
Innate Immunity in Mucopolysaccharide Diseases



Abstract
:1. The Mucopolysaccharidoses
2. Pathology in MPSs
2.1. Primary Storage of GAGs
2.2. Lysosomal Stress
2.3. Secondary Molecules
3. Innate Immunity in Mucopolysaccharidoses
3.1. TLR Signalling
3.2. Inflammatory Cytokines
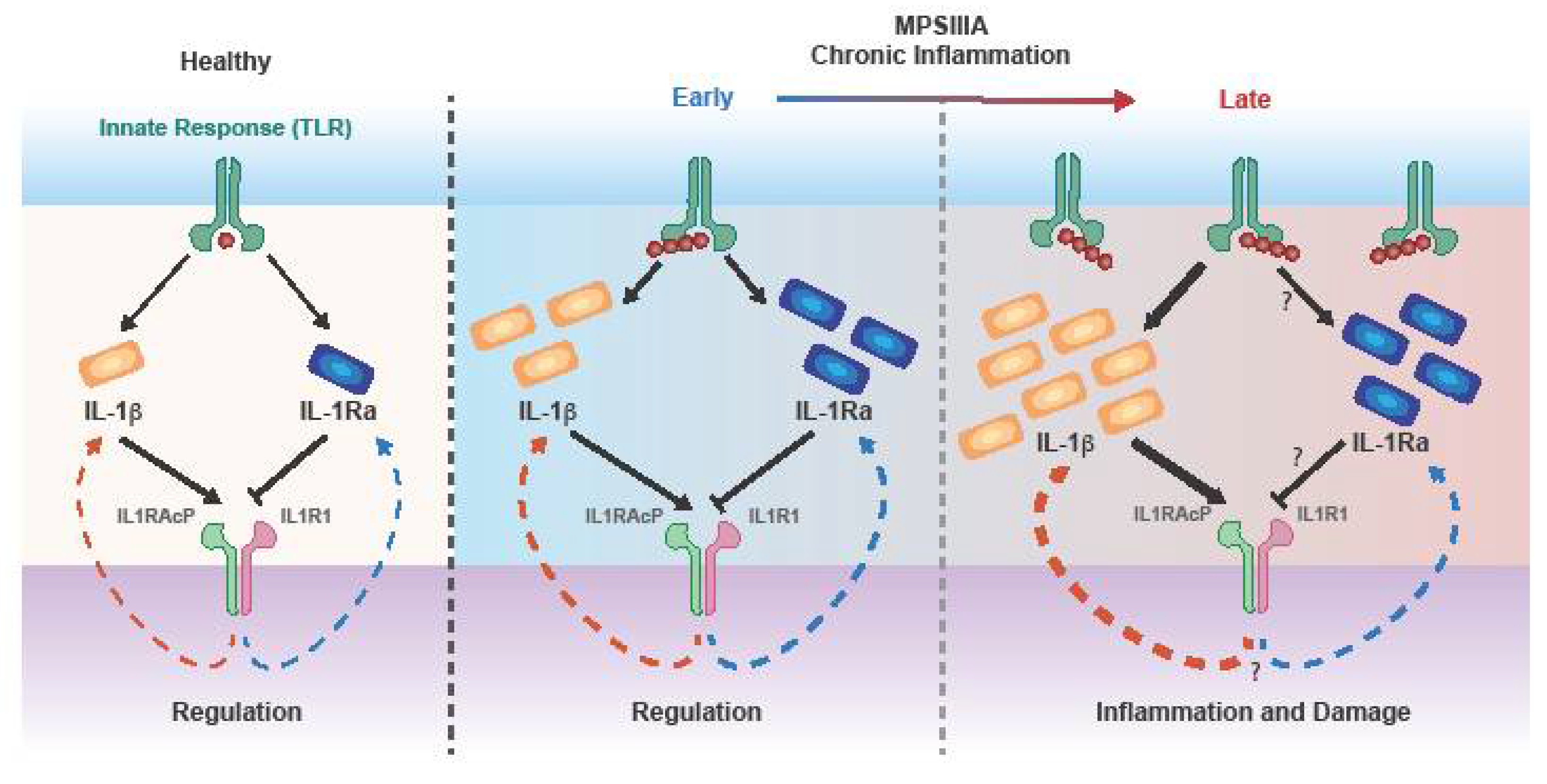
3.3. The Role of Cascade Initiating Cytokines TNFα and IL-1
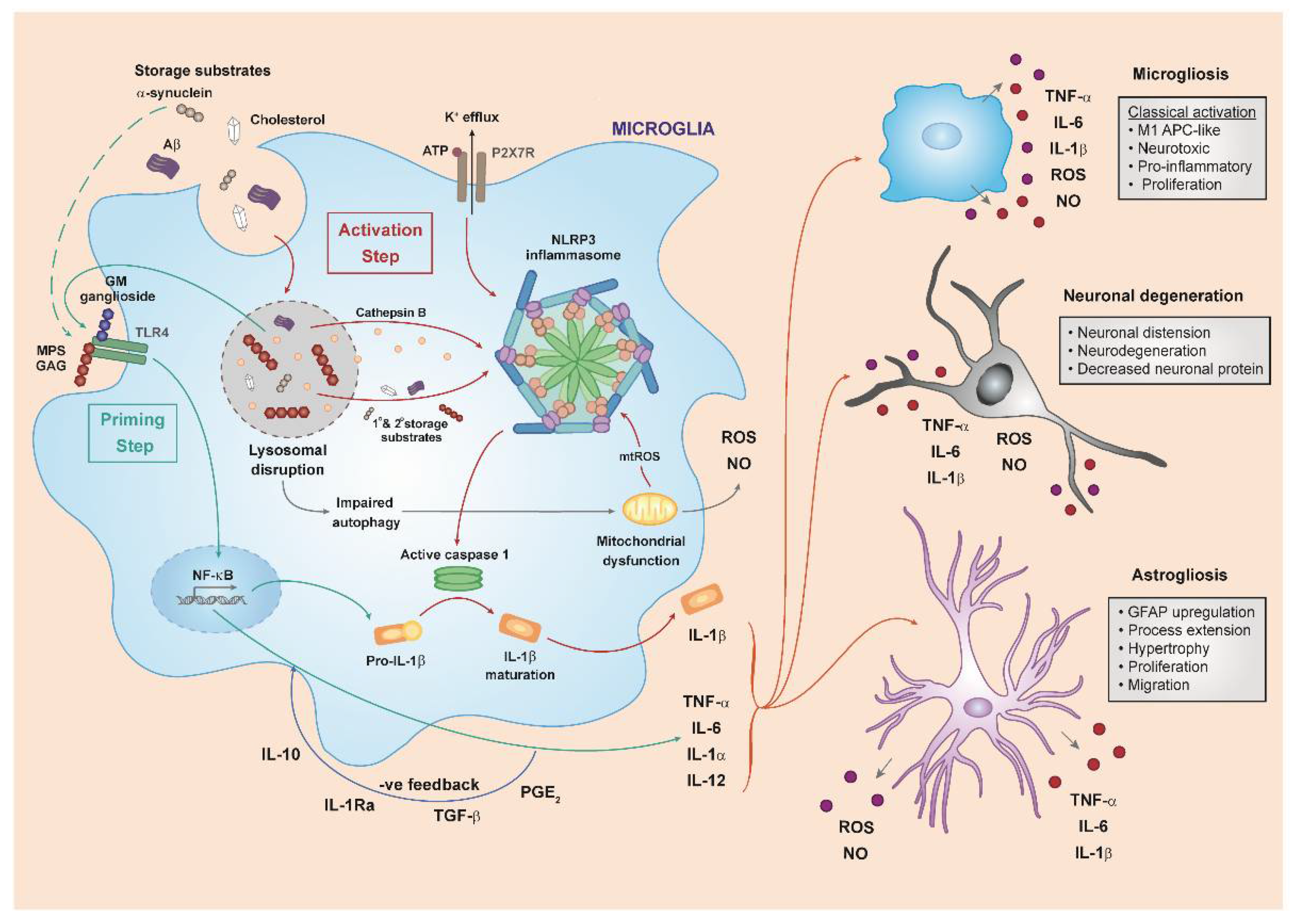
3.4. The NLRP3 Inflammasome and IL-1
3.5. Cell Death
3.6. Innate Immunity in Mucopolysaccharide Diseases
4. Anti-Inflammatory Therapies in MPS Diseases
4.1. CNS-Targeting Therapies
4.2. Periphery-Targeting Therapies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Mucopolysaccharidoses–NORD (National Organization for Rare Disorders). Available online: https://rarediseases.org/rare-diseases/mucopolysaccharidoses/ (accessed on 19 November 2019).
