
Current and Future Treatment of Mucopolysaccharidosis (MPS) Type II: Is Brain-Targeted Stem Cell Gene Therapy the Solution for This Devastating Disorder?



Abstract
:1. Introduction
2. Current Therapies for MPSII
2.1. Enzyme Replacement Therapy
2.2. Intrathecal or Brain Targeted Enzyme Replacement Therapy
2.3. Substrate Reduction Therapy
3. Bone Marrow Transplant for MPSII
3.1. Introduction
3.2. Summary of Bone Marrow Transplant Outcomes in Hunter Syndrome
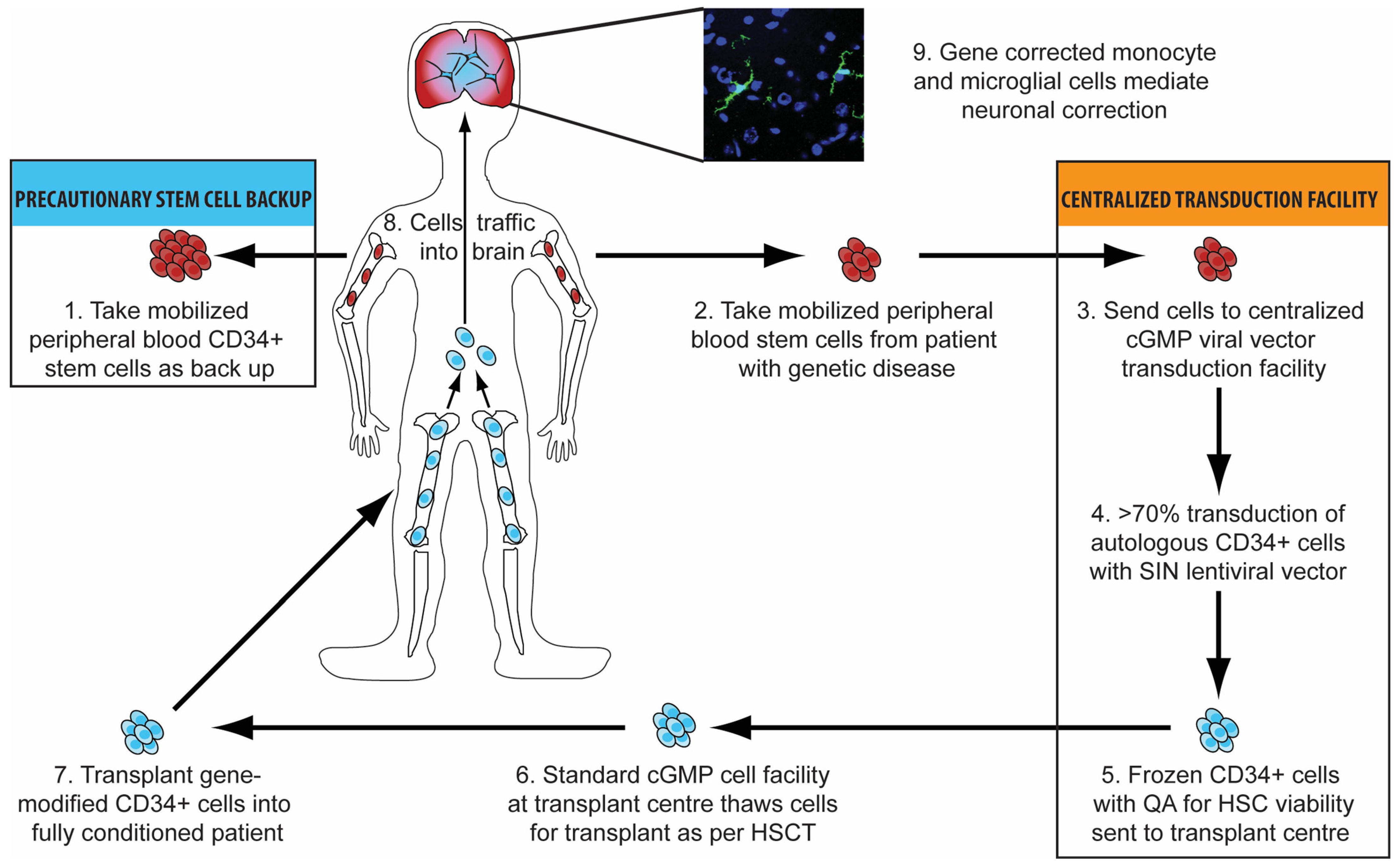
3.3. Haematopoietic Stem Cell Gene Therapy
3.4. Advantages of Haematopoietic Stem Cell Gene Therapy
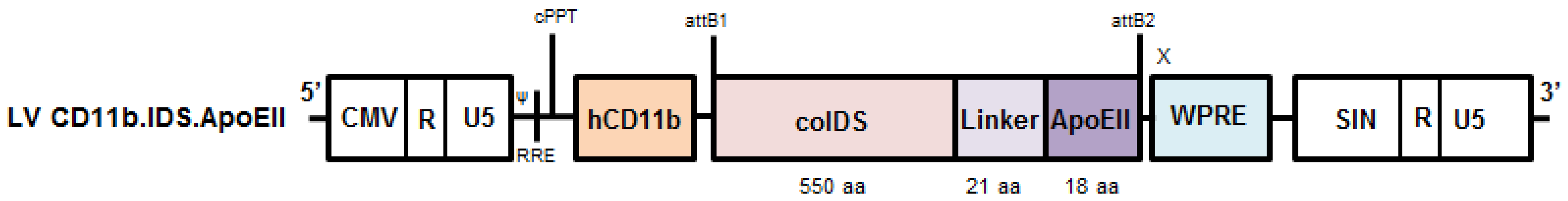
3.5. Vectors
3.6. Brain-Targeted Stem Cell Gene Therapy
3.7. Adeno-Associated Virus Gene Therapy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sawamoto, K.; Stapleton, M.; Almeciga-Diaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments. Drugs 2019, 79, 1103–1134. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, E.; Jakobkiewicz-Banecka, J.; Baranska, S.; Tylki-Szymanska, A.; Czartoryska, B.; Wegrzyn, A.; Wegrzyn, G. Genistein-mediated inhibition of glycosaminoglycan synthesis as a basis for gene expression-targeted isoflavone therapy for mucopolysaccharidoses. Eur. J. Hum. Genet. 2006, 14, 846–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friso, A.; Tomanin, R.; Salvalaio, M.; Scarpa, M. Genistein reduces glycosaminoglycan levels in a mouse model of mucopolysaccharidosis type II. Br. J. Pharmacol. 2010, 159, 1082–1091. [Google Scholar] [CrossRef] [Green Version]
- Marucha, J.; Tylki-Szymanska, A.; Jakobkiewicz-Banecka, J.; Piotrowska, E.; Kloska, A.; Czartoryska, B.; Wegrzyn, G. Improvement in the range of joint motion in seven patients with mucopolysaccharidosis type II during experimental gene expression-targeted isoflavone therapy (GET IT). Am. J. Med. Genet. Part A 2011, 155, 2257–2262. [Google Scholar] [CrossRef]
- Delgadillo, V.; O’Callaghan Mdel, M.; Artuch, R.; Montero, R.; Pineda, M. Genistein supplementation in patients affected by Sanfilippo disease. J. Inherit. Metab. Dis. 2011, 34, 1039–1044. [Google Scholar] [CrossRef]
- Ghosh, A.; Rust, S.; Langford-Smith, K.; Weisberg, D.; Canal, M.; Breen, C.; Hepburn, M.; Tylee, K.; Vaz, F.M.; Vail, A.; et al. High dose genistein in Sanfilippo syndrome: A randomised controlled trial. J. Inherit. Metab. Dis. 2021, 44, 1248–1262. [Google Scholar] [CrossRef] [PubMed]
- Fratantoni, J.C.; Hall, C.W.; Neufeld, E.F. Hurler and Hunter syndromes: Mutual correction of the defect in cultured fibroblasts. Science 1968, 162, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, J.R.; Hugh-Jones, K.; Barrett, A.J.; Byrom, N.; Chambers, D.; Henry, K.; James, D.C.; Lucas, C.F.; Rogers, T.R.; Benson, P.F.; et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 1981, 2, 709–712. [Google Scholar] [CrossRef]
