
Genome-Wide Identification, Classification and Expression Analysis of m6A Gene Family in Solanum lycopersicum


Abstract
:1. Introduction
2. Results
2.1. Genome-Wide Identification of m6A Gene Family in Tomato
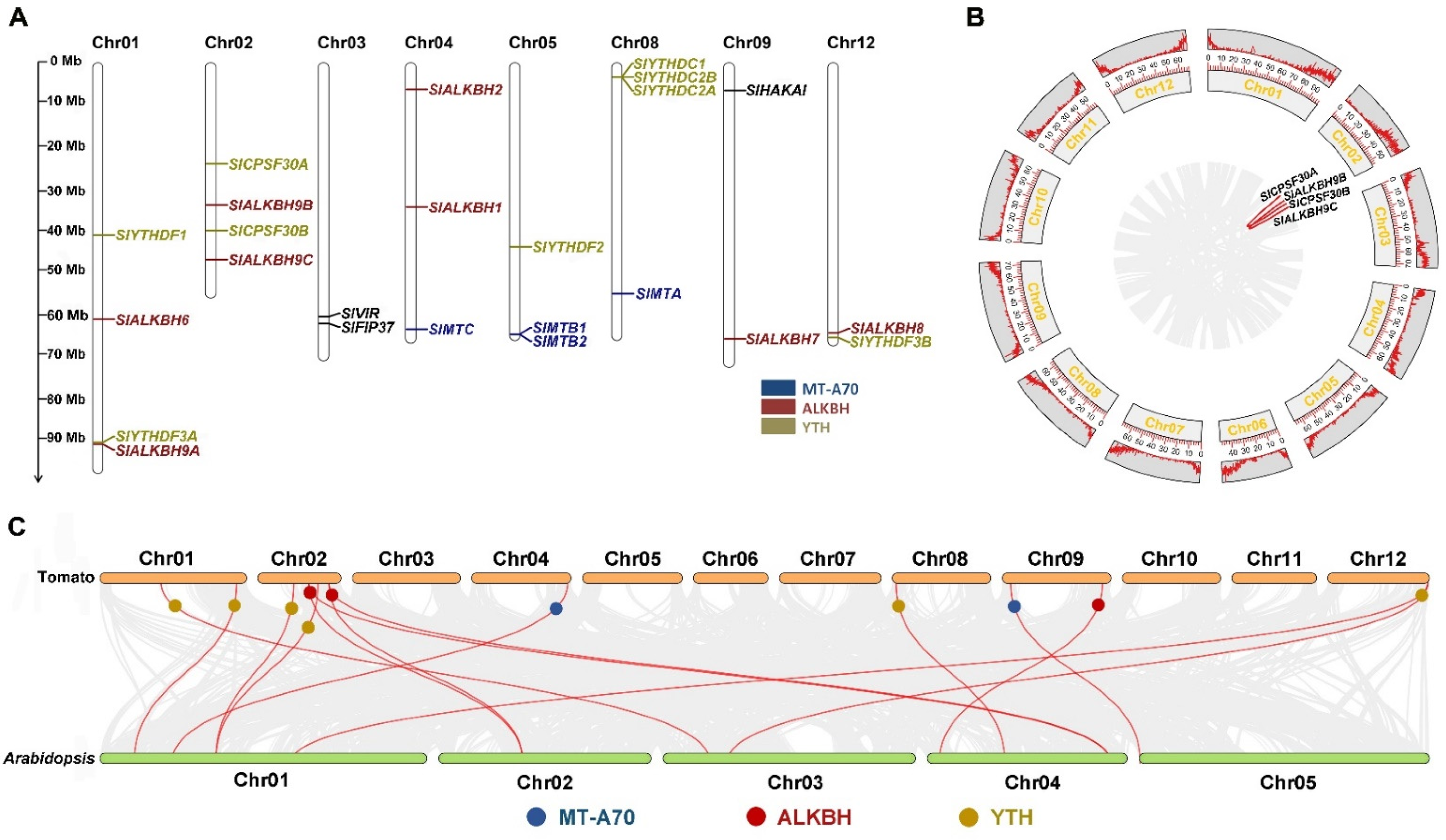
2.2. Chromosomal Location and Collinearity Analysis of m6A Gene Family in Tomato
2.3. Evolutionary and Structure Analyses of MT-A70 Family in Tomato
2.4. Evolutionary and Structure Analyses of ALKBH Family in Tomato
2.5. Evolutionary and Structure Analysis of YTH Family in Tomato
2.6. The Tissue Expression of m6A Genes and Their Family Genes in Tomato
2.7. Analysis of m6A Components and Their Family Genes under Abiotic Stress Treatments
2.8. Detection of RNA Modifications by LC-MS/MS
3. Discussion
4. Materials and Methods
4.1. Identification of m6A Components and Their Protein Families in Tomato
4.2. Chromosome Location, Synteny Analysis, and Ka/Ks Calculation
4.3. Alignment and Phylogenetic Analysis
4.4. Structure Construction by Homology Modeling
4.5. Gene Structure, Conserved Domain, Conserved Motif, and Cis-element Analyses
4.6. Digital Gene Expression and STEM Analysis
4.7. Plant Materials, Growth Conditions, and Stress Treatments
4.8. Total RNA Extraction and qPCR Analysis
4.9. Detection of RNA Modifications
4.10. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tuck, M.T. The formation of internal 6-methyladenine residues in eucaryotic mssenger RNA. Int. J. Biochem. 1992, 24, 379–386. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; He, C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013, 29, 108–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, X.-L.; Sun, B.-F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.-J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.-S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.-F.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.-Q.; Yang, Y.; Yi, C.-Q.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Zheng, G.-Q.; Dahl, J.A.; Niu, Y.-M.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.-F.; et al. N6-Methyladenosine dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.-S.; Roundtree, I.A.; Lu, Z.; Han, D.-L.; Ma, H.-H.; Weng, X.-C.; Chen, K.; Shi, H.L.; He, C. N6-Methyladenosine modulates messenger RNA translation efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wang, X.; Liu, K.; Roundtree, I.A.; Tempel, W.; Li, Y.-J.; Lu, Z.-K.; He, C.; Min, J.-R. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat. Chem. Biol. 2014, 10, 927–929. [Google Scholar] [CrossRef]
- Batista, P.J.; Molinie, B.; Wang, J.-K.; Qu, K.; Zhang, J.-J.; Li, L.-J.; Bouley, D.M.; Lujan, E.; Haddad, B.; Daneshvar, K.; et al. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014, 15, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Fustin, J.M.; Doi, M.; Yamaguchi, Y.; Hida, H.; Nishimura, S.; Yoshida, M.; Isagawa, T.; Morioka, M.S.; Kakeya, H.; Manabe, I.; et al. RNA methylation-dependent RNA processing controls the speed of the circadian clock. Cell 2013, 155, 793–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-B.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-Z.; Samanta, D.; Lu, H.-Q.; Bullen, J.W.; Zhang, H.-M.; Chen, I.; He, X.-S.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [PubMed] [Green Version]