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenzer, J. The mucopolysaccharidoses: A heterogeneous group of disorders with variable pediatric presentations. J. Pediatr. 2004, 144, S27–S34. [Google Scholar] [CrossRef] [PubMed]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 2009; ISBN 9780879697709. [Google Scholar]
- Meyer, K. Biochemistry and biology of mucopolysaccharides. Am. J. Med. 1969, 47, 664–672. [Google Scholar] [CrossRef]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology 2011, 50, v4–v12. [Google Scholar] [CrossRef] [Green Version]
- Rasalkar, D.D.; Chu, W.C.W.; Hui, J.; Chu, C.-M.; Paunipagar, B.K.; Li, C.-K. Pictorial review of mucopolysaccharidosis with emphasis on MRI features of brain and spine. Br. J. Radiol. 2011, 84, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Simonaro, C.M.; Ge, Y.; Eliyahu, E.; He, X.; Jepsen, K.J.; Schuchman, E.H. Involvement of the Toll-like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proc. Natl. Acad. Sci. USA 2010, 107, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.; Ellison, S.M.; Holley, R.J.; O’Leary, C.; Liao, A.; Asadi, J.; Glover, E.; Ghosh, A.; Jones, S.; Wilkinson, F.L.; et al. Haematopoietic stem cell gene therapy with IL-1Ra rescues cognitive loss in mucopolysaccharidosis IIIA. EMBO Mol. Med. 2020, 12, e11185. [Google Scholar] [CrossRef]
- Jeyakumar, M.; Thomas, R.; Elliot-Smith, E.; Smith, D.A.; van der Spoel, A.C.; d’Azzo, A.; Hugh Perry, V.; Butters, T.D.; Dwek, R.A.; Platt, F.M. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 2003, 126, 974–987. [Google Scholar] [CrossRef] [Green Version]
- Vitner, E.B.; Platt, F.M.; Futerman, A.H. Common and uncommon pathogenic cascades in lysosomal storage diseases. J. Biol. Chem. 2010, 285, 20423–20427. [Google Scholar] [CrossRef] [Green Version]
- de Ruijter, J.; de Ru, M.H.; Wagemans, T.; Ijlst, L.; Lund, A.M.; Orchard, P.J.; Schaefer, G.B.; Wijburg, F.A.; van Vlies, N. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol. Genet. Metab. 2012, 107, 705–710. [Google Scholar] [CrossRef]
- Holley, R.J.; Ellison, S.M.; Fil, D.; O’Leary, C.; McDermott, J.; Senthivel, N.; Langford-Smith, A.W.W.; Wilkinson, F.L.; D’Souza, Z.; Parker, H.; et al. Macrophage enzyme and reduced inflammation drive brain correction of mucopolysaccharidosis IIIB by stem cell gene therapy. Brain 2018, 141, 99–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkley, S.U. Pathogenic cascades in lysosomal disease—Why so complex? J. Inherit. Metab. Dis. 2009, 32, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, R.; Brown, J.R.; Al-Mafraji, K.; Lamanna, W.C.; Beitel, J.R.; Boons, G.-J.; Esko, J.D.; Crawford, B.E. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat. Chem. Biol. 2012, 8, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holley, R.J.; Deligny, A.; Wei, W.; Watson, H.A.; Niñ Onuevo, M.R.; Dagälv, A.; Leary, J.A.; Bigger, B.W.; Kjellén, L.; Merry, C.L.R. Mucopolysaccharidosis Type I, Unique Structure of Accumulated Heparan Sulfate and Increased N-Sulfotransferase Activity in Mice Lacking-L-iduronidase. J. Biol. Chem. 2011, 286, 37515–37524. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, C.A.; Scudder, S.L.; Lin, Y.; Dozier, L.E.; Phan, D.; Allen, N.J.; Patrick, G.N.; Esko, J.D. Neurodevelopmental Changes in Excitatory Synaptic Structure and Function in the Cerebral Cortex of Sanfilippo Syndrome IIIA Mice. Sci. Rep. 2017, 7, 46576. [Google Scholar] [CrossRef]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef]
- Ledin, J.; Ringvall, M.; Thuveson, M.; Eriksson, I.; Wilé, M.; Kusche-Gullberg, M.; Forsberg, E.; Kjellén, L. Enzymatically Active N-Deacetylase/N-Sulfotransferase-2 Is Present in Liver but Does Not Contribute to Heparan Sulfate N-Sulfation. J. Biol. Chem. 2006, 281, 35727–35734. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, N.S.; Mancera, R.L. The Structure of Glycosaminoglycans and their Interactions with Proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef]
- Watson, H.A.; Holley, R.J.; Langford-Smith, K.J.; Wilkinson, F.L.; van Kuppevelt, T.H.; Wynn, R.F.; Wraith, J.E.; Merry, C.L.R.; Bigger, B.W. Heparan sulfate inhibits hematopoietic stem and progenitor cell migration and engraftment in mucopolysaccharidosis I. J. Biol. Chem. 2014, 289, 36194–36203. [Google Scholar] [CrossRef] [Green Version]
- Amara, A.; Lorthioir, O.; Valenzuela, A.; Magerus, A.; Thelen, M.; Montes, M.; Virelizier, J.L.; Delepierre, M.; Baleux, F.; Lortat-Jacob, H.; et al. Stromal cell-derived factor-1alpha associates with heparan sulfates through the first beta-strand of the chemokine. J. Biol. Chem. 1999, 274, 23916–23925. [Google Scholar] [CrossRef] [Green Version]