- Bartelink, I.H.; van Reij, E.M.; Gerhardt, C.E.; van Maarseveen, E.M.; de Wildt, A.; Versluys, B.; Lindemans, C.A.; Bierings, M.B.; Boelens, J.J. Fludarabine and exposure-targeted busulfan compares favorably with busulfan/cyclophosphamide-based regimens in pediatric hematopoietic cell transplantation: Maintaining efficacy with less toxicity. Biol. Blood Marrow Transplant. 2014, 20, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, E.Y.; Boelens, J.J.; Jones, S.A.; Wynn, R.F. Hematopoietic Stem Cell Transplantation in Inborn Errors of Metabolism. Front. Pediatr. 2019, 7, 433. [Google Scholar] [CrossRef] [Green Version]
- Church, H.; Tylee, K.; Cooper, A.; Thornley, M.; Mercer, J.; Wraith, E.; Carr, T.; O’Meara, A.; Wynn, R.F. Biochemical monitoring after haemopoietic stem cell transplant for Hurler syndrome (MPSIH): Implications for functional outcome after transplant in metabolic disease. Bone Marrow Transplant. 2007, 39, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boelens, J.J.; Aldenhoven, M.; Purtill, D.; Ruggeri, A.; Defor, T.; Wynn, R.; Wraith, E.; Cavazzana-Calvo, M.; Rovelli, A.; Fischer, A.; et al. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood 2013, 121, 3981–3987. [Google Scholar] [CrossRef]
- Lum, S.H.; Miller, W.P.; Jones, S.; Poulton, K.; Ogden, W.; Lee, H.; Logan, A.; Bonney, D.; Lund, T.C.; Orchard, P.J.; et al. Changes in the incidence, patterns and outcomes of graft failure following hematopoietic stem cell transplantation for Hurler syndrome. Bone Marrow Transplant. 2017, 52, 846–853. [Google Scholar] [CrossRef]
- Nataraj, R.; Hiwarkar, P.; Bonney, D.; Campbell, H.; Jones, S.; Deambrosis, D.; Evans, P.; Poulton, K.; van Hasselt, P.M.; Bierings, M.B.; et al. B-cell depletion abrogates immune mediated cytopenia and rejection of cord blood transplantation in Hurler syndrome. Bone Marrow Transplant. 2021, 57, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A.; International Consensus Panel on, M.; Treatment of Mucopolysaccharidosis, I. Mucopolysaccharidosis I: Management and treatment guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Araya, K.; Sakai, N.; Mohri, I.; Kagitani-Shimono, K.; Okinaga, T.; Hashii, Y.; Ohta, H.; Nakamichi, I.; Aozasa, K.; Taniike, M.; et al. Localized donor cells in brain of a Hunter disease patient after cord blood stem cell transplantation. Mol. Genet. Metab. 2009, 98, 255–263. [Google Scholar] [CrossRef]
- Barth, A.L.; Horovitz, D.D.G. Hematopoietic Stem Cell Transplantation in Mucopolysaccharidosis Type II: A Literature Review and Critical Analysis. J. Inborn Errors Metab. Screen. 2018, 6, 2326409818779097. [Google Scholar] [CrossRef]