- Bertero, A.; Brown, S.; Madrigal, P.; Osnato, A.; Ortmann, D.; Yiangou, L.; Kadiwala, J.; Hubner, N.C.; de Los Mozos, I.R.; Sadée, C.; et al. The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature 2018, 555, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; SalmonDivon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, L.-Q.; Zhao, Y.-L.; Yang, C.-G.; Roundtree, I.A.; Zhang, Z.-J.; Ren, J.; Xie, W.; He, C.; Luo, G.-Z. Single-base mapping of m6A by an antibody-independent method. Sci. Adv. 2019, 5, eaax0250. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.-L.; Li, H.-Y.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R.G. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008, 20, 1278–1288. [Google Scholar] [CrossRef] [Green Version]
- Bodi, Z.; Zhong, S.-L.; Mehra, S.; Song, J.; Graham, N.; Li, H.-Y.; May, S.; Fray, R.G. Adenosine methylation in Arabidopsis mRNA is associated with the 3′ end and reduced levels cause developmental defects. Front. Plant Sci. 2012, 3, 48. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.-S.; Liang, Z.; Gu, X.-F.; Chen, Y.; Teo, Z.W.; Hou, X.-L.; Cai, W.M.; Dedon, P.C.; Liu, L.; Yu, H. N6-Methyladenosine RNA modification regulates shoot stem cell fate in Arabidopsis. Dev. Cell 2016, 38, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, H.-C.; Wei, L.-H.; Zhang, C.; Wang, Y.; Chen, L.; Lu, Z.-K.; Chen, P.R.; He, C.; Jia, G.-F. ALKBH10B Is an RNA N6-Methyladenosine Demethylase Affecting Arabidopsis Floral Transition. Plant Cell 2017, 29, 2995–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arribas-Hernandez, L.; Bressendorff, S.; Hansen, M.H.; Poulsen, C.; Erdmann, S.; Brodersen, P. An m6A-YTH module controls developmental timing and morphogenesis in Arabidopsis. Plant Cell 2018, 30, 952–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scutenaire, J.; Deragon, J.M.; Jean, V.; Benhamed, M.; Raynaud, C.; Favory, J.J.; Merret, R.; Bousquet-Antonelli, C. The YTH domain protein ECT2 is an m6A reader required for normal trichome branching in Arabidopsis. Plant Cell 2018, 30, 986–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.H.; Song, P.-Z.; Wang, Y.; Lu, Z.-K.; Tang, Q.; Yu, Q.; Xiao, Y.; Zhang, X.; Duan, H.-C.; Jia, G.-F. The m6A reader ECT2 controls trichome morphology by affecting mRNA stability in Arabidopsis. Plant Cell 2018, 30, 968–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.-Z.; Cai, J.; Umme, A.; Chen, Y.; Xu, T.; Kang, H. Unique features of mRNA m6A methylomes during expansion of Tomato (Solanum lycopersicum) fruits. Plant Physiol. 2021, 3, kiab509. [Google Scholar] [CrossRef]