- Pye, D.A.; Vivès, R.R.; Hyde, P.; Gallagher, J.T. Regulation of FGF-1 mitogenic activity by heparan sulfate oligosaccharides is dependent on specific structural features: Differential requirements for the modulation of FGF-1 and FGF-2. Glycobiology 2000, 10, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Yu, Y.; Leary, J.A. Effects of sulfate position on heparin octasaccharide binding to CCL2 examined by tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 1114–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pye, D.A.; Vives, R.R.; Turnbull, J.E.; Hyde, P.; Gallagher, J.T. Heparan sulfate oligosaccharides require 6-O-sulfation for promotion of basic fibroblast growth factor mitogenic activity. J. Biol. Chem. 1998, 273, 22936–22942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodbury, M.E.; Ikezu, T. Fibroblast growth factor-2 signaling in neurogenesis and neurodegeneration. J. Neuroimmune Pharmacol. 2014, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Dhoot, G.K.; Gustafsson, M.K.; Ai, X.; Sun, W.; Standiford, D.M.; Emerson, J. Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science 2001, 293, 1663–1666. [Google Scholar] [CrossRef]
- Malmström, A.; Bartolini, B.; Thelin, M.A.; Pacheco, B.; Maccarana, M. Iduronic acid in chondroitin/dermatan sulfate: Biosynthesis and biological function. J. Histochem. Cytochem. 2012, 60, 916–925. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.A. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: Glycosaminoglycan storage is merely the instigator. Rheumatology 2011, 50, v13–v18. [Google Scholar] [CrossRef] [Green Version]
- Simmons, M.A.; Bruce, I.A.; Penney, S.; Wraith, E.; Rothera, M.P. Otorhinolaryngological manifestations of the mucopolysaccharidoses. Int. J. Pediatr. Otorhinolaryngol. 2005, 69, 589–595. [Google Scholar] [CrossRef]
- Pal, A.R.; Mercer, J.; Jones, S.A.; Bruce, I.A.; Bigger, B.W. Substrate accumulation and extracellular matrix remodelling promote persistent upper airway disease in mucopolysaccharidosis patients on enzyme replacement therapy. PLoS ONE 2018, 13, e0203216. [Google Scholar] [CrossRef] [Green Version]
- Simonaro, C.M.; D’Angelo, M.; Haskins, M.E.; Schuchman, E.H. Joint and bone disease in mucopolysaccharidoses VI and VII: Identification of new therapeutic targets and biomarkers using animal models. Pediatr. Res. 2005, 57, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Donida, B.; Marchetti, D.P.; Biancini, G.B.; Deon, M.; Manini, P.R.; da Rosa, H.T.; Moura, D.J.; Saffi, J.; Bender, F.; Burin, M.G.; et al. Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futerman, A.H.; van Meer, G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 2004, 5, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Gleitz, H.F.; Liao, A.Y.; Cook, J.R.; Rowlston, S.F.; Forte, G.M.; D’Souza, Z.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 2018, 10, e8730. [Google Scholar] [CrossRef] [PubMed]
- Swaroop, M.; Brooks, M.J.; Gieser, L.; Swaroop, A.; Zheng, W. Patient iPSC-derived neural stem cells exhibit phenotypes in concordance with the clinical severity of mucopolysaccharidosis I. Hum. Mol. Genet. 2018, 27, 3612–3626. [Google Scholar] [CrossRef]
- Kobolák, J.; Molnár, K.; Varga, E.; Bock, I.; Jezsó, B.; Téglási, A.; Zhou, S.; Lo Giudice, M.; Hoogeveen-Westerveld, M.; Pijnappel, W.P.; et al. Modelling the neuropathology of lysosomal storage disorders through disease-specific human induced pluripotent stem cells. Exp. Cell Res. 2019, 380, 216–233. [Google Scholar] [CrossRef]
- Pereira, V.G.; Gazarini, M.L.; Rodrigues, L.C.; da Silva, F.H.; Han, S.W.; Martins, A.M.; Tersariol, I.L.S.; D’Almeida, V. Evidence of lysosomal membrane permeabilization in mucopolysaccharidosis type I: Rupture of calcium and proton homeostasis. J. Cell. Physiol. 2010, 223, 335–342. [Google Scholar] [CrossRef]
- Kawai, A.; Uchiyama, H.; Takano, S.; Nakamura, N.; Ohkuma, S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 2007, 3, 154–157. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Tooze, S.A.; Abada, A.; Elazar, Z. Endocytosis and autophagy: Exploitation or cooperation? Cold Spring Harb. Perspect. Biol. 2014, 6, a018358. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef]
- Beard, H.; Hassiotis, S.; Gai, W.-P.; Parkinson-Lawrence, E.; Hopwood, J.J.; Hemsley, K.M. Axonal dystrophy in the brain of mice with Sanfilippo syndrome. Exp. Neurol. 2017, 295, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V.G.; Martins, A.M.; Micheletti, C.; D’Almeida, V. Mutational and oxidative stress analysis in patients with mucopolysaccharidosis type I undergoing enzyme replacement therapy. Clin. Chim. Acta 2008, 387, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Filippon, L.; Vanzin, C.S.; Biancini, G.B.; Pereira, I.N.; Manfredini, V.; Sitta, A.; Peralba, M.C.R.; Schwartz, I.V.D.; Giugliani, R.; Vargas, C.R. Oxidative stress in patients with mucopolysaccharidosis type II before and during enzyme replacement therapy. Mol. Genet. Metab. 2011, 103, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Arfi, A.; Richard, M.; Gandolphe, C.; Bonnefont-Rousselot, D.; Thérond, P.; Scherman, D. Neuroinflammatory and oxidative stress phenomena in MPS IIIA mouse model: The positive effect of long-term aspirin treatment. Mol. Genet. Metab. 2011, 103, 18–25. [Google Scholar] [CrossRef]