- Warkentin, P.I.; Dixon, M.S., Jr.; Schafer, I.; Strandjord, S.E.; Coccia, P.F. Bone marrow transplantation in Hunter syndrome: A preliminary report. Birth Defects Orig. Artic Ser. 1986, 22, 31–39. [Google Scholar] [PubMed]
- Bergstrom, S.K.; Quinn, J.J.; Greenstein, R.; Ascensao, J. Long-term follow-up of a patient transplanted for Hunter’s disease type IIB: A case report and literature review. Bone Marrow Transplant. 1994, 14, 653–658. [Google Scholar] [PubMed]
- Imaizumi, M.; Gushi, K.; Kurobane, I.; Inoue, S.; Suzuki, J.; Koizumi, Y.; Suzuki, H.; Sato, A.; Gotoh, Y.; Haginoya, K.; et al. Long-term effects of bone marrow transplantation for inborn errors of metabolism: A study of four patients with lysosomal storage diseases. Pediatr. Int. 1994, 36, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Coppa, G.V.; Gabrielli, O.; Zampini, L.; Pierani, P.; Giorgi, P.L.; Jezequel, A.M.; Orlandi, F.; Miniero, R.; Busca, A.; De Luca, T.; et al. Bone marrow transplantation in Hunter syndrome (mucopolysaccharidosis type II): Two-year follow-up of the first Italian patient and review of the literature. Pediatr. Med. Chir. 1995, 17, 227–235. [Google Scholar]
- McKinnis, E.J.; Sulzbacher, S.; Rutledge, J.C.; Sanders, J.; Scott, C.R. Bone marrow transplantation in Hunter syndrome. J. Pediatr. 1996, 129, 145–148. [Google Scholar] [CrossRef]
- Li, P.; Thompson, J.N.; Hug, G.; Huffman, P.; Chuck, G. Biochemical and molecular analysis in a patient with the severe form of Hunter syndrome after bone marrow transplantation. Am. J. Med. Genet. 1996, 64, 531–535. [Google Scholar] [CrossRef]
- Vellodi, A.; Young, E.; Cooper, A.; Lidchi, V.; Winchester, B.; Wraith, J.E. Long-term follow-up following bone marrow transplantation for Hunter disease. J. Inherit. Metab. Dis. 1999, 22, 638–648. [Google Scholar] [CrossRef]
- Maria, E.L.; Michele, P.D.; Vinod, P.; Joanne, K.; Paul, S. 169: Neurodevelopmental outcomes of children with MPS II after unrelated umbilical cord blood transplantation. Biol. Blood Marrow Transplant. 2007, 13, 63. [Google Scholar] [CrossRef] [Green Version]
- Guffon, N.; Bertrand, Y.; Forest, I.; Fouilhoux, A.; Froissart, R. Bone marrow transplantation in children with Hunter syndrome: Outcome after 7 to 17 years. J. Pediatr. 2009, 154, 733–737. [Google Scholar] [CrossRef]
- Poe, M.; Escolar, M.; Kurtzberg, J.; Szabolcs, P.; Prasad, V.; Parikh, S.; Holt, J. MPS II: Developmental outcomes after hematopoietic stem cell transplantation. Mol. Genet. Metab. 2011, 102, S35–S36. [Google Scholar] [CrossRef]
- Escolar, M.; Poe, M.; Rajan, D.; Szabolcs, P. Longterm outcomes of patients receiving umbilical blood stem cell transplantation for MPS II. Mol. Genet. Metab. 2013, 108, S37–S38. [Google Scholar] [CrossRef]