- Zhou, L.-L.; Tian, S.-P.; Qin, G.-Z. RNA methylomes reveal the m6A mediated regulation of DNA demethylase gene SlDML2 in tomato fruit ripening. Genome Biol. 2019, 20, 156. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.-L.; Tang, R.-K.; Li, X.-J.; Tian, S.-P.; Li, B.-B.; Qin, G.-Z. N6-Methyladenosine RNA modification regulates strawberry fruit ripening in an ABA-dependent manner. Genome Biol. 2021, 22, 168. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Y.-C.; Liao, J.-Y.; Yu, Y.; Zhou, Y.-F.; Feng, Y.-Z.; Yang, Y.-W.; Lei, M.-Q.; Bai, M.; Wu, H.; et al. The subunit of RNA N6-methyladenosine methyltransferase OsFIP regulates early degeneration of microspores in rice. PLoS Genet. 2019, 15, e1008120. [Google Scholar] [CrossRef] [Green Version]
- Miao, Z.-Y.; Zhang, T.; Qi, Y.-H.; Song, J.; Han, Z.-X.; Ma, C. Evolution of the RNA N6-methyladenosine methylome mediated by genomic duplication. Plant Physiol. 2020, 182, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.-X.; Sun, X.; Li, J.-L.; Song, Y.-S.; Song, J.; Wang, F.; Liu, L.-N.; Zhang, X.-S.; Sui, N. Analysis of N6-methyladenosine reveals a new important mechanism regulating the salt tolerance of sweet sorghum. Plant Sci. 2021, 304, 110801. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.J.; Kramer, M.C.; Gosai, S.J.; Yu, X.; Vandivier, L.E.; Nelson, A.D.L.; Anderson, Z.D.; Beilstein, M.A.; Fray, R.G.; Lyons, E.; et al. N6-methyladenosine inhibits local ribonucleolytic cleavage to stabilize mRNAs in Arabidopsis. Cell Rep. 2018, 25, 1146–1157.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huong, T.T.; Ngoc, L.N.-T.; Kang, H. Functional characterization of a putative RNA demethylase ALKBH6 in Arabidopsis growth and abiotic stress responses. Int. J. Mol. Sci. 2020, 21, 6707. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Nie, X.-J.; Yan, Z.-G.; Weining, S. N6-Methyladenosine regulatory machinery in plants: Composition, function and evolution. Plant Biotechnol. J. 2019, 17, 1194–1208. [Google Scholar] [CrossRef]
- Sun, J.; Bie, X.-M.; Wang, N.; Zhang, X.-S.; Gao, X.-Q. Genome-wide identification and expression analysis of YTH domain-containing RNA-binding protein family in common wheat. BMC Plant Biol. 2020, 20, 351. [Google Scholar] [CrossRef]
- Yin, S.-Q.; Ao, Q.-J.; Tan, C.-Y.; Yang, Y.-W. Genome-wide identification and characterization of YTH domain-containing genes, encoding the m6A readers, and their expression in tomato. Plant Cell Rep. 2021, 40, 1229–1245. [Google Scholar] [CrossRef]
- Liang, Z.; Geng, Y.-K.; Gu, X.-F. Adenine methylation: New epigenetic marker of DNA and mRNA. Mol. Plant 2018, 11, 1219–1221. [Google Scholar] [CrossRef] [Green Version]
- Iyer, L.M.; Zhang, D.-P.; Aravind, L. Adenine methylation in eukaryotes: Apprehending the complex evolutionary history and functional potential of an epigenetic modification. Bioessays 2016, 38, 27–40. [Google Scholar] [CrossRef]