- Zalfa, C.; Verpelli, C.; D’Avanzo, F.; Tomanin, R.; Vicidomini, C.; Cajola, L.; Manara, R.; Sala, C.; Scarpa, M.; Vescovi, A.L.; et al. Glial degeneration with oxidative damage drives neuronal demise in MPSII disease. Cell Death Dis. 2016, 7, e2331. [Google Scholar] [CrossRef]
- Bigger, B.W.; Begley, D.J.; Virgintino, D.; Pshezhetsky, A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018, 125, 322–331. [Google Scholar] [CrossRef]
- Reolon, G.K.; Reinke, A.; de Oliveira, M.R.; Braga, L.M.; Camassola, M.; Andrades, M.É.; Moreira, J.C.F.; Nardi, N.B.; Roesler, R.; Dal-Pizzol, F. Alterations in Oxidative Markers in the Cerebellum and Peripheral Organs in MPS I Mice. Cell. Mol. Neurobiol. 2009, 29, 443–448. [Google Scholar] [CrossRef]
- Salvalaio, M.; D’Avanzo, F.; Rigon, L.; Zanetti, A.; D’Angelo, M.; Valle, G.; Scarpa, M.; Tomanin, R. Brain RNA-seq profiling of the mucopolysaccharidosis type II mouse model. Int. J. Mol. Sci. 2017, 18, 1072. [Google Scholar] [CrossRef] [Green Version]
- Russell, C.; Hendson, G.; Jevon, G.; Matlock, T.; Yu, J.; Aklujkar, M.; Ng, K.Y.; Clarke, L.A. Murine MPS I: Insights into the Pathogenesis of Hurler Syndrome. Clin. Genet. 1998, 53, 349–361. [Google Scholar] [CrossRef]
- McGlynn, R.; Dobrenis, K.; Walkley, S.U. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp. Neurol. 2004, 480, 415–426. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.-T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides--An overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmaris, N.; Verot, L.; Puech, J.P.; Caillaud, C.; Vanier, M.T.; Heard, J.M. Prevention of neuropathology in the mouse model of hurler syndrome. Ann. Neurol. 2004, 56, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.K.; Tsai, Y.-T.; Ariga, T. Functional roles of gangliosides in neurodevelopment—An overview of recent advances. Neurochem. Res. 2012, 37, 1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkley, S.U.; Vanier, M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta 2009, 1793, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Walkley, S.U. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin. Cell Dev. Biol. 2004, 15, 433–444. [Google Scholar] [CrossRef]
- Son, M.-Y.; Kwak, J.E.; Seol, B.; Lee, D.Y.; Jeon, H.; Cho, Y.S. A novel human model of the neurodegenerative disease GM1 gangliosidosis using induced pluripotent stem cells demonstrates inflammasome activation. J. Pathol. 2015, 237, 98–110. [Google Scholar] [CrossRef]
- Ohmi, K.; Kudo, L.C.; Ryazantsev, S.; Zhao, H.Z.; Karsten, S.L.; Neufeld, E.F. Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 8332–8337. [Google Scholar] [CrossRef] [Green Version]
- Ohmi, K.; Zhao, H.Z.; Neufeld, E.F. Defects in the medial entorhinal cortex and dentate gyrus in the mouse model of Sanfilippo syndrome type B. PLoS ONE 2011, 6, e27461. [Google Scholar] [CrossRef]
- Liu, C.C.; Zhao, N.; Yamaguchi, Y.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-β clearance and aggregation in Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 332ra44. [Google Scholar] [CrossRef] [Green Version]
- Martins, C.; Hůlková, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef] [Green Version]
- Stefanis, L. Alpha-Synuclein and Lewy pathology in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 4, a009399. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hato, T.; Dagher, P.C. How the Innate Immune System Senses Trouble and Causes Trouble. Clin. J. Am. Soc. Nephrol. 2015, 10, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, P. Understanding Immunology; Pearson Canada: North York, ON, Canada, 2011. [Google Scholar]
- Nguyen, M.D.; Julien, J.-P.; Rivest, S. Innate immunity: The missing link in neuroprotection and neurodegeneration? Nat. Rev. Neurosci. 2002, 3, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.-I. Gene Expression Profiles of M1 and M2 Microglia Characterized by Comparative Analysis of Public Datasets. Clin. Exp. Neuroimmunol. 2018, 9, 124–138. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Fernandez-Lizarbe, S.; Pascual, M.; Guerri, C. Critical role of TLR4 response in the activation of microglia induced by ethanol. J. Immunol. 2009, 183, 4733–4744. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.; Bigger, B.W. The role of innate immunity in mucopolysaccharide diseases. J. Neurochem. 2018, 148, 639–651. [Google Scholar] [CrossRef] [Green Version]