- Tanaka, A.; Okuyama, T.; Suzuki, Y.; Sakai, N.; Takakura, H.; Sawada, T.; Tanaka, T.; Otomo, T.; Ohashi, T.; Ishige-Wada, M.; et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: A nationwide survey in Japan. Mol. Genet. Metab. 2012, 107, 513–520. [Google Scholar] [CrossRef]
- Annibali, R.; Caponi, L.; Morganti, A.; Manna, M.; Gabrielli, O.; Ficcadenti, A. Hunter syndrome (Mucopolysaccharidosis type II), severe phenotype: Long term follow-up on patients undergone to hematopoietic stem cell transplantation. Minerva Pediatr. 2013, 65, 487–496. [Google Scholar]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17, 1065. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.; Zensi, A.; Wien, S.L.; Tschickardt, S.E.; Maier, W.; Vogel, T.; Worek, F.; Pietrzik, C.U.; Kreuter, J.; von Briesen, H. Uptake mechanism of ApoE-modified nanoparticles on brain capillary endothelial cells as a blood-brain barrier model. PLoS ONE 2012, 7, e32568. [Google Scholar] [CrossRef]
- Wang, D.; El-Amouri, S.S.; Dai, M.; Kuan, C.Y.; Hui, D.Y.; Brady, R.O.; Pan, D. Engineering a lysosomal enzyme with a derivative of receptor-binding domain of apoE enables delivery across the blood-brain barrier. Proc. Natl. Acad. Sci. USA 2013, 110, 2999–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockenhoff, A.; Cramer, S.; Wolte, P.; Knieling, S.; Wohlenberg, C.; Gieselmann, V.; Galla, H.J.; Matzner, U. Comparison of five peptide vectors for improved brain delivery of the lysosomal enzyme arylsulfatase A. J. Neurosci. 2014, 34, 3122–3129. [Google Scholar] [CrossRef] [PubMed]
- Boelens, J.J.; Wynn, R.F.; O’Meara, A.; Veys, P.; Bertrand, Y.; Souillet, G.; Wraith, J.E.; Fischer, A.; Cavazzana-Calvo, M.; Sykora, K.W.; et al. Outcomes of hematopoietic stem cell transplantation for Hurler’s syndrome in Europe: A risk factor analysis for graft failure. Bone Marrow Transplant. 2007, 40, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Joseph, R.; DiCesare, E.B.; Miller, A. Hunter Syndrome: Is It Time to Make It Part of Newborn Screening? Adv. Neonatal Care 2018, 18, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, B.; Jayandharan, G.R. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr. Gene Ther. 2014, 14, 86–100. [Google Scholar] [CrossRef]
- McCarty, D.M. Self-complementary AAV vectors; advances and applications. Mol. Ther. 2008, 16, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.; Polito, V.A.; Pepe, S.; Mann, L.; D’Azzo, A.; Auricchio, A.; Ballabio, A.; Cosma, M.P. Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum. Mol. Genet. 2006, 15, 1225–1236. [Google Scholar] [CrossRef]
