
Molecular Mechanisms of Tumor Immunomodulation in the Microenvironment of Colorectal Cancer

Abstract
:1. Introduction
2. Overview of Colorectal Tumor Subtypes and Associated Immune Microenvironments
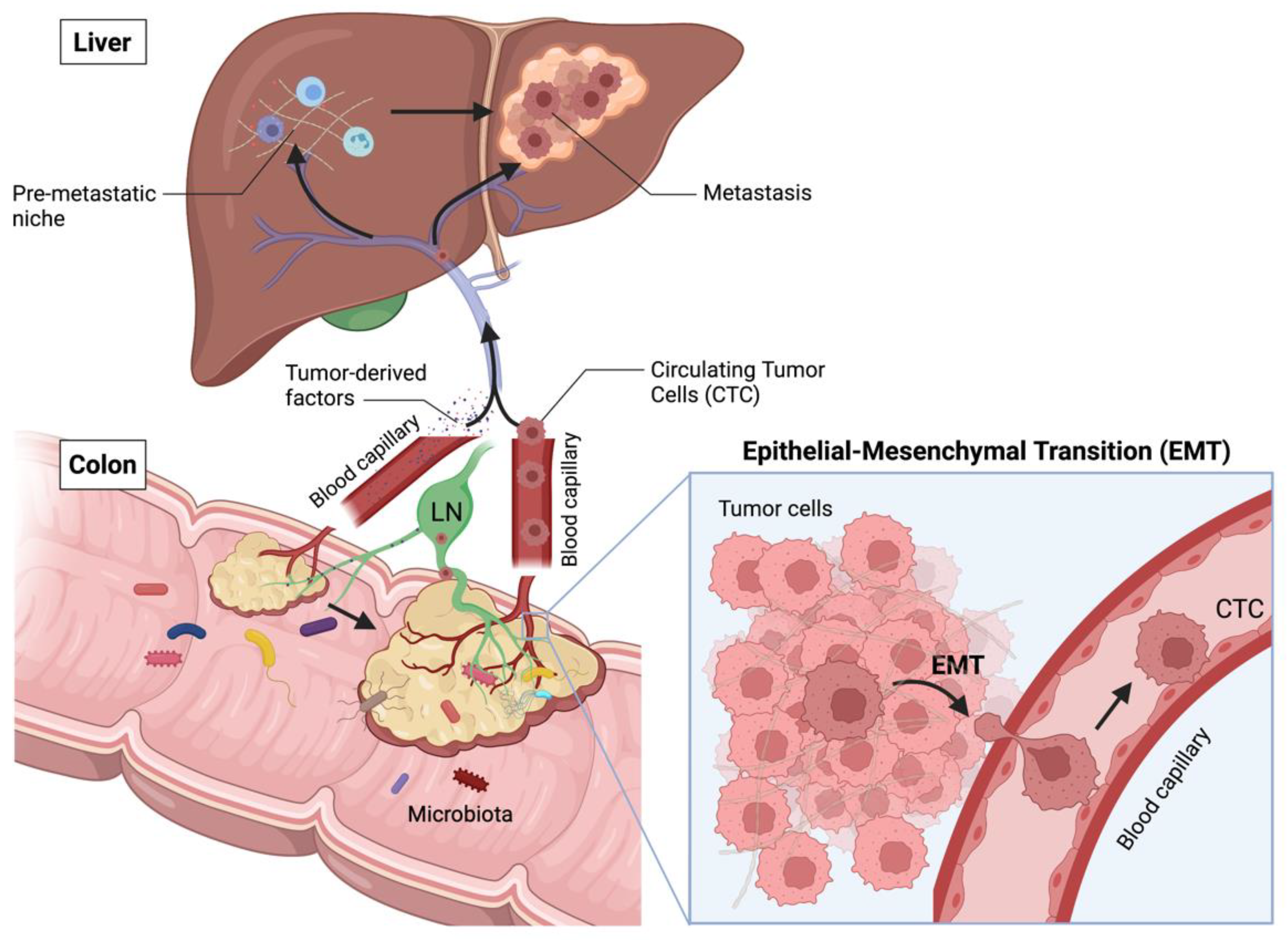
2.1. Main Stages of CRC Progression
2.2. CRC Consensus Molecular Subgroups (CMS) and Associated Immune Landscapes
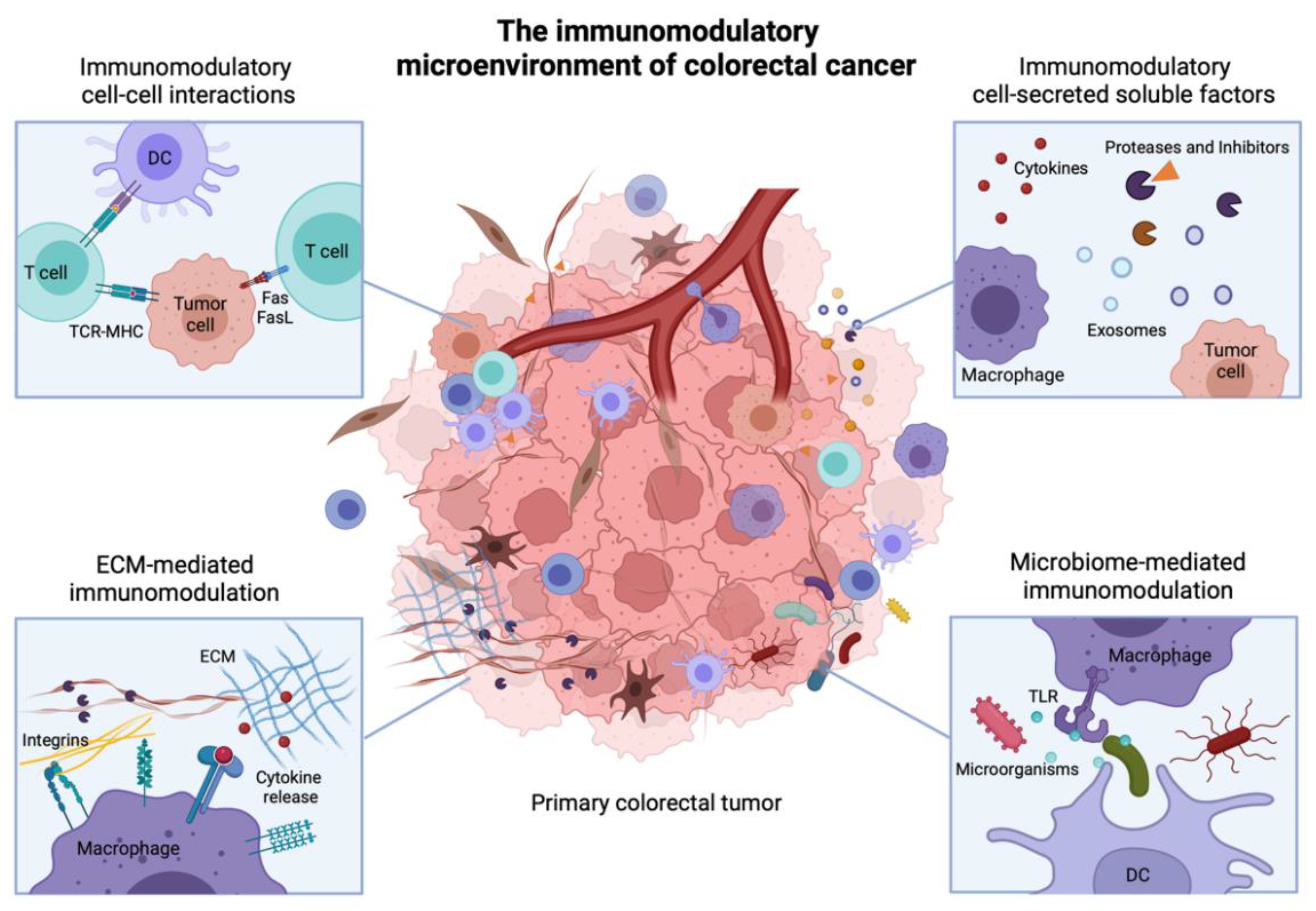
3. Immunomodulatory Cell-Cell Interactions in the TME
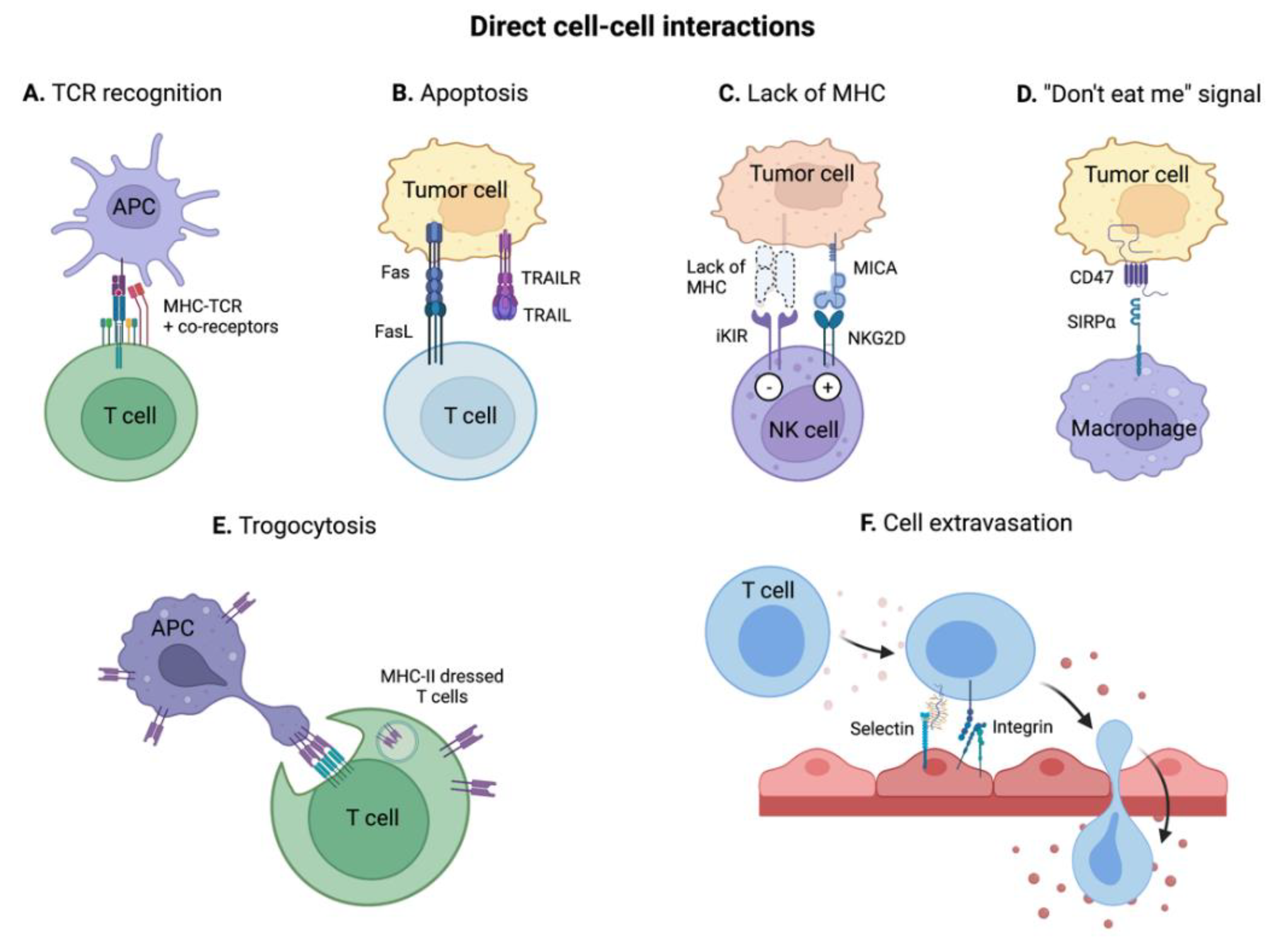
3.1. Direct Cell-Cell Receptor Interactions
3.1.1. MHC-T Cell Receptor (TCR) Interaction and Co-Receptors
3.1.2. Induction of Apoptosis via Fas/FasL and TRAIL/TRAIL-R Interactions
3.1.3. Detection of the Lack of MHC I and of Oncogenic Stress by NK Cells
3.1.4. “Do Not Eat Me” Signals: Escaping Phagocytosis
3.1.5. Trogocytosis: Receptor Transfer between Tumor and Immune Cells
3.1.6. Immune Cell Recruitment in the TME
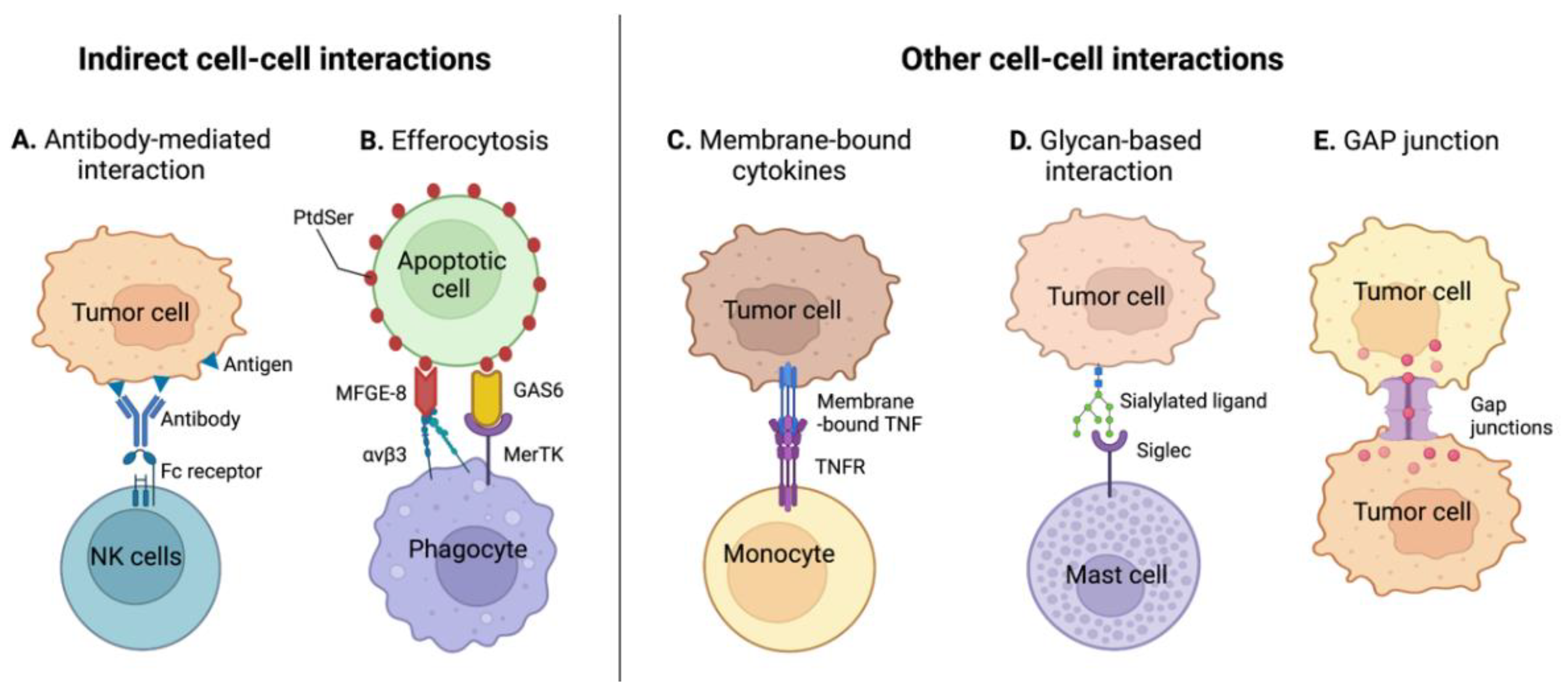
3.2. Indirect Cell-Cell Receptor Interactions
3.2.1. Antibody-Mediated Cell-Cell Interactions
3.2.2. Tumor Efferocytosis
3.3. Other Immunomodulatory Cell-Cell Interactions
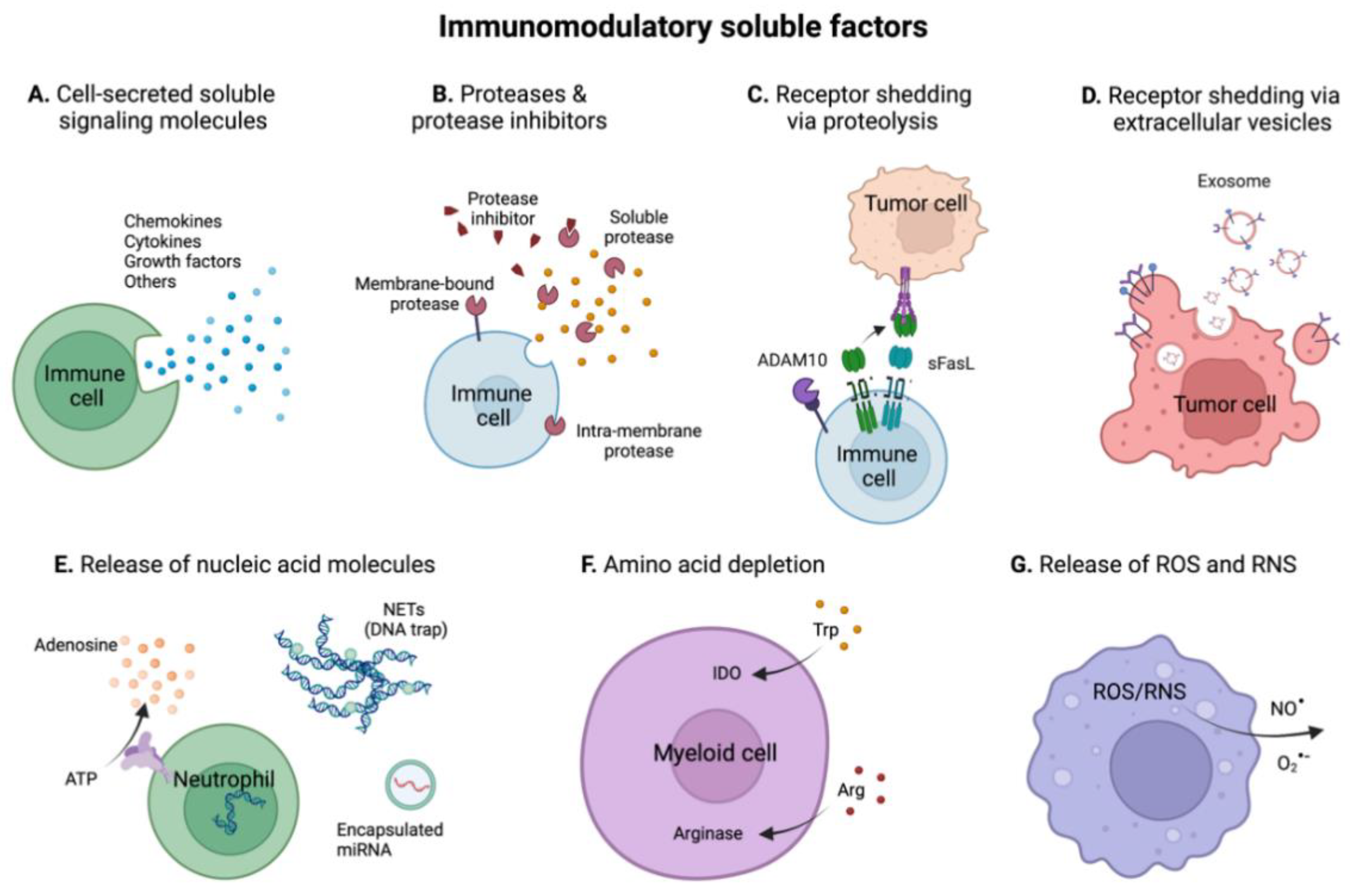
4. Immunomodulatory Soluble Factors in the TME
4.1. Cytokines
4.1.1. Chemokines
4.1.2. ILs
4.1.3. IFNs
4.1.4. TNF Superfamily (TNFSF)
4.1.5. CSF Superfamily
4.1.6. TGF-β Superfamily
4.1.7. Other Cell-Secreted Soluble Signaling Proteins
4.2. Proteases and Protease Inhibitors
4.3. Receptor Shedding in the TME
4.4. Immunomodulation by Nucleic Acids in the TME
4.5. Immunomodulation by Depletion of Amino Acids in the TME
4.6. Reactive Oxygen and Nitrogen Species (ROS and RNS)
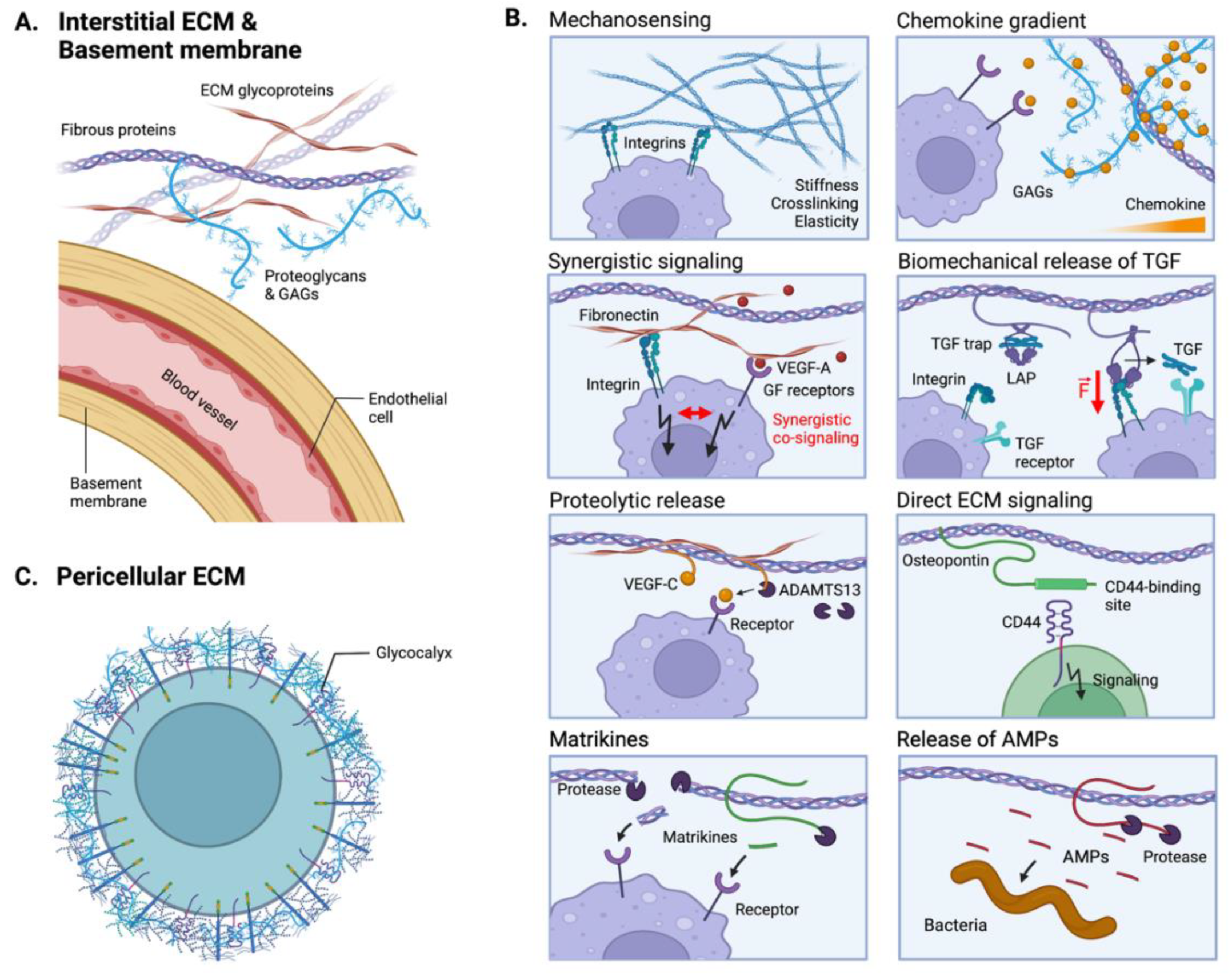
5. ECM-Mediated Tumor Immunomodulation
5.1. The ECM as an Immunomodulatory Biomechanical Scaffold
5.2. The ECM as a Reservoir of Immunomodulatory Proteins
5.3. Direct ECM Signaling to Immunoreceptors
5.4. Indirect ECM-Mediated Immunomodulation via Anti-Microbial Activities
5.5. The Emergent Role of the Cell Glycocalyx in Cancer Immunity
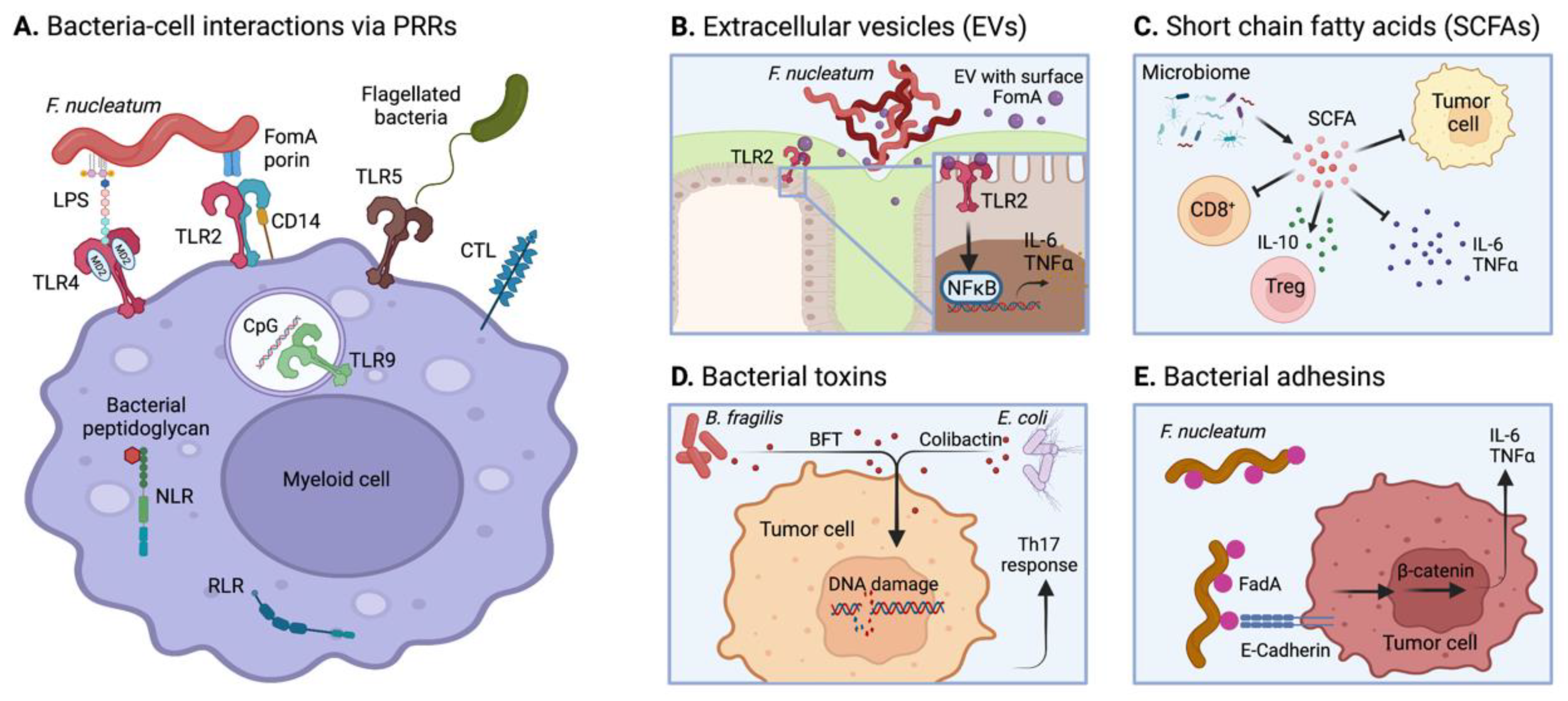
6. Microbiome-Mediated Tumor Immunomodulation
6.1. Intestinal Microbiota and Gut Immune Homeostasis
6.2. Pro- and Anti-Tumorigenic Microbial Inflammation
6.3. Microbial Modulation of Tumor Inflammation
6.3.1. PRR Activation
6.3.2. Microbial EVs
6.3.3. Short-Chain Fatty Acids (SCFAs)
6.3.4. Microbial Toxins
6.3.5. Bacterial Adhesins
7. Conclusions: The Integrated Immunomodulatory Microenvironment of Colorectal Tumors and Therapeutic Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henderson, R.H.; French, D.; Maughan, T.; Adams, R.; Allemani, C.; Minicozzi, P.; Coleman, M.P.; McFerran, E.; Sullivan, R.; Lawler, M. The economic burden of colorectal cancer across Europe: A population-based cost-of-illness study. Lancet Gastroenterol. Hepatol. 2021, 6, 709–722. [Google Scholar] [CrossRef]
- Holch, J.W.; Ricard, I.; Stintzing, S.; Modest, D.P.; Heinemann, V. The relevance of primary tumour location in patients with metastatic colorectal cancer: A meta-analysis of first-line clinical trials. Eur. J. Cancer 2017, 70, 87–98. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Rumpold, H.; Niedersüß-Beke, D.; Heiler, C.; Falch, D.; Wundsam, H.V.; Metz-Gercek, S.; Piringer, G.; Thaler, J. Prediction of mortality in metastatic colorectal cancer in a real-life population: A multicenter explorative analysis. BMC Cancer 2020, 20, 1–9. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Kesselring, R.; Glaesner, J.; Hiergeist, A.; Naschberger, E.; Neumann, H.; Brunner, S.M.; Wege, A.K.; Seebauer, C.; Köhl, G.; Merkl, S.; et al. IRAK-M Expression in Tumor Cells Supports Colorectal Cancer Progression through Reduction of Antimicrobial Defense and Stabilization of STAT3. Cell 2016, 29, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Erler, T.; Bennewith, L.; Cox, R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.; Giaccia, J. Hypoxia-induced lysis oxidase is a critical mediator of bone marrow cell recruitment to form the pre-metastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, S.; Watanabe, A.; Sakurai, Y.; Akashi-Takamura, S.; Ishibashi, S.; Miyake, K.; Shibuya, M.; Akira, S.; Aburatani, H.; Maru, Y. The S100A8–serum amyloid A3–TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 2008, 10, 1349–1355. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seubert, B.; Grünwald, B.; Kobuch, J.; Cui, H.; Schelter, F.; Schaten, S.; Jams, S.; Lim, N.; Nagase, H.; Simonavicius, N.; et al. TIMP-1 creates a pre-metastatic niche in the liver through SDF-1/ CXCR4-dependent neutrophil recruitment in mice. Hepatology 2015, 61, 238–248. [Google Scholar] [CrossRef]
- Dong, Q.; Liu, X.; Cheng, K.; Sheng, J.; Kong, J.; Liu, T. Pre-metastatic Niche Formation in Different Organs Induced by Tumor Extracellular Vesicles. Front. Cell Dev. Biol. 2021, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R. The Hallmarks of Cancer. Med. Lav. 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Brabletz, T. To differentiate or not-routes towards metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef]
- Kamal, Y.; Schmit, S.L.; Frost, H.R.; Amos, C.I. The tumor microenvironment of colorectal cancer metastases: Opportunities in cancer immunotherapy. Immunotherapy 2020, 12, 1083–1100. [Google Scholar] [CrossRef]
- Bertocchi, A.; Carloni, S.; Ravenda, P.S.; Bertalot, G.; Spadoni, I.; Lo Cascio, A.; Gandini, S.; Lizier, M.; Braga, D.; Asnicar, F.; et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell 2021, 39, 708–724.e11. [Google Scholar] [CrossRef]