- Termeer, C.; Benedix, F.; Sleeman, J.; Fieber, C.; Voith, U.; Ahrens, T.; Miyake, K.; Freudenberg, M.; Galanos, C.; Simon, J.C. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J. Exp. Med. 2002, 195, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Simonaro, C.M.; D’Angelo, M.; He, X.; Eliyahu, E.; Shtraizent, N.; Haskins, M.E.; Schuchman, E.H. Mechanism of glycosaminoglycan-mediated bone and joint disease: Implications for the mucopolysaccharidoses and other connective tissue diseases. Am. J. Pathol. 2008, 172, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ausseil, J.; Desmaris, N.; Bigou, S.; Attali, R.; Corbineau, S.; Vitry, S.; Parent, M.; Cheillan, D.; Fuller, M.; Maire, I.; et al. Early Neurodegeneration Progresses Independently of Microglial Activation by Heparan Sulfate in the Brain of Mucopolysaccharidosis IIIB Mice. PLoS ONE 2008, 3, e2296. [Google Scholar] [CrossRef] [PubMed]
- Casula, M.; Iyer, A.M.; Spliet, W.G.M.; Anink, J.J.; Steentjes, K.; Sta, M.; Troost, D.; Aronica, E. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience 2011, 179, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.M.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Yue, Z. A Peek into Parkinson’s Disease Progression through Human Dopamine Neurons in a Dish. Trends Neurosci. 2018, 41, 74–76. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Galvin, J.E.; Lee, V.M.; Rorke, L.B.; Dickson, D.W.; Wolfe, J.H.; Jones, M.Z.; Trojanowski, J.Q. Accumulation of intracellular amyloid-beta peptide (A beta 1-40) in mucopolysaccharidosis brains 74. J. Neuropathol. Exp. Neurol. 1999, 58, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Hamano, K.; Hayashi, M.; Shioda, K.; Fukatsu, R.; Mizutani, S. Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: Analysis of human brain tissue. Acta Neuropathol. 2008, 115, 547–559. [Google Scholar] [CrossRef]
- Shapiro, E.G.; Rudser, K.; Ahmed, A.; Steiner, R.D.; Delaney, K.A.; Yund, B.; King, K.; Kunin-Batson, A.; Eisengart, J.; Whitley, C.B. A longitudinal study of emotional adjustment, quality of life and adaptive function in attenuated MPS II. Mol. Genet. Metab. Rep. 2016, 7, 32–39. [Google Scholar] [CrossRef]
- Monaco, A.; Fraldi, A. Protein Aggregation and Dysfunction of Autophagy-Lysosomal Pathway: A Vicious Cycle in Lysosomal Storage Diseases. Front. Mol. Neurosci. 2020, 13, 37. [Google Scholar] [CrossRef]
- Winder-Rhodes, S.E.; Garcia-Reitböck, P.; Ban, M.; Evans, J.R.; Jacques, T.S.; Kemppinen, A.; Foltynie, T.; Williams-Gray, C.H.; Chinnery, P.F.; Hudson, G.; et al. Genetic and pathological links between Parkinson’s disease and the lysosomal disorder Sanfilippo syndrome. Mov. Disord. 2012, 27, 312–315. [Google Scholar] [CrossRef]
- Béraud, D.; Maguire-Zeiss, K.A. Misfolded α-synuclein and Toll-like receptors: Therapeutic targets for Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. S1), S17–S20. [Google Scholar] [CrossRef] [Green Version]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, M.J.; Kelvin, D.J. Cytokines, Chemokines and Their Receptors–Madame Curie Bioscience Database; NCBI Bookshelf; Landes Bioscience: Austin, TX, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6294/ (accessed on 15 January 2020).
- Subramaniam, S.; Stansberg, C.; Cunningham, C. The interleukin 1 receptor family. Dev. Comp. Immunol. 2004, 28, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.O.; Feldmann, M.; Maini, R.N. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 1992, 89, 9784. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; He, C. TNF-α and IL-6: The Link between Immune and Bone System. Curr. Drug Targets 2019, 21, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Huang, Z.; Sun, X.; Zhu, X.; Zhou, L.; Li, M.; Cheng, B.; Liu, X.; He, C. Microglia Polarization with M1/M2 Phenotype Changes in rd1 Mouse Model of Retinal Degeneration. Front. Neuroanat. 2017, 11, 77. [Google Scholar] [CrossRef] [Green Version]
- Fujitsuka, H.; Sawamoto, K.; Peracha, H.; Mason, R.W.; Mackenzie, W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Biomarkers in patients with mucopolysaccharidosis type II and IV. Mol. Genet. Metab. Rep. 2019, 19, 100455. [Google Scholar] [CrossRef]
- Ohmi, K.; Greenberg, D.S.; Rajavel, K.S.; Ryazantsev, S.; Li, H.H.; Neufeld, E.F. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. USA 2003, 100, 1902–1907. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2019, 281, 8–27. [Google Scholar] [CrossRef]
- IL1R2 Interleukin 1 Receptor Type 2 [Homo Sapiens (Human)]–Gene–NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=Gene&Cmd=DetailsSearch&Term=7850 (accessed on 16 January 2020).