- Motas, S.; Haurigot, V.; Garcia, M.; Marco, S.; Ribera, A.; Roca, C.; Sanchez, X.; Sanchez, V.; Molas, M.; Bertolin, J.; et al. CNS-directed gene therapy for the treatment of neurologic and somatic mucopolysaccharidosis type II (Hunter syndrome). JCI Insight 2016, 1, e86696. [Google Scholar] [CrossRef]
- Laoharawee, K.; Podetz-Pedersen, K.M.; Nguyen, T.T.; Evenstar, L.B.; Kitto, K.F.; Nan, Z.; Fairbanks, C.A.; Low, W.C.; Kozarsky, K.F.; McIvor, R.S. Prevention of Neurocognitive Deficiency in Mucopolysaccharidosis Type II Mice by Central Nervous System-Directed, AAV9-Mediated Iduronate Sulfatase Gene Transfer. Hum. Gene Ther. 2017, 28, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luan, Z.; Jiang, H.; Fang, J.; Qin, M.; Lee, V.; Chen, J. Allogeneic Hematopoietic Stem Cell Transplantation in Thirty-Four Pediatric Cases of Mucopolysaccharidosis-A Ten-Year Report from the China Children Transplant Group. Biol. Blood Marrow Transplant. 2016, 22, 2104–2108. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; Yabe, H.; Suzuki, Y.; Seto, T.; Hamazaki, T.; Mason, R.W.; Xie, L.; Onsten, T.G.H.; Leistner-Segal, S.; Giugliani, R.; et al. Hematopoietic Stem Cell Transplantation for Patients with Mucopolysaccharidosis II. Biol. Blood Marrow Transplant. 2017, 23, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Selvanathan, A.; Ellaway, C.; Wilson, C.; Owens, P.; Shaw, P.J.; Bhattacharya, K. Effectiveness of Early Hematopoietic Stem Cell Transplantation in Preventing Neurocognitive Decline in Mucopolysaccharidosis Type II: A Case Series. JIMD Rep. 2018, 41, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Tanjuakio, J.; Suzuki, Y.; Patel, P.; Yasuda, E.; Kubaski, F.; Tanaka, A.; Yabe, H.; Mason, R.W.; Montano, A.M.; Orii, K.E.; et al. Activities of daily living in patients with Hunter syndrome: Impact of enzyme replacement therapy and hematopoietic stem cell transplantation. Mol. Genet. Metab. 2015, 114, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Suzuki, Y.; Tanaka, A.; Yabe, H.; Kato, S.; Shimada, T.; Mason, R.W.; Orii, K.E.; Fukao, T.; Orii, T.; et al. Impact of Enzyme Replacement Therapy and Hematopoietic Stem Cell Therapy on Growth in Patients with Hunter Syndrome. Mol. Genet. Metab. Rep. 2014, 1, 184–196. [Google Scholar] [CrossRef]
- Ito, K.; Ochiai, T.; Suzuki, H.; Chin, M.; Shichino, H.; Mugishima, H. The effect of haematopoietic stem cell transplant on papules with ‘pebbly’ appearance in Hunter’s syndrome. Br. J. Derm. 2004, 151, 207–211. [Google Scholar] [CrossRef]
- Bigger, B.W.; Wynn, R.F. Novel approaches and mechanisms in hematopoietic stem cell gene therapy. Discov. Med. 2014, 17, 207–215. [Google Scholar]
- Celik, B.; Tomatsu, S.C.; Tomatsu, S.; Khan, S.A. Epidemiology of Mucopolysaccharidoses Update. Diagnostics 2021, 11, 273. [Google Scholar] [CrossRef]