- Greer, E.L.; Blanco, M.A.; Gu, L.; Sendinc, E.; Liu, J.-Z.; Aristizábal-Corrales, D.; Hsu, C.H.; Aravind, L.; He, C.; Shi, Y. DNA methylation on N (6)-adenine in C. elegans. Cell 2015, 161, 868–878. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Yang, Y.; Sun, B.-F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.-J.; Ping, X.-L.; Chen, Y.-S.; Wang, W.-J.; et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef]
- Tang, C.; Klukovich, R.; Peng, H.-Y.; Wang, Z.-Q.; Yu, T.; Zhang, Y.; Zheng, H.-L.; Klungland, A.; Yan, W. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3’-UTR mRNAs in male germ cells. Proc. Natl. Acad. Sci. USA 2018, 115, E325–E333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcinkowski, M.; Pilžys, T.; Garbicz, D.; Steciuk, J.; Zugaj, D.; Mielecki, D.; Sarnowski, T.J.; Grzesiuk, E. Human and Arabidopsis alpha-ketoglutarate-dependent dioxygenase homolog proteins-new players in important regulatory processes. IUBMB Life 2020, 72, 1126–1144. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Riaz, A.; Chachar, S.; Ding, Y.-K.; Du, H.; Gu, X.-F. Epigenetic modifications of mRNA and DNA in Plants. Mol. Plant 2020, 13, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, C.B.; Yu, D.; Hajian, T.; Li, J.; Huang, Y.; Dai, N.; Corrêa, I.R., Jr.; Wu, T.; Vedadi, M.; Zhang, X.; et al. Human MettL3-MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discov. 2019, 5, 63. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, S.-M.; Nelakanti, R.; Zhao, W.-T.; Liu, G.-C.; Li, Z.; Liu, X.-H.; Wu, T.; Xiao, A.; Li, H. Mammalian ALKBH1 serves as an N6-mA demethylase of unpairing DNA. Cell Res. 2020, 30, 197–210. [Google Scholar] [CrossRef]
- Tian, L.-F.; Liu, Y.-P.; Chen, L.-Q.; Tang, Q.; Wu, W.; Sun, W.; Chen, Z.-Z.; Yan, X.-X. Structural basis of nucleic acid recognition and 6mA demethylation by human ALKBH1. Cell Res. 2020, 30, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, C.B.; Horton, J.R.; Zhou, J.-J.; Bedford, M.T.; Blumenthal, R.M.; Zhang, X.; Cheng, X.-D. Biochemical and structural basis for YTH domain of human YTHDC1 binding to methylated adenine in DNA. Nucleic Acids Res. 2020, 48, 10329–10341. [Google Scholar] [CrossRef]
- Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar] [CrossRef] [Green Version]
- Thüring, K.; Schmid, K.; Keller, P.; Helm, M. LC-MS analysis of methylated RNA. Methods Mol. Biol. 2017, 1562, 3–18. [Google Scholar]
- Sugita, A.; Kuruma, S.; Yanagisawa, N.; Ishiguro, H.; Kano, R.; Ohkuma, Y.; Hirose, Y. The cap-specific m6A methyltransferase, PCIF1/CAPAM, is dynamically recruited to the gene promoter in a transcription-dependent manner. J. Biochem. 2021, 170, 203–213. [Google Scholar] [CrossRef]
- Akichika, S.; Hirano, S.; Shichino, Y.; Suzuki, T.; Nishimasu, H.; Ishitani, R.; Sugita, A.; Hirose, Y.; Iwasaki, S.; Nureki, O.; et al. Cap-specific terminal N6-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science 2019, 363, eaav0080. [Google Scholar] [CrossRef]