- Zhang, J.-M.; An, J. Cytokines. Inflamm. Pain Int. Anesthesiol. Clin. 2009, 45, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Arend, W.P. The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev. 2002, 13, 323–340. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Polgreen, L.E.; Vehe, R.K.; Rudser, K.; Kunin-Batson, A.; Utz, J.J.; Dickson, P.; Shapiro, E.; Whitley, C.B. Elevated TNF-α is associated with pain and physical disability in mucopolysaccharidosis types I, II, and VI. Mol. Genet. Metab. 2016, 117, 427–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliyahu, E.; Wolfson, T.; Ge, Y.; Jepsen, K.J.; Schuchman, E.H.; Simonaro, C.M. Anti-TNF-Alpha Therapy Enhances the Effects of Enzyme Replacement Therapy in Rats with Mucopolysaccharidosis Type VI. PLoS ONE 2011, 6, e22447. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Song, N.; Liu, Z.-S.; Xue, W.; Bai, Z.-F.; Wang, Q.-Y.; Dai, J.; Liu, X.; Huang, Y.-J.; Cai, H.; Zhan, X.-Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185–197.e6. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsnelson, M.A.; Lozada-Soto, K.M.; Russo, H.M.; Miller, B.A.; Dubyak, G.R. NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: Roles for K+ efflux and Ca2+ influx. Am. J. Physiol. Physiol. 2016, 311, C83–C100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, H.; Yang, B.; Yu, W.; Xiao, Y.; Yu, D.; Zhang, Q. Cathepsin B links oxidative stress to the activation of NLRP3 inflammasome. Exp. Cell Res. 2018, 362, 180–187. [Google Scholar] [CrossRef]
- Aflaki, E.; Moaven, N.; Borger, D.K.; Lopez, G.; Westbroek, W.; Chae, J.J.; Marugan, J.; Patnaik, S.; Maniwang, E.; Gonzalez, A.N.; et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 2016, 15, 77–88. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Cell Death Dis. 2014, 5, e1382. [Google Scholar] [CrossRef] [Green Version]
- Kingham, P.J.; Pocock, J.M. Microglial secreted cathepsin B induces neuronal apoptosis. J. Neurochem. 2001, 76, 1475–1484. [Google Scholar] [CrossRef] [Green Version]
- Baldo, G.; Lorenzini, D.M.; Santos, D.S.; Mayer, F.Q.; Vitry, S.; Bigou, S.; Heard, J.M.; Matte, U.; Giugliani, R. Shotgun proteomics reveals possible mechanisms for cognitive impairment in Mucopolysaccharidosis I mice. Mol. Genet. Metab. 2015, 114, 138–145. [Google Scholar] [CrossRef]
- Azambuja, A.S.; Pimentel-Vera, L.N.; Gonzalez, E.A.; Poletto, E.; Pinheiro, C.V.; Matte, U.; Giugliani, R.; Baldo, G. Evidence for inflammasome activation in the brain of mucopolysaccharidosis type II mice. Metab. Brain Dis. 2020, 35, 1231–1236. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-α Induces Two Distinct Caspase-8 Activation Pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonaro, C.M.; Haskins, M.E.; Schuchman, E.H. Articular chondrocytes from animals with a dermatan sulfate storage disease undergo a high rate of apoptosis and release nitric oxide and inflammatory cytokines: A possible mechanism underlying degenerative joint disease in the mucopolysaccharidoses. Lab. Investig. 2001, 81, 1319–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adigun, R.; Bhimji, S.S. Necrosis, Cell (Liquefactive, Coagulative, Caseous, Fat, Fibrinoid, and Gangrenous); StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Abe, J.-I.; Morrell, C. Pyroptosis as a regulated form of necrosis. PI + /Annexin V − /high caspase 1/low caspase 9 activity in cells = pyroptosis? Circ. Res. 2016, 118, 1457–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.B.; Salomon, R.; Farfel-Becker, T.; Meshcheriakova, A.; Ali, M.; Klein, A.D.; Platt, F.M.; Cox, T.M.; Futerman, A.H. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med. 2014, 20, 204–208. [Google Scholar] [CrossRef]
- Moriwaki, K.; Ka, F.; Chan, M. The inflammatory signal adaptor RIPK3: Functions beyond necroptosis. Int. Rev. Cell Mol. Biol. 2017, 328, 253–275. [Google Scholar] [CrossRef] [Green Version]
- Gaffke, L.; Pierzynowska, K.; Piotrowska, E.; Węgrzyn, G. How close are we to therapies for Sanfilippo disease? Metab. Brain Dis. 2018, 33, 1. [Google Scholar] [CrossRef] [Green Version]
- Malinowska, M.; Wilkinson, F.L.; Langford-Smith, K.J.; Langford-Smith, A.; Brown, J.R.; Crawford, B.E.; Vanier, M.T.; Grynkiewicz, G.; Wynn, R.F.; Ed Wraith, J.; et al. Genistein improves neuropathology and corrects behaviour in a mouse model of neurodegenerative metabolic disease. PLoS ONE 2010, 5, e14192. [Google Scholar] [CrossRef] [Green Version]
- Piotrowska, E.; Jakóbkiewicz-Banecka, J.; Tylki-Szymanska, A.; Liberek, A.; Maryniak, A.; Malinowska, M.; Czartoryska, B.; Puk, E.; Kloska, A.; Liberek, T.; et al. Genistin-rich soy isoflavone extract in substrate reduction therapy for Sanfilippo syndrome: An open-label, pilot study in 10 pediatric patients. Curr. Ther. Res. 2008, 69, 166–179. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Rust, S.; Langford-Smith, K.; Weisberg, D.; Canal, M.; Breen, C.; Hepburn, M.; Tylee, K.; Vaz, F.M.; Vail, A.; et al. High dose genistein in Sanfilippo syndrome: A randomised controlled trial. J. Inherit. Metab. Dis. 2021, 44, 1248–1262. [Google Scholar] [CrossRef]
- DiRosario, J.; Divers, E.; Wang, C.; Etter, J.; Charrier, A.; Jukkola, P.; Auer, H.; Best, V.; Newsom, D.L.; McCarty, D.M.; et al. Innate and adaptive immune activation in the brain of MPS IIIB mouse model. J. Neurosci. Res. 2009, 87, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Open-Label Study of Anakinra in MPS III–Full Text View–ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04018755?term=ANAKINRA+MPS&draw=2&rank=1 (accessed on 18 November 2021).