- Mukherjee, S.; Thrasher, A.J. Gene therapy for PIDs: Progress, pitfalls and prospects. Gene 2013, 525, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, R.F.; Wraith, J.E.; Mercer, J.; O’Meara, A.; Tylee, K.; Thornley, M.; Church, H.J.; Bigger, B.W. Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J. Pediatr. 2009, 154, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; De Palma, M.; Quattrini, A.; Del Carro, U.; Amadio, S.; Visigalli, I.; Sessa, M.; Fasano, S.; Brambilla, R.; Marchesini, S.; et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Investig. 2004, 113, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- Visigalli, I.; Delai, S.; Politi, L.S.; Di Domenico, C.; Cerri, F.; Mrak, E.; D’Isa, R.; Ungaro, D.; Stok, M.; Sanvito, F.; et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood 2010, 116, 5130–5139. [Google Scholar] [CrossRef]
- Langford-Smith, A.; Wilkinson, F.L.; Langford-Smith, K.J.; Holley, R.J.; Sergijenko, A.; Howe, S.J.; Bennett, W.R.; Jones, S.A.; Wraith, J.; Merry, C.L.; et al. Hematopoietic stem cell and gene therapy corrects primary neuropathology and behavior in mucopolysaccharidosis IIIA mice. Mol. Ther. 2012, 20, 1610–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergijenko, A.; Langford-Smith, A.; Liao, A.Y.; Pickford, C.E.; McDermott, J.; Nowinski, G.; Langford-Smith, K.J.; Merry, C.L.; Jones, S.A.; Wraith, J.E.; et al. Myeloid/Microglial driven autologous hematopoietic stem cell gene therapy corrects a neuronopathic lysosomal disease. Mol. Ther. 2013, 21, 1938–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holley, R.J.; Ellison, S.M.; Fil, D.; O’Leary, C.; McDermott, J.; Senthivel, N.; Langford-Smith, A.W.W.; Wilkinson, F.L.; D’Souza, Z.; Parker, H.; et al. Macrophage enzyme and reduced inflammation drive brain correction of mucopolysaccharidosis IIIB by stem cell gene therapy. Brain 2018, 141, 99–116. [Google Scholar] [CrossRef] [Green Version]
- Gleitz, H.F.; Liao, A.Y.; Cook, J.R.; Rowlston, S.F.; Forte, G.M.; D’Souza, Z.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 2018, 10, e8730. [Google Scholar] [CrossRef] [PubMed]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Fumagalli, F.; Calbi, V.; Sessa, M.; Zambon, A.; Baldoli, C.; Rancoita, P.M.V.; Acquati, S.; de Mattia, F.; Tucci, F.; Gallo, V.; et al. Lentiviral hematopoietic stem and progenitor cell gene therapy (HSPC-GT) for metachromatic leukodystrophy (MLD): Clinical outcomes from 33 patients. Mol. Genet. Metab. 2020, 129, S59. [Google Scholar] [CrossRef]
- England, N. NHS to Roll out Life-Saving Gene Therapy for Rare Disease Affecting Babies. 2022. Available online: https://www.england.nhs.uk/2022/02/nhs-to-roll-out-life-saving-gene-therapy-for-rare-disease-affecting-babies/ (accessed on 5 April 2022).