- Sendinc, E.; Valle-Garcia, D.; Dhall, A.; Chen, H.; Henriques, T.; Navarrete-Perea, J.; Sheng, W.-Q.; Gygi, S.P.; Adelman, K.; Shi, Y. PCIF1 Catalyzes m6Am mRNA methylation to regulate gene expression. Mol. Cell 2019, 75, 620–630.e9. [Google Scholar] [CrossRef] [PubMed]
- Boulias, K.; Toczydłowska-Socha, D.; Hawley, B.R.; Liberman, N.; Takashima, K.; Zaccara, S.; Guez, T.; Vasseur, J.J.; Debart, F.; Aravind, L.; et al. Identification of the m6Am methyltransferase PCIF1 reveals the location and functions of m6Am in the transcriptome. Mol. Cell 2019, 75, 631–643.e8. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.C.; Purugganan, M.D. The early stages of duplicate gene evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 15682–15687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, H.-Z.; Landherr, L.L.; Frohlich, M.W.; Leebens-Mack, J.; Ma, H.; DePamphilis, C.W. Patterns of gene duplication in the plant SKP1 gene family in angiosperms: Evidence for multiple mechanisms of rapid gene birth. Plant J. 2007, 50, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.-L.; Wong, C.-E.; Shen, L.-S.; Yu, H. N6-Methyladenosine modification underlies messenger RNA metabolism and plant development. Curr. Opin. Plant Biol. 2021, 63, 102047. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Shen, L.-S.; Cui, X.-A.; Bao, S.-J.; Geng, Y.-K.; Yu, G.-L.; Liang, F.; Xie, S.; Lu, T.-G.; Gu, X.-F.; et al. DNA N6-Adenine methylation in Arabidopsis thaliana. Dev. Cell 2018, 45, 406–416.e3. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Liang, Z.; Cui, X.; Ji, C.; Li, Y.; Zhang, P.; Liu, J.; Riaz, A.; Yao, P.; Liu, M.; et al. N6-methyladenine DNA methylation in Japonica and Indica rice genomes and its association with gene expression, plant development, and stress responses. Mol Plant. 2018, 11, 1492–1508. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.-Z.; Cai, J.; Park, S.J.; Lee, K.; Li, Y.-X.; Chen, Y.; Yun, J.-Y.; Xu, T.; Kang, H.-S. N6-Methyladenosine mRNA methylation is important for salt stress tolerance in Arabidopsis. Plant J. 2021, 106, 1759–1775. [Google Scholar] [CrossRef]
- Yang, D.-D.; Xu, H.-C.; Liu, Y.; Li, M.-Z.; Ali, M.; Xu, X.-Y.; Lu, G. RNA N6-methyladenosine responds to low-temperature stress in tomato anthers. Front. Plant Sci. 2021, 12, 687826. [Google Scholar] [CrossRef]
- Chen, C.-J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.-H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-P.; Tang, H.-B.; Debarry, J.D.; Tan, X.; Li, J.-P.; Wang, X.-Y.; Lee, T.H.; Jin, H.-Z.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, M.A.; Haubold, B.; Mitchell-Olds, T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 2000, 17, 1483–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [Green Version]
- Expósito-Rodríguez, M.; Borges, A.A.; Borges-Pérez, A.; Pérez, J.A. Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biol. 2008, 8, 131. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Family | Gene Name | Gene ID | Protein Length (aa) | Molecular Weight (KD) | Theoretical pI |