- MPS III | MPS Society. Available online: https://www.mpssociety.org.uk/mps-iii (accessed on 18 November 2021).
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stylianou, E. INTERLEUKINS | IL-1 and IL-18; Elsevier: Amsterdam, The Netherlands, 2006; pp. 350–354. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets NLRP 3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef] [PubMed]
- Fecarotta, S.; Gasperini, S.; Parenti, G. New treatments for the mucopolysaccharidoses: From pathophysiology to therapy. Ital. J. Pediatr. 2018, 44, 124. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Ge, Y.; Lai, A.; Borisov, Y.; Faillace, M.; Eliyahu, E.; He, X.; Iatridis, J.; Vlassara, H.; Striker, G.; et al. Pentosan Polysulfate: A Novel Therapy for the Mucopolysaccharidoses. PLoS ONE 2013, 8, e54459. [Google Scholar] [CrossRef] [Green Version]
- Simonaro, C.M.; Tomatsu, S.; Sikora, T.; Kubaski, F.; Frohbergh, M.; Guevara, J.M.; Wang, R.Y.; Vera, M.; Kang, J.L.; Smith, L.J.; et al. Pentosan Polysulfate: Oral Versus Subcutaneous Injection in Mucopolysaccharidosis Type I Dogs. PLoS ONE 2016, 11, e0153136. [Google Scholar] [CrossRef] [Green Version]
- Hennermann, J.B.; Gökce, S.; Solyom, A.; Mengel, E.; Schuchman, E.H.; Simonaro, C.M. Treatment with pentosan polysulphate in patients with MPS I: Results from an open label, randomized, monocentric phase II study. J. Inherit. Metab. Dis. 2016, 39, 831–837. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Category | Disease | Deficient Enzyme | Accumulating Substance | Gene |
|---|---|---|---|---|
| MPS I-S | Scheie | DS, HS | IDUA | |
| MPS I-HS | Hurler-Scheie | α-l-iduronidase | ||
| MPS I-H | Hurler | |||
| MPS II | Hunter | Iduronate 2-sulfatase | DS, HS | IDS |
| MPS IIIA | Sanfilippo | Heparan N-Sulfatase | HS | SGSH |
| MPS IIIB | α-N-acetylglucosaminidase Acetyl-CoA-α | NAGLU | ||
| MPS IIIC | glucosaminidase | HGSNAT | ||
| MPS IIID | N-acetylglucosamine-6-sufatase | GNS | ||
| MPS IVA | Morquio A | Galactosamine-6-sulfatase | KS, CS | GALNS |
| MPS IVB | Morquio B | Β-galactosidase | KS | GBL1 |
| MPS VI | Maroteaux-Lamy | N-acetylgalactosamine-4-sulfatase | DS | ARSB |
| MPS VII | Sly | β-Glucuronidase | DS, HS, CS | GUSB |
| MPS IX | Natowicz | Hyaluronidase | HA | HYAL1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandolfo, O.; Parker, H.; Bigger, B. Innate Immunity in Mucopolysaccharide Diseases. Int. J. Mol. Sci. 2022, 23, 1999. https://doi.org/10.3390/ijms23041999
Mandolfo O, Parker H, Bigger B. Innate Immunity in Mucopolysaccharide Diseases. International Journal of Molecular Sciences. 2022; 23(4):1999. https://doi.org/10.3390/ijms23041999
Chicago/Turabian StyleMandolfo, Oriana, Helen Parker, and Brian Bigger. 2022. "Innate Immunity in Mucopolysaccharide Diseases" International Journal of Molecular Sciences 23, no. 4: 1999. https://doi.org/10.3390/ijms23041999