- Gentner, B.; Tucci, F.; Galimberti, S.; Fumagalli, F.; De Pellegrin, M.; Silvani, P.; Camesasca, C.; Pontesilli, S.; Darin, S.; Ciotti, F.; et al. Hematopoietic Stem- and Progenitor-Cell Gene Therapy for Hurler Syndrome. N. Engl. J. Med. 2021, 385, 1929–1940. [Google Scholar] [CrossRef]
- Kinsella, J.L.; Jones, S.; Thrasher, A.J.; Booth, C.; Buckland, K.F.; Izotova, N.; Rust, S.; Weisberg, D.; Church, H.J.; Tylee, K.L.; et al. Ex-vivo autologous stem cell gene therapy clinical trial for mucopolysaccharidosis type IIIA: Update on phase I/II clinical trial. Mol. Genet. Metab. 2021, 132, S56–S57. [Google Scholar] [CrossRef]
- Tucci, F.; Scaramuzza, S.; Aiuti, A.; Mortellaro, A. Update on Clinical Ex Vivo Hematopoietic Stem Cell Gene Therapy for Inherited Monogenic Diseases. Mol. Ther. 2021, 29, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.T.; Aiuti, A.; Cavazzana, M. Autologous Stem-Cell-Based Gene Therapy for Inherited Disorders: State of the Art and Perspectives. Front. Pediatr. 2019, 7, 443. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Shimada, Y.; Akiyama, K.; Higuchi, T.; Fukuda, T.; Kobayashi, H.; Eto, Y.; Ida, H.; Ohashi, T. Hematopoietic Stem Cell Gene Therapy Corrects Neuropathic Phenotype in Murine Model of Mucopolysaccharidosis Type II. Hum. Gene Ther. 2015, 26, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, M.; Zerah, M.; Husson, B.; de Bournonville, S.; Deiva, K.; Adamsbaum, C.; Vincent, F.; Hocquemiller, M.; Broissand, C.; Furlan, V.; et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum. Gene Ther. 2014, 25, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, M.; Zerah, M.; Gougeon, M.L.; Ausseil, J.; de Bournonville, S.; Husson, B.; Zafeiriou, D.; Parenti, G.; Bourget, P.; Poirier, B.; et al. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: An uncontrolled phase 1/2 clinical trial. Lancet Neurol. 2017, 16, 712–720. [Google Scholar] [CrossRef]
- Laoharawee, K.; DeKelver, R.C.; Podetz-Pedersen, K.M.; Rohde, M.; Sproul, S.; Nguyen, H.O.; Nguyen, T.; St Martin, S.J.; Ou, L.; Tom, S.; et al. Dose-Dependent Prevention of Metabolic and Neurologic Disease in Murine MPS II by ZFN-Mediated In Vivo Genome Editing. Mol. Ther. 2018, 26, 1127–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National MPS Society. Advisory Committee on Heritable Disorders in Newborns and Children Votes to Approve MPS II for Recommended Uniform Screening Panel; National MPS Society: Durham, NC, USA, 2022. [Google Scholar]



{kind=link}
{kind=link}
| Author and Year | Number of Patients and Age at HSCT | Conclusions | Issues |
|---|---|---|---|
| Vellodi et al., 1999 [32] | 10 patients Age: 10 months–5 years 1 month |