|---|---|---|---|---|---|
| MT-A70 | SlMTA | Solyc08g066730.3 | 739 | 81.47 | 6.43 |
| SlMTB1 | Solyc05g056210.2 | 1094 | 122.62 | 6.34 | |
| SlMTB2 | Solyc05g056220.2 | 1091 | 121.98 | 6.39 | |
| SlMTC | Solyc04g079950.3 | 376 | 48.09 | 6.73 | |
| ALKBH | SlALKBH1 | Solyc04g045590.3 | 354 | 39.91 | 5.59 |
| SlALKBH2 | Solyc04g015080.3 | 253 | 29.10 | 9.02 | |
| SlALKBH6 | Solyc01g057570.3 | 261 | 29.47 | 6.70 | |
| SlALKBH7 | Solyc09g074920.3 | 259 | 29.47 | 4.86 | |
| SlALKBH8 | Solyc12g096230.2 | 342 | 38.59 | 6.32 | |
| SlALKBH9A | Solyc01g104130.3 | 445 | 50.47 | 8.76 | |
| SlALKBH9B | Solyc02g062180.3 | 643 | 71.18 | 5.87 | |
| SlALKBH9C | Solyc02g083960.3 | 538 | 60.30 | 6.37 | |
| YTH | SlYTHDF1 | Solyc01g028860.3 | 706 | 77.22 | 6.95 |
| SlYTHDF2 | Solyc05g032850.3 | 604 | 65.97 | 5.31 | |
| SlYTHDF3A | Solyc01g103540.3 | 570 | 63.29 | 8.49 | |
| SlYTHDF3B | Solyc12g099090.2 | 728 | 79.40 | 6.05 | |
| SlYTHDC1 | Solyc08g007740.2 | 395 | 44.21 | 6.39 | |
| SlYTHDC2A | Solyc08g007760.3 | 394 | 44.27 | 6.10 | |
| SlYTHDC2B | Solyc08g007750.3 | 389 | 43.16 | 6.17 | |
| SlCPSF30A | Solyc02g021760.3 | 689 | 75.93 | 6.24 | |
| SlCOSF30B | Solyc02g070240.3 | 671 | 73.77 | 6.10 | |
| SlFIP37 * | Solyc03g112520.3 | 342 | 38.64 | 4.86 | |
| SlVIR * | Solyc03g020020.3 | 2196 | 240.80 | 5.49 | |
| SlHAKAI * | Solyc09g013120.3 | 424 | 46.47 | 6.80 |
| Duplicated Gene Pair | Ka | Ks | Ka/Ks | Duplication Type | Selection | Time (MYA) |
|---|---|---|---|---|---|---|
| SlMTB1/SlMTB2 | 0.083561 | 0.291480 | 0.286679 | Tandem | Purifying | 9.71 |
| SlYTHDC1/SlYTHDC2A | 0.793902 | 2.605968 | 0.304647 | Tandem | Purifying | 86.86 |
| SlYTHDC2A/SlYTHDC2B | 0.879281 | 2.117476 | 0.415249 | Tandem | Purifying | 70.58 |
| SlALKBH9B/SlALKBH9C | 0.187651 | 0.724894 | 0.258866 | Segment | Purifying | 24.16 |
| SlCPSF30A/SlCPSF30B | 0.11570 | 0.585231 | 0.197709 | Segment | Purifying | 19.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, H.; Luo, B.; Wang, Y.; Li, J.; Hu, Z.; Xie, Q.; Wu, T.; Chen, G. Genome-Wide Identification, Classification and Expression Analysis of m6A Gene Family in Solanum lycopersicum. Int. J. Mol. Sci. 2022, 23, 4522. https://doi.org/10.3390/ijms23094522
Shen H, Luo B, Wang Y, Li J, Hu Z, Xie Q, Wu T, Chen G. Genome-Wide Identification, Classification and Expression Analysis of m6A Gene Family in Solanum lycopersicum. International Journal of Molecular Sciences. 2022; 23(9):4522. https://doi.org/10.3390/ijms23094522
Chicago/Turabian StyleShen, Hui, Baobing Luo, Yunshu Wang, Jing Li, Zongli Hu, Qiaoli Xie, Ting Wu, and Guoping Chen. 2022. "Genome-Wide Identification, Classification and Expression Analysis of m6A Gene Family in Solanum lycopersicum" International Journal of Molecular Sciences 23, no. 9: 4522. https://doi.org/10.3390/ijms23094522