- Barbirou, M.; Woldu, H.G.; Sghaier, I.; Bedoui, S.A.; Mokrani, A.; Aami, R.; Mezlini, A.; Yacoubi-Loueslati, B.; Tonellato, P.J.; Bouhaouala-Zahar, B. Western influenced lifestyle and Kv2.1 association as predicted biomarkers for Tunisian colorectal cancer. BMC Cancer 2020, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Ren, C.; Liu, Y.; Lan, H.; Wang, Z. An update on colorectal cancer microenvironment, epigenetic and immunotherapy. Int. Immunopharmacol. 2020, 89, 107041. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, S.; Lartey, D.A.; D’haens, G.R.; Grootjans, J. The role of the immune system in ibd-associated colorectal cancer: From pro to anti-tumorigenic mechanisms. Int. J. Mol. Sci. 2021, 22, 12739. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Picard, E.; Verschoor, C.P.; Ma, G.W.; Pawelec, G. Relationships Between Immune Landscapes, Genetic Subtypes and Responses to Immunotherapy in Colorectal Cancer. Front. Immunol. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Fabian, K.P.; Wolfson, B.; Hodge, J.W. From Immunogenic Cell Death to Immunogenic Modulation: Select Chemotherapy Regimens Induce a Spectrum of Immune-Enhancing Activities in the Tumor Microenvironment. Front. Oncol. 2021, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Fang, T.; Yin, X.; Wang, Y.; Yang, Y.; Zhang, L.; Xue, Y. Serum CD4 Is Associated with the Infiltration of CD4+T Cells in the Tumor Microenvironment of Gastric Cancer. J. Immunol. Res. 2021, 2021, 1–13. [Google Scholar] [CrossRef]
- Eissner, G.; Kirchner, S.; Lindner, H.; Kolch, W.; Janosch, P.; Grell, M.; Scheurich, P.; Andreesen, R.; Holler, E. Reverse Signaling Through Transmembrane TNF Confers Resistance to Lipopolysaccharide in Human Monocytes and Macrophages. J. Immunol. 2000, 164, 6193–6198. [Google Scholar] [CrossRef] [PubMed]
- Briquez, P.S.; Hauert, S.; de Titta, A.; Gray, L.T.; Alpar, A.T.; Swartz, M.A.; Hubbell, J.A. Engineering Targeting Materials for Therapeutic Cancer Vaccines. Front. Bioeng. Biotechnol. 2020, 8, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Mullins, C.S.; Linnebacher, M. Colorectal cancer vaccines: Tumor-associated antigens vs neoantigens. World J. Gastroenterol. 2018, 24, 5418–5432. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, T.; Yin, L.; Zuo, D.; Lin, Y.; Wang, L. Prognostic and clinicopathological value of MUC1 expression in colorectal cancer; A meta-analysis. Medicine 2019, 98, 1–8. [Google Scholar]
- Simons, K.H.; de Jong, A.; Jukema, J.W.; de Vries, M.R.; Arens, R.; Quax, P.H.A. T cell co-stimulation and co-inhibition in cardiovascular disease: A double-edged sword. Nat. Rev. Cardiol. 2019, 16, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H.; Bronte, V. Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 2016, 39, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sznol, M.; Chen, L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin. Cancer Res. 2013, 19, 1021–1034. [Google Scholar] [CrossRef] [Green Version]
- Oyewole-Said, D.; Konduri, V.; Vazquez-Perez, J.; Weldon, S.A.; Levitt, J.M.; Decker, W.K. Beyond T-Cells: Functional Characterization of CTLA-4 Expression in Immune and Non-Immune Cell Types. Front. Immunol. 2020, 11, 3168. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, Y.; Du, J.; Zhan, Y.; Sun, D.; Zhao, J.; Zhang, S.; Li, J.; He, K. Both light-induced SA accumulation and ETI mediators contribute to the cell death regulated by BAK1 and BKK1. Front. Plant Sci. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houston, J.P. Apoptosis and autophagy. Cytom. Part A 2019, 95, 655–656. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.; Fais, S.; Iero, M.; Lugini, L.; Canese, P.; Squarcina, P.; Zaccheddu, A.; Colone, M.; Arancia, G.; Gentile, M.; et al. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: Role in immune escape. Gastroenterology 2005, 128, 1796–1804. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.R.; Kim, T.D.; Choi, I. Understanding of molecular mechanisms in natural killer cell therapy. Exp. Mol. Med. 2015, 47, e141. [Google Scholar] [CrossRef] [PubMed]
- McGilvray, R.W.; Eagle, R.A.; Watson, N.F.S.; Al-Attar, A.; Ball, G.; Jafferji, I.; Trowsdale, J.; Durrant, L.G. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin. Cancer Res. 2009, 15, 6993–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.P.; Weissman, I.L.; Majeti, R. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front. Oncol. 2020, 9, 1380. [Google Scholar] [CrossRef]
- Banerjea, A.; Ahmed, S.; Hands, R.E.; Huang, F.; Han, X.; Shaw, P.M.; Feakins, R.; Bustin, S.A.; Dorudi, S. Colorectal cancers with microsatellite instability display mRNA expression signatures characteristic of increased immunogenicity. Mol. Cancer 2004, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara-Tani, R.; Sasaki, T.; Ohmori, H.; Luo, Y.; Goto, K.; Nishiguchi, Y.; Mori, S.; Nakashima, C.; Mori, T.; Miyagawa, Y.; et al. Concurrent Expression of CD47 and CD44 in Colorectal Cancer Promotes Malignancy. Pathobiology 2019, 86, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Mohme, M.; Riethdorf, S.; Pantel, K. Circulating and disseminated tumour cells-mechanisms of immune surveillance and escape. Nat. Rev. Clin. Oncol. 2017, 14, 155–167. [Google Scholar] [CrossRef]
- Barkal, A.A.; Weiskopf, K.; Kao, K.S.; Gordon, S.R.; Rosental, B.; Yiu, Y.Y.; George, B.M.; Markovic, M.; Ring, N.G.; Tsai, J.M.; et al. Engagement of MHC class i by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy article. Nat. Immunol. 2018, 19, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Feng, K.; Guo, Y.; Dai, H.; Wang, Y.; Li, X.; Jia, H.; Han, W. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci. China Life Sci. 2016, 59, 468–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.H.; Jeong, J.; Maher, S.E.; Lee, H.-W.; Lim, J.; Bothwell, A.L.M. Colon cancer cells acquire immune regulatory molecules from tumor-infiltrating lymphocytes by trogocytosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2110241118. [Google Scholar] [CrossRef]
- Sabzevari, H.; Kantor, J.; Jaigirdar, A.; Tagaya, Y.; Naramura, M.; Hodge, J.W.; Bernon, J.; Schlom, J. Acquisition of CD80 (B7-1) by T Cells. J. Immunol. 2001, 166, 2505–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dopfer, E.P.; Minguet, S.; Schamel, W.W.A. A New Vampire Saga: The Molecular Mechanism of T Cell Trogocytosis. Immunity 2011, 35, 151–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnoor, M.; Lai, F.P.L.; Zarbock, A.; Kläver, R.; Polaschegg, C.; Schulte, D.; Weich, H.A.; Oelkers, J.M.; Rottner, K.; Vestweber, D. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J. Exp. Med. 2011, 208, 1721–1735. [Google Scholar] [CrossRef] [Green Version]
- Strell, C.; Paulsson, J.; Jin, S.B.; Tobin, N.P.; Mezheyeuski, A.; Roswall, P.; Mutgan, C.; Mitsios, N.; Johansson, H.; Wickberg, S.M.; et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J. Natl. Cancer Inst. 2019, 111, 983–995. [Google Scholar] [CrossRef]
- Thomas, S.N.; Zhu, F.; Schnaar, R.L.; Alves, C.S.; Konstantopoulos, K. Carcinoembryonic antigen and CD44 variant isoforms cooperate to mediate colon carcinoma cell adhesion to E- and L-selectin in shear flow. J. Biol. Chem. 2008, 283, 15647–15655. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gül, N.; van Egmond, M. Antibody-dependent phagocytosis of tumor cells by Macrophages: A Potent effector mechanism of monoclonal antibody therapy of cancer. Cancer Res. 2015, 75, 5008–5013. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.; Ishii, Y.; Ochiai, H.; Fukuda, K.; Akimoto, S.; Hayashida, T.; Okabayashi, K.; Tsuruta, M.; Hasegawa, H.; Kitagawa, Y. Cetuximab-mediated ADCC activity is correlated with the cell surface expression level of EGFR but not with the KRAS/BRAF mutational status in colorectal cancer. Oncol. Rep. 2014, 31, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veluchamy, J.P.; Spanholtz, J.; Tordoir, M.; Thijssen, V.L.; Heideman, D.A.M.; Verheul, H.M.W.; De Gruijl, T.D.; Van Der Vliet, H.J. Combination of NK cells and cetuximab to enhance anti-tumor responses in RAS mutant metastatic colorectal cancer. PLoS ONE 2016, 11, e0157830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdes, C.A.; Nicolini, V.G.; Herter, S.; Van Puijenbroek, E.; Lang, S.; Roemmele, M.; Moessner, E.; Freytag, O.; Friess, T.; Ries, C.H.; et al. GA201 (RG7160): A novel, humanized, glycoengineered anti—EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin. Cancer Res. 2013, 19, 1126–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, G. Phosphatidylserine is the signal for TAM receptors and their ligands. Trends Biochem. Sci. 2017, 42, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werfel, A.; Cook, S. Efferocytosis in the Tumor Microenvironment. Encycl. Cell Biol. 2016, 3, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Akitake-Kawano, R.; Seno, H.; Nakatsuji, M.; Kimura, Y.; Nakanishi, Y.; Yoshioka, T.; Kanda, K.; Kawada, M.; Kawada, K.; Sakai, Y.; et al. Inhibitory role of Gas6 in intestinal tumorigenesis. Carcinogenesis 2013, 34, 1567–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, M.; Yao, H.; Chen, C.; Wang, Y.; Wang, H.; Cui, T.; Zhu, J. Prognostic Correlation Between MFG-E8 Expression Level and Colorectal Cancer. Arch. Med. Res. 2017, 48, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yao, Y.; Deng, Y.; Shao, A. Regulation of efferocytosis as a novel cancer therapy. Cell Commun. Signal. 2020, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Josephs, S.F.; Ichim, T.E.; Prince, S.M.; Kesari, S.; Marincola, F.M.; Escobedo, A.R.; Jafri, A. Unleashing endogenous TNF-alpha as a cancer immunotherapeutic. J. Transl. Med. 2018, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, S.; Deskins, D.L.; Young, P.P. Membrane TNF-alpha-activated programmed necrosis is mediated by Ceramide-induced reactive oxygen species. J. Mol. Signal. 2013, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Blokhuis, B.R.J.; Diks, M.A.P.; Keshavarzian, A.; Garssen, J.; Redegeld, F.A. Functional inhibitory siglec-6 is upregulated in human colorectal cancer-associated mast cells. Front. Immunol. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph receptors and ephrins engage in cellular cannibalism. J. Cell Biol. 2019, 218, 3168–3170. [Google Scholar] [CrossRef] [Green Version]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bristol Myers Squibb. Opdivo (Nivolumab) in Combination with Yervoy (Ipilimumab) Demonstrates Clinical Activity in Previously Treated Patients with dMMR or MSI-H Metastatic Colorectal Cancer. 2018; Press Release. New York, NY, USA. [Google Scholar]
- Dinarello, C.A. Historical Review of Cytokines. Eur. J. Immunol. 2007, 37 (Suppl. S1), S34–S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. The Chemokine Superfamily Revisited NIH Public Access. Bone 2011, 23, 1–7. [Google Scholar]
- Franca-Koh, J.; Kamimura, Y.; Devreotes, P. Navigating signaling networks: Chemotaxis in Dictyostelium discoideum. Curr. Opin. Genet. Dev. 2006, 16, 333–338. [Google Scholar] [CrossRef]
- Jin, T.; Xu, X.; Hereld, D. Chemotaxis, chemokine receptors and human disease. Cytokine 2008, 44, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2021, 29, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Karin, N.; Wildbaum, G. The role of chemokines in shaping the balance between CD4+ T cell subsets and its therapeutic implications in autoimmune and cancer diseases. Front. Immunol. 2015, 6, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, M.; Hauschild, R.; Schwarz, J.; Moussion, C.; De Vries, I.; Legler, D.F.; Luther, S.A.; Bollenbach, T.; Sixt, M. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 2013, 339, 328–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günther, K.; Leier, J.; Henning, G.; Dimmler, A.; Weißbach, R.; Hohenberger, W.; Förster, R. Prediction of lymph node metastasis in colorectal carcinoma by expression of chemokine receptor CCR7. Int. J. Cancer 2005, 116, 726–733. [Google Scholar] [CrossRef]
- Mantovani, A.; Savino, B.; Locati, M.; Zammataro, L.; Allavena, P.; Bonecchi, R. The chemokine system in cancer biology and therapy. Cytokine Growth Factor Rev. 2010, 21, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Brocker, C.; Thompson, D.; Matsumoto, A.; Nebert, D.W.; Vasiliou, V. Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum. Genom. 2010, 5, 30–55. [Google Scholar] [CrossRef]
- Briukhovetska, D.; Dörr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in cancer: From biology to therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, L.; Zhao, H.; Yan, Y.; Lu, J. The role of interleukins in colorectal cancer. Int. J. Biol. Sci. 2020, 16, 2323–2339. [Google Scholar] [CrossRef] [PubMed]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R.; et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef] [Green Version]
- Spolski, R.; Li, P.; Leonard, W.J. Biology and regulation of IL-2: From molecular mechanisms to human therapy. Nat. Rev. Immunol. 2018, 18, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.M.; Magram, J.; Ferrante, J.; Orme, I.M. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with mycobacterium tuberculosis. J. Exp. Med. 1997, 186, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pagès, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, Th2, Treg, Th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.G.; Vrabel, M.R.; Mantooth, S.M.; Hopkins, J.J.; Wagner, E.S.; Gabaldon, T.A.; Zaharoff, D.A. Localized Interleukin-12 for Cancer Immunotherapy. Front. Immunol. 2020, 11, 1–36. [Google Scholar]
- Lin, X.; Wang, S.; Sun, M.; Zhang, C.; Wei, C.; Yang, C.; Dou, R.; Liu, Q.; Xiong, B. MiR-195-5p/NOTCH2-mediated EMT modulates IL-4 secretion in colorectal cancer to affect M2-like TAM polarization. J. Hematol. Oncol. 2019, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Junttila, I.S.; Mizukami, K.; Dickensheets, H.; Meier-Schellersheim, M.; Yamane, H.; Donnelly, R.P.; Paul, W.E. Tuning sensitivity to IL-4 and IL-13: Differential expression of IL-4Rα, IL-13Ra1, and γc regulates relative cytokine sensitivity. J. Exp. Med. 2008, 205, 2595–2608. [Google Scholar] [CrossRef] [Green Version]
- Gamez-Belmonte, R.; Erkert, L.; Wirtz, S.; Becker, C. The regulation of intestinal inflammation and cancer development by type 2 immune responses. Int. J. Mol. Sci. 2020, 21, 9772. [Google Scholar] [CrossRef]
- De Simone, V.; Franzè, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Tang, C.; Zhang, G.; Wan, X. DAMPs released by pyroptotic cells as major contributors and therapeutic targets for CAR-T-related toxicities. Cell Death Dis. 2021, 12, 10–13. [Google Scholar] [CrossRef]
- Ivashkiv, B.; Donlin, T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, M.R. The Role of Structure in the Biology of Interferon Signaling. Front. Immunol. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Budhwani, M.; Mazzieri, R.; Dolcetti, R. Plasticity of type I interferon-mediated responses in cancer therapy: From anti-tumor immunity to resistance. Front. Oncol. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johdi, N.A.; Sukor, N.F. Colorectal Cancer Immunotherapy: Options and Strategies. Front. Immunol. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 2018, 9, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, M.A.; Elliott, J.M.; Keyel, P.A.; Yang, L.; Carrero, J.A.; Yokoyama, W.M. Cytokine-induced memory-like natural killer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 1915–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Alvarado, D.M.; Iticovici, M.; Kau, N.S.; Park, H.; Parikh, P.J.; Thotala, D.; Ciorba, M.A. Therapeutic Target in Colorectal Cancer. World J. Gastrointest. Oncol. 2020, 8, 451–464. [Google Scholar]
- Croft, M.; Benedict, C.A.; Ware, C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013, 12, 147–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.H.; Seo, D.; Lim, S.G.; Suk, K. Reverse Signaling of Tumor Necrosis Factor Superfamily Proteins in Factor Superfamily Proteins in Macrophages and microgia: Superfamily portrait in the neuroimmune interface. Front. Immunol. 2019, 10, 1–19. [Google Scholar]
- Bremer, E. Targeting of the Tumor Necrosis Factor Receptor Superfamily for Cancer Immunotherapy. ISRN Oncol. 2013, 2013, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. TNF-α in promotion and progression of cancer. Cancer Metastasis Rev. 2006, 25, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Ségui, B. The TNF paradox in cancer progression and immunotherapy. Front. Immunol. 2019, 10, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Rong, L.; Zhao, X.; Li, X.; Liu, X.; Deng, J.; Wu, H.; Xu, X.; Erben, U.; Wu, P.; et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Investig. 2012, 122, 4094–4104. [Google Scholar] [CrossRef] [PubMed]
- Al Obeed, O.A.; Alkhayal, K.A.; Al Sheikh, A.; Zubaidi, A.M.; Vaali-Mohammed, M.A.; Boushey, R.; Mckerrow, J.H.; Abdulla, M.H. Increased expression of tumor necrosis factor-α is associated with advanced colorectal cancer stages. World J. Gastroenterol. 2014, 20, 18390–18396. [Google Scholar] [CrossRef]
- Metcalf, D.; Nicola, N.A. Proliferative effects of purified granulocyte colony-stimulating factor (G-CSF) on normal mouse hemopoietic cells. J. Cell. Physiol. 1983, 116, 198–206. [Google Scholar] [CrossRef]
- Hamilton, J.A. GM-CSF in inflammation. J. Exp. Med. 2019, 217, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Zeng, R.Y.; Tan, H.S.W.; et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: What we do and don’t know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Nebiker, C.A.; Han, J.; Eppenberger-Castori, S.; Iezzi, G.; Hirt, C.; Amicarella, F.; Cremonesi, E.; Huber, X.; Padovan, E.; Angrisani, B.; et al. GM-CSF production by tumor cells is associated with improved survival in colorectal cancer. Clin. Cancer Res. 2014, 20, 3094–3106. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Han, G.; Shen, B.; Li, Y. GM-CSF facilitates the development of inflammation-associated colorectal carcinoma. Oncoimmunology 2014, 3, 2–3. [Google Scholar] [CrossRef]
- Wakefield, L.M.; Kondaiah, P.; Hollands, R.S.; Winokur, T.S.; Sporn, M.B. Addition of a C-Terminal Extension Sequence to Transforming Growth Factor-pl Interferes with Biosynthetic Processing and Abolishes Biological Activity. Growth Factors 1991, 5, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Gunaltay, S.; McEntee, C.P.; Shuttleworth, E.E.; Smedley, C.; Houston, S.A.; Fenton, T.M.; Levison, S.; Mann, E.R.; Travis, M.A. Human monocytes and macrophages regulate immune tolerance via integrin αvβ8-mediated TGFβ activation. J. Exp. Med. 2018, 215, 2725–2736. [Google Scholar] [CrossRef] [Green Version]
- Tacconi, C.; Ungaro, F.; Correale, C.; Arena, V.; Massimino, L.; Detmar, M.; Spinelli, A.; Carvello, M.; Mazzone, M.; Oliveira, A.I.; et al. Activation of the VEGFC/VEGFR3 pathway induces tumor immune escape in colorectal cancer. Cancer Res. 2019, 79, 4196–4210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, V.K.; Long, T.; Long, R.; Figley, C.; Smith, D.M.C.; Ostrand-Rosenberg, S. Frontline Science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J. Leukoc. Biol. 2018, 103, 395–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abolhassani, M.; Aloulou, N.; Chaumette, M.T.; Aparicio, T.; Martin-Garcia, N.; Mansour, H.; Le Gouvello, S.; Delchier, J.C.; Sobhani, I. Leptin receptor-related immune response in colorectal tumors: The role of colonocytes and interleukin-8. Cancer Res. 2008, 68, 9423–9432. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [Green Version]
- Afonina, I.S.; Müller, C.; Martin, S.J.; Beyaert, R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 2015, 42, 991–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyrillou, K.; Burzynski, L.C.; Clarke, M.C.H. Alternative Pathways of IL-1 Activation, and Its Role in Health and Disease. Front. Immunol. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Oskeritzian, C.A.; Pozez, A.L.; Schwartz, L.B. Cytokine Production by Skin-Derived Mast Cells: Endogenous Proteases Are Responsible for Degradation of Cytokines. J. Immunol. 2005, 175, 2635–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, J.D.; Fleury, M.E.; Yong, C.; Tomei, A.A.; Randolph, G.J.; Swartz, M.A. Autologous Chemotaxis as a Mechanism of Tumor Cell Homing to Lymphatics via Interstitial Flow and Autocrine CCR7 Signaling. Cancer Cell 2007, 11, 526–538. [Google Scholar] [CrossRef] [Green Version]
- Fleury, M.E.; Boardman, K.C.; Swartz, M.A. Autologous morphogen gradients by subtle interstitial flow and matrix interactions. Biophys. J. 2006, 91, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumann, K.; Lämmermann, T.; Bruckner, M.; Legler, D.F.; Polleux, J.; Spatz, J.P.; Schuler, G.; Förster, R.; Lutz, M.B.; Sorokin, L.; et al. Immobilized Chemokine Fields and Soluble Chemokine Gradients Cooperatively Shape Migration Patterns of Dendritic Cells. Immun. 2010, 32, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, N.; Loef, E.J.; Kelch, I.D.; Verdon, D.J.; Black, M.M.; Middleditch, M.J.; Greenwood, D.R.; Graham, E.S.; Brooks, A.E.S.; Dunbar, P.R.; et al. Plasmin and regulators of plasmin activity control the migratory capacity and adhesion of human T cells and dendritic cells by regulating cleavage of the chemokine CCL21. Immunol. Cell Biol. 2016, 94, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Said, A.H.; Raufman, J.P.; Xie, G. The role of matrix metalloproteinases in colorectal cancer. Cancers 2014, 6, 366–375. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Matrisian, L.M. Emerging roles of porteases in tumour suppression. Tumour Microenviron. Opin. 2007, 7, 800–808. [Google Scholar]
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2017, 17, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015, 44–46, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Joos, S.; Zörnig, M. The role of Bcl-2 family members in tumorigenesis. Biochim. Biophys. Acta Mol. Cell Res. 2004, 3, 229–249. [Google Scholar] [CrossRef]
- Schulte, M.; Reiss, K.; Lettau, M.; Maretzky, T.; Ludwig, A.; Hartmann, D.; de Strooper, B.; Janssen, O.; Saftig, P. ADAM10 regulates FasL cell surface expression and modulates FasL-induced cytotoxicity and activation-induced cell death. Cell Death Differ. 2007, 14, 1040–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, E.; Chen, J.; Ouyang, N.; Su, F.; Wang, M.; Heemann, U. Soluble Fas ligand released by colon adenocarcinoma cells induces host lymphocyte apoptosis: An active mode of immune evasion in colon cancer. Br. J. Cancer 2001, 85, 1047–1054. [Google Scholar] [CrossRef] [Green Version]
- Barros, F.M.; Carneiro, F.; Machado, J.C.; Melo, S.A. Exosomes and immune response in cancer: Friends or foes? Front. Immunol. 2018, 9, 730. [Google Scholar] [CrossRef]
- Shah, R.; Patel, T.; Freedman, E. Circulating Extracellular Vesicles in Human Disease. N. Engl. J. Med. 2018, 379, 2178–2179. [Google Scholar] [CrossRef]
- Mannavola, F.; Salerno, T.; Passarelli, A.; Tucci, M.; Internò, V.; Silvestris, F. Revisiting the role of exosomes in colorectal cancer: Where are we now? Front. Oncol. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mimoto, F.; Tatsumi, K.; Shimizu, S.; Kadono, S.; Haraya, K.; Nagayasu, M.; Suzuki, Y.; Fujii, E.; Kamimura, M.; Hayasaka, A.; et al. Exploitation of Elevated Extracellular ATP to Specifically Direct Antibody to Tumor Microenvironment. Cell Rep. 2020, 33, 108542. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajizadeh, F.; Masjedi, A.; Heydarzedeh Asl, S.; Karoon Kiani, F.; Peydaveisi, M.; Ghalamfarsa, G.; Jadidi-Niaragh, F.; Sevbitov, A. Adenosine and adenosine receptors in colorectal cancer. Int. Immunopharmacol. 2020, 87, 106853. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nie, J.; Mei, Q.; Han, W.D. MicroRNAs: Novel immunotherapeutic targets in colorectal carcinoma. World J. Gastroenterol. 2016, 22, 5317–5331. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Chen, T.; Zheng, X.; Yang, S.; Xu, K.; Chen, X.; Xu, F.; Wang, L.; Shen, Y.; Wang, T.; et al. Colorectal Cancer-derived Small Extracellular Cesicles Establish an Inflammatory Pre-metastatic Niche in Liver Metastasisi. Carcinogenesis 2018, 39, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Masucci, M.T.; Minopoli, M.; del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1–7. [Google Scholar] [CrossRef] [PubMed]
- SenGupta, S.; Subramnian, C.; Parent, A. Getting TANned: How the tumor microenvironment drive neutrophil recruitment. J. Leukoc. Biol. 2019, 105, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Teijeira, Á.; Garasa, S.; Gato, M.; Alfaro, C.; Migueliz, I.; Cirella, A.; de Andrea, C.; Ochoa, M.C.; Otano, I.; Etxeberria, I.; et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 2020, 52, 856–871.e8. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef]
- Timosenko, E.; Hadjinicolaou, V.; Cerundolo, V. Modulation of cancer-specific immune responses by amino acid degrading enzymes. Immunotherapy 2017, 9, 83–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11, 1–24. [Google Scholar] [CrossRef]
- Munn, D.H. Indoleamine 2,3-dioxygenase, Tregs and Cancer. Curr. Med. Chem. 2012, 18, 2240–2246. [Google Scholar] [CrossRef]
- Benedetti, S.; Nuvoli, B.; Catalani, S.; Galati, R. Reactive oxygen species a double-edged sword for mesothelioma. Oncotarget 2015, 6, 16848–16865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive oxygen species and antitumor immunity—from surveillance to evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tseng, H.; Chen, Y.; Tanzih, A.; Haq, A.; Hwang, P.; Hsu, H. Oligo-Fucoidan Prevents M2 Macrophage Differentiation and HCT116 Tumor Progression. Cancer 2020, 12, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and cancer: Insight into tumor progression and immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Nardy, A.F.F.R.; Freire-de-Lima, L.; Freire-de-Lima, C.G.; Morrot, A. The sweet side of immune evasion: Role of Glycans in the Mechanisms of Cancer Progression. Front. Oncol. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briquez, P.S.; Hubbell, J.A.; Martino, M.M. Extracellular Matrix-Inspired Growth Factor Delivery Systems for Skin Wound Healing. Adv. Wound Care 2015, 4, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Birk, J.W.; Tadros, M.; Moezardalan, K.; Nadyarnykh, O.; Forouhar, F.; Anderson, J.; Campagnola, P. Second harmonic generation imaging distinguishes both high-grade dysplasia and cancer from normal colonic mucosa. Dig. Dis. Sci. 2014, 59, 1529–1534. [Google Scholar] [CrossRef]
- Coulson-Thomas, V.J.; Coulson-Thomas, Y.M.; Gesteira, T.F.; De Paula, C.A.A.; Mader, A.M.; Waisberg, J.; Pinhal, M.A.; Friedl, A.; Toma, L.; Nader, H.B. Colorectal cancer desmoplastic reaction up-regulates collagen synthesis and restricts cancer cell invasion. Cell Tissue Res. 2011, 346, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.M.; Bird, D.; Welti, J.C.; Gourlaouen, M.; Lang, G.; Murray, G.I.; Reynolds, A.R.; Cox, T.R.; Erler, J.T. Lysyl oxidase plays a critical role in endothelial cell stimulation to drive tumor angiogenesis. Cancer Res. 2013, 73, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Kuczek, D.E.; Larsen, A.M.H.; Carretta, M.; Kalvisa, A.; Siersbæk, M.S.; Simões, A.M.C.; Roslindn, A.; Engelholm, L.H.; Donia, M.; Svane, I.M.; et al. Collagen density regulates the activity of tumor-infiltrating T cells. bioRxiv 2018, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.; Shibutani, M.; Nagahara, H.; Fukuoka, T.; Iseki, Y.; Okazaki, Y.; Kashiwagi, S.; Tanaka, H.; Maeda, K.; Hirakawa, K.; et al. Abundant intratumoral fibrosis prevents lymphocyte infiltration into peritoneal metastases of colorectal cancer. PLoS ONE 2021, 16, e0255049. [Google Scholar] [CrossRef]
- Gordon-Weeks, A.; Lim, Y.; Yuzhalin, E.; Jones, K.; Markelc, B.; Buzelli, N.; Fokas, E.; Cao, Y.; Smart, S.; Muschel, R. Neutrophils Promote Hepatic Metastasis Growth Through fibroblast growth factor (FGF)2-dependent Angiogenesis. Hepatology 2017, 65, 1920–1935. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.; Artym, V.V.; Green, J.A.; Yamada, K.M. The matrix reorganized: Extracellular matrix remodeling and integrin signaling. Curr. Opin. Cell Biol. 2006, 18, 463–471. [Google Scholar] [CrossRef]
- Liang, Y.; Lv, Z.; Huang, G.; Qin, J.; Li, H.; Nong, F.; Wen, B. Prognostic significance of abnormal matrix collagen remodeling in colorectal cancer based on histologic and bioinformatics analysis. Oncol. Rep. 2020, 44, 1671–1685. [Google Scholar] [CrossRef]
- Fagerholm, S.C.; Guenther, C.; Asens, M.L.; Savinko, T.; Uotila, L.M. Beta2-Integins and interacting proteins in leukocyte trafficking, immune supression, and immunodeficiency disease. Front. Immunol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammi, M.I.; Oikari, S.; Pasonen-Seppänen, S.; Rilla, K.; Auvinen, P.; Tammi, R.H. Activated hyaluronan metabolism in the tumor matrix—Causes and consequences. Matrix Biol. 2019, 78–79, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Lu, R.; Wu, M.; Liu, Y.; He, Y.; Xu, J.; Yang, C.; Du, Y.; Gao, F. Colorectal cancer-associated ~ 6 kDa hyaluronan serves as a novel biomarker for cancer progression and metastasis. FEBS J. 2019, 286, 3148–3163. [Google Scholar] [CrossRef] [PubMed]
- Spaderna, S.; Schmalhofer, O.; Hlubek, F.; Berx, G.; Eger, A.; Merkel, S.; Jung, A.; Kirchner, T.; Brabletz, T. A Transient, EMT-Linked Loss of Basement Membranes Indicates Metastasis and Poor Survival in Colorectal Cancer. Gastroenterology 2006, 131, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Chaudhuri, O. Beyond proteases: Basement membrane mechanics and cancer invasion. J. Cell Biol. 2019, 218, 2456–2469. [Google Scholar] [CrossRef] [Green Version]
- Kai, F.B.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Li, L.; Song, J.; Chuquisana, O.; Hannocks, M.J.; Loismann, S.; Vogl, T.; Roth, J.; Hallmann, R.; Sorokin, L. Endothelial Basement Membrane Laminins as an Environmental Cue in Monocyte Differentiation to Macrophages. Front. Immunol. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Haessler, U.; Pisano, M.; Wu, M.; Swartz, M.A. Dendritic cell chemotaxis in 3D under defined chemokine gradients reveals differential response to ligands CCL21 and CCL19. Proc. Natl. Acad. Sci. USA 2011, 108, 5614–5619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proudfoot, A.E.I.; Uguccioni, M. Modulation of chemokine responses: Synergy and cooperativity. Front. Immunol. 2016, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Franitza, S.; Hershkoviz, R.; Kam, N.; Lichtenstein, N.; Vaday, G.G.; Alon, R.; Lider, O. TNF-α Associated with Extracellular Matrix Fibronectin Provides a Stop Signal for Chemotactically Migrating T Cells. J. Immunol. 2000, 165, 2738–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, M.M.; Mochizuki, M.; Rothenfluh, D.A.; Rempel, S.A.; Hubbell, J.A.; Barker, T.H. Controlling integrin specificity and stem cell differentiation in 2D and 3D environments through regulation of fibronectin domain stability. Biomaterials 2009, 30, 1089–1097. [Google Scholar] [CrossRef] [Green Version]
- Villalba, M.; Evans, S.R.; Vidal-Vanaclocha, F.; Calvo, A. Role of TGF-β in metastatic colon cancer: It is finally time for targeted therapy. Cell Tissue Res. 2017, 370, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G.; Discher, D.E. Matrix elasticity, cytoskeletal tension, and TGF-β: The insoluble and soluble meet. Sci. Signal. 2008, 1, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscemi, L.; Ramonet, D.; Klingberg, F.; Formey, A.; Smith-Clerc, J.; Meister, J.J.; Hinz, B. The single-molecule mechanics of the latent TGF-β1 complex. Curr. Biol. 2011, 21, 2046–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, S.L. Integrin-mediated transforming growth factor-βactivation, a potential therapeutic target in fibrogenic disorders. Am. J. Pathol. 2009, 175, 1362–1370. [Google Scholar] [CrossRef] [Green Version]
- Jha, S.K.; Rauniyar, K.; Karpanen, T.; Leppänen, V.M.; Brouillard, P.; Vikkula, M.; Alitalo, K.; Jeltsch, M. Efficient activation of the lymphangiogenic growth factor VEGF-C requires the C-terminal domain of VEGF-C and the N-terminal domain of CCBE1. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Ikeda, Y.; Miyazaki, M.; Abe, T.; Kinoshita, J.; Maehara, Y.; Sugimachi, K. Vascular endothelial growth factor-C (VEGF-C) expression in human colorectal cancer tissues. Br. J. Cancer 2000, 83, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Güç, E.; Briquez, P.S.; Foretay, D.; Fankhauser, M.A.; Hubbell, J.A.; Kilarski, W.W.; Swartz, M.A. Local induction of lymphangiogenesis with engineered fibrin-binding VEGF-C promotes wound healing by increasing immune cell trafficking and matrix remodeling. Biomaterials 2017, 131, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Fankhauser, M.; Broggi, M.A.S.; Potin, L.; Bordry, N.; Jeanbart, L.; Lund, A.W.; Da Costa, E.; Hauert, S.; Rincon-Restrepo, M.; Tremblay, C.; et al. Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci. Transl. Med. 2017, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Skobe, M.; Hamberg, L.M.; Hawighorst, T.; Schirner, M.; Wolf, G.L.; Alitalo, K.; Detmar, M. Concurrent induction of lymphangiogenesis, angiogenesis, and macrophage recruitment by vascular endothelial growth factor-C in melanoma. Am. J. Pathol. 2001, 159, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Castello, L.M.; Raineri, D.; Salmi, L.; Clemente, N.; Vaschetto, R.; Quaglia, M.; Garzaro, M.; Gentilli, S.; Navalesi, P.; Cantaluppi, V.; et al. Osteopontin at the Crossroads of Inflammation and Tumor Progression. Mediat. Inflamm. 2017, 2017, 912. [Google Scholar] [CrossRef]
- Klement, J.D.; Paschall, A.V.; Redd, P.S.; Ibrahim, M.L.; Lu, C.; Yang, D.; Celis, E.; Abrams, S.I.; Ozato, K.; Liu, K. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Invest. 2018, 128, 5549–5560. [Google Scholar] [CrossRef] [PubMed]
- Shurin, M.R. Osteopontin controls immunosuppression in the tumor microenvironment. J. Clin. Investig. 2018, 128, 5209–5212. [Google Scholar] [CrossRef]
- Julier, Z.; Martino, M.M.; de Titta, A.; Jeanbart, L.; Hubbell, J.A. The TLR4 agonist fibronectin extra domain a is cryptic, Exposed by elastase-2; Use in a fibrin matrix cancer vaccine. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hope, C.; Emmerich, P.B.; Papadas, A.; Pagenkopf, A.; Matkowskyj, K.A.; Van De Hey, D.R.; Payne, S.N.; Clipson, L.; Callander, N.S.; Hematti, P.; et al. Versican-Derived Matrikines Regulate Batf3–Dendritic Cell Differentiation and Promote T Cell Infiltration in Colorectal Cancer. J. Immunol. 2017, 199, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Oldberg, Å.; Kalamajski, S.; Salnikov, A.V.; Stuhr, L.; Mörgelin, M.; Reed, R.K.; Heldin, N.E.; Rubin, K. Collagen-binding proteoglycan fibromodulin can determine stroma matrix structure and fluid balance in experimental carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 13966–13971. [Google Scholar] [CrossRef] [Green Version]
- Sjöberg, P.; Manderson, A.; Mögelin, M.; Heinegard, D.; Mlom, M. Short leucine-rich glycoproteins of the extracellular matrix display diverse patterns of complement interaction and activation. Mol. Immunol. 2010, 46, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Gastélum, G.R.; Aguilar-Medina, E.M.; Soto-Sainz, E.; Ramos-Payán, R.; Silva-Benítez, E.L. Antimicrobial Properties of Extracellular Matrix Scaffolds for Tissue Engineering. BioMed Res. Int. 2019, 2019, 1–9. [Google Scholar] [CrossRef]
- Alfano, M.; Canducci, F.; Nebuloni, M.; Clementi, M.; Montorsi, F.; Salonia, A. The interplay of extracellular matrix and microbiome in urothelial bladder cancer. Nat. Rev. Urol. 2016, 13, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Villalba, N.; Baby, S.; Yuan, S.Y. The Endothelial Glycocalyx as a Double-Edged Sword in Microvascular Homeostasis and Pathogenesis. Front. Cell Dev. Biol. 2021, 9, 1–9. [Google Scholar] [CrossRef]
- Möckl, L.; Pedram, K.; Roy, A.R.; Krishnan, V.; Gustavsson, A.K.; Dorigo, O.; Bertozzi, C.R.; Moerner, W.E. Quantitative Super-Resolution Microscopy of the Mammalian Glycocalyx. Dev. Cell 2019, 50, 57–72.e6. [Google Scholar] [CrossRef] [Green Version]
- Paszek, M.J.; Dufort, C.C.; Rossier, O.; Bainer, R.; Mouw, J.K.; Godula, K.; Hudak, J.E.; Lakins, J.N.; Wijekoon, A.C.; Cassereau, L.; et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 2014, 511, 319–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanyo, N.; Kovacs, K.D.; Saftics, A.; Szekacs, I.; Peter, B.; Santa-Maria, A.R.; Walter, F.R.; Dér, A.; Deli, M.A.; Horvath, R. Glycocalyx regulates the strength and kinetics of cancer cell adhesion revealed by biophysical models based on high resolution label-free optical data. Sci. Rep. 2020, 10, 1–20. [Google Scholar] [CrossRef]
- Ghasempour, S.; Freeman, S.A. The glycocalyx and immune evasion in cancer. FEBS J. 2021, 289, 1–11. [Google Scholar] [CrossRef]
- Hu, Z.; Cano, I.; D’Amore, P.A. Update on the Role of the Endothelial Glycocalyx in Angiogenesis and Vascular Inflammation. Front. Cell Dev. Biol. 2021, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mensah, S.A.; Harding, I.C.; Zhang, M.; Jaeggli, M.P.; Torchilin, V.P.; Niedre, M.J.; Ebong, E.E. Metastatic cancer cell attachment to endothelium is promoted by endothelial glycocalyx sialic acid degradation. AIChE J. 2019, 65, e16634. [Google Scholar] [CrossRef] [Green Version]
- Mensah, S.A.; Nersesyan, A.A.; Harding, I.C.; Lee, C.I.; Tan, X.; Banerjee, S.; Niedre, M.; Torchilin, V.P.; Ebong, E.E. Flow-regulated endothelial glycocalyx determines metastatic cancer cell activity. FASEB J. 2020, 34, 6166–6184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwnag, Y.; Geller, T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Brody, H. The gut microbiome. Nature 2020, 577, S5. [Google Scholar] [CrossRef] [PubMed]
- Thorbeck, G.J. Some Histological and Functional Aspects of Lymphoid Tissue in Germ Free Animals: I Morphological Studies. Ann. N. Y. Acad. Sci. 1959, 79, 237–246. [Google Scholar]
- Hill, A.; Artis, D. Intestinal Bacteria and the Regulation of Immune Cell Homeostasis. Annu. Rev. Immunol 2010, 28, 623–667. [Google Scholar] [CrossRef] [Green Version]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Oka, A.; Liu, B.; Herzog, J.W.; Eun, C.S.; Fan, T.J.; Bulik-Sullivan, E.; Carroll, I.M.; Hansen, J.J.; Chen, L.; et al. Microbiota maintain colonic homeostasis by activating TLR2/MyD88/PI3K signaling in IL-10-producing regulatory B cells. J. Clin. Invest. 2019, 129, 3702–3716. [Google Scholar] [CrossRef] [Green Version]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Kayama, H.; Takeda, K. Polysaccharide A of bacteroides fragilis: Actions on dendritic cells and T cells. Mol. Cell 2014, 54, 206–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Xu, E.; Ai, D. Inferring bacterial infiltration in primary colorectal tumors from host whole genome sequencing data. Front. Genet. 2019, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Abed, J.; Emgård, J.E.M.; Zamir, G.; Faroja, M.; Almogy, G.; Grenov, A.; Sol, A.; Naor, R.; Pikarsky, E.; Atlan, K.A.; et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe 2016, 20, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, C.W.; Ma, X.; Paranjpe, A.; Jewett, A.; Lux, R.; Kinder-Haake, S.; Shi, W. Fusobacterium nucleatum outer membrane proteins Fap2 and RadD induce cell death in human lymphocytes. Infect. Immun. 2010, 78, 4773–4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.E.; Kim, J.H.; Cho, N.Y.; Lee, H.S.; Kang, G.H. Intratumoral Fusobacterium nucleatum abundance correlates with macrophage infiltration and CDKN2A methylation in microsatellite-unstable colorectal carcinoma. Virchows Arch. 2017, 471, 329–336. [Google Scholar] [CrossRef]
- Hamada, T.; Zhang, X.; Mima, K.; Bullman, S.; Sukawa, Y.; Nowak, J