| High TRM, variable age at HSCT, variable clinical phenotype, 1 of the 3 surviving patients transplanted with carrier donor |
| Maria et al., 2007 [33] | 5 patients Age: 3 months– 3 years 4 month |
| Long term follow-up data needed |
| Guffon et al., 2009 [34] | 8 patients Age: 3 years–16 years |
| Patients with severe phenotype had significant cognitive impairment at time of HSCT, all patents aged over 3 years at time of transplant, 2 patients with severe form transplanted from heterozygous siblings |
| Poe et al., 2011 [35] | 9 patients Age: 1.5 months–3 years 11 months |
| Unclear neurological status pre-HSCT and whether any correlation between age at time of transplant, expected phenotype and outcome |
| Escolar et al., 2012 [36] | 9 patients Age: 1.5 to 47 months |
| Data only available up until patients 8 years old, more consistent data needed |
| Tanaka et al., 2012 [37] | 21 patients Age: 2 years to 19 years 8 months |
| Retrospective data, all patients aged over 2 years at HSCT |
| Annibali et al., 2012 [38] | 4 patients Age: 2 years 6 months to 2 years 11 months |
| Patients had mild to moderate mental retardation prior to HSCT |
| Wang et al., 2016 [39] | 12 patients Age: 2–6 years |
| Short follow up (only 2 years), patients evaluated as part of larger MPS cohort so making conclusions applicable to MPSII challenging |
| Kubaski et al., 2017 [40] | 27 patients Age: 2–21.4 years |
| Major limitation of study age at time of transplant, need more data on patients transplanted under 2 years old |
| Selvanathan et al., 2017 [41] | 4 patients Age: 8 months–3 years, 8 months |
| Varying pre-HSCT baselines make it difficult to draw any significant conclusions |
| Current and Potential Problems with the Therapeutic Strategies for MPSII | Potential Solutions |
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horgan, C.; Jones, S.A.; Bigger, B.W.; Wynn, R. Current and Future Treatment of Mucopolysaccharidosis (MPS) Type II: Is Brain-Targeted Stem Cell Gene Therapy the Solution for This Devastating Disorder? Int. J. Mol. Sci. 2022, 23, 4854. https://doi.org/10.3390/ijms23094854
Horgan C, Jones SA, Bigger BW, Wynn R. Current and Future Treatment of Mucopolysaccharidosis (MPS) Type II: Is Brain-Targeted Stem Cell Gene Therapy the Solution for This Devastating Disorder? International Journal of Molecular Sciences. 2022; 23(9):4854. https://doi.org/10.3390/ijms23094854
Chicago/Turabian StyleHorgan, Claire, Simon A. Jones, Brian W. Bigger, and Robert Wynn. 2022. "Current and Future Treatment of Mucopolysaccharidosis (MPS) Type II: Is Brain-Targeted Stem Cell Gene Therapy the Solution for This Devastating Disorder?" International Journal of Molecular Sciences 23, no. 9: 4854. https://doi.org/10.3390/ijms23094854