-N-MC and HEK293 | Deletion of either of the two enhancers reduced expression of FOXP2 and its targets in SK-N-MC, but no significant changes were observed in HEK293. | [40] |
| DLG2 | CRISPR/Cas9/gene knockout in hESCs | DGL2 deletion causes deregulation of several transcriptional programs, resulting in delayed neurogenesis, abnormal morphology, migration and action potential generation of differentiated cortical neurons | [41] |
| CUL9 | CRISPR/Cas9/single-nucleotide insertions in CUL9 leading to frame shift mutations | Deletion or depletion of the CUL9 protein in hiPSCs causes aberrant neuronal rosette formation in an in vitro model of early neuralization | [42] |
| FURIN | CRISPR/Cas9/allelic conversion from rs4702 AA to GG | NGN2-induced neurons carrying rs4702 GG showed significantly shorter neurite length and significantly shorter average burst duration compared to isogenic controls | [12] |
| GRIN2A | CRISPR/Cas9/A>G nucleotide mutation leading to S644G substitution | Homozygous and heterozygous mutant mice exhibited altered hippocampal morphology at 2 weeks of age, and all homozygotes exhibited lethal tonic-clonic seizures by mid-third week. Heterozygous adults exhibited susceptibility to induced generalized seizures, hyperactivity, repetitive and reduced anxious behavior. | [43] |
| Gria3 | CRISPR/Cas9/an orthologous mutation A647T in the mouse Gria3 gene | The mutation results in an occlusion of the pore in the channel and a deficit in the activation of the corresponding ionotropic glutamate receptor. The mutant mice exhibited slight changes in sleep and activity patterns, as well as increased sensitivity to constant light. | [44] |
| KANSL1 | CRISPR/Cas9/a frameshift mutation leading to a premature stop codon in exon 2 | KANSL1 (KAT8 Regulatory NSL Complex Subunit 1) deficiency is associated with increased oxidative stress and autophagy in iPSCs and iNeurons, resulting in reduced synaptic connectivity and neuronal activity both at the individual cell and network level. The observed neuronal phenotype can be restored by treatment with the antioxidant apocynin. | [45] |
| SETD1A | CRISPR/Cas9/heterozygous indels in exon 7 in iPSC-derived glutamatergic/GABAergic neuronal cultures | SETD1A loss-of-function mutations result in a morphological increase in dendrite complexity and a functional increase in bursting activity. | [46] |
| Stag1 and Stag2 | CRISPR/Cas9/Stag1 or Stag2 gene knockouts in mESC | Inactivation of each of these genes causes a severe depletion of cohesin in chromatin, followed by widespread transcriptome dysregulation and reduced cell proliferation. | [47] |
| Bdnf | CRISPR/Cas9/intronic enhancer deletion in mESC | Strongly reduces both basal and stimulus-dependent levels of exon I-, IIc- and III-containing transcripts of Bdnf | [48] |
| FMR1 | CRISPR/Cas9/gene knockout | FMR1-KO organoids show increased size and number of astrocytes | [49] |
| AUTS2 (Autism Susceptibility Gene 2) | CRISPR/Cas9/correction of the de novo missense mutation c.1600 A>C | Restoration of NPCs proliferative activity and cerebral organoid growth | [50] |
| Target | Epigenome Editor/Model | Observations | References |
|---|---|---|---|
| PTEN | CRISPR/dCas-KRAB/9HEK293T, hIPSC-derived neurons | Efficient PTEN repression causes increased lengths of neurites | [109] |
| GRM2, Tent5b, Fos, Sstr2 and Gadd45b | SVI-DIO-dCas9-VPR, SVI-DIO-dCas9-KRAB-MeCP2/HEK293, rat primary neurons | Improved CRE-dependent CRISPR activator targeting with no leaky gene induction | [110] |
| BDNF | dCas9-KRAB or VP64-dCas9-VP64/rat primary cortical astrocytes and neurons | Novel intronic enhancer controlling the expression of neuron-specific Bdnf transcripts was identified | [48] |
| BDNF | dCas9-KRAB-MeCP2/primary rat hippocampal neuron culture | An improved epigenetic editor provides transcript-selective suppression of Bdnf at near-knockout levels | [111] |
| SCN1A | dCas9-VP160/primary hippocampal neurons | Upregulation of the SCN1A gene and subsequent increase in Nav1.1 protein level in primary Dravet neurons, which is sufficient to restore the firing rate of GAD67+ GABAergic Dravet interneurons | [112] |
| TSNARE1 and SNAP91 | dCas9-VPR and dCas9-KRAB/NGN2-induced neurons | SNAP91 and TSNARE1 deregulation leads to a reduction in synaptic puncta number and size and reciprocal changes in spontaneous excitatory postsynaptic currents | [12] |
| FMR1 | dCas9-Tet1/FXS patient-derived iPSCs, neurons | Epigenetic editing activated FMR1 expression and reversed spontaneous hyperactivity associated with FXS neurons | [113] |
| PAX6, ARX | dCas9-DNMT3A/hESC with DNMT3A knockout | Restoration of the differentiation trajectory of DNMT3A knockout hESCs: rescue of motor neurogenesis and suppression of floor plate induction. | [114] |
| ATP6V1A | dCas9-KRAB/hiPSC-derived NGN2-induced glutamatergic neurons | Neurons with suppressed ATP6V1A show a significant decrease in the number of SYN1+ punctures, neuronal activity, expression of various volt-generated subunits of sodium channels (e.g., SCN3A, SCN2A and SCN4B), the number of full action potentials and an increase in immature spikes. | [115] |
| KCTD13, TAOK2, NRXN1, SNAP91, CLCN3 | dCas9-KRAB, dCas9-VP64 and dCas9-VPR/hiPSC-derived NPCs, neurons, and astrocytes | The authors characterized the discrepancies and difficulties in the application of epigenetic CRISPR tools in different cell types | [116] |
| Syt1 | dCas9-KRAB/cultured hippocampal neurons, glutamatergic and GABAergic neurons in the dentate gyrus of the mouse hippocampus | Conditional inactivation of Syt1 shifts the excitation-inhibition (E-I) balance in the dentate gyrus. Shifting the E-I balance toward excitation improved the animals′ ability to spatial distinction. The learning ability of the animals could be bidirectionally regulated, but the mice always exhibited anxious- and depressive-like behavior. | [117] |
| TCF4 | Three-component lentiSAMv2 system containing dCas9-VP64, MS2-P65-HSF1 and sgRNA-MS2/brain cortical organoids | TCF4 overexpression reverses molecular and phenotypic abnormalities associated with PTHS | [118] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurishev, A.O.; Karpov, D.S.; Nadolinskaia, N.I.; Goncharenko, A.V.; Golimbet, V.E. CRISPR/Cas-Based Approaches to Study Schizophrenia and Other Neurodevelopmental Disorders. Int. J. Mol. Sci. 2023, 24, 241. https://doi.org/10.3390/ijms24010241
Kurishev AO, Karpov DS, Nadolinskaia NI, Goncharenko AV, Golimbet VE. CRISPR/Cas-Based Approaches to Study Schizophrenia and Other Neurodevelopmental Disorders. International Journal of Molecular Sciences. 2023; 24(1):241. https://doi.org/10.3390/ijms24010241
Chicago/Turabian StyleKurishev, Artemiy O., Dmitry S. Karpov, Nonna I. Nadolinskaia, Anna V. Goncharenko, and Vera E. Golimbet. 2023. "CRISPR/Cas-Based Approaches to Study Schizophrenia and Other Neurodevelopmental Disorders" International Journal of Molecular Sciences 24, no. 1: 241. https://doi.org/10.3390/ijms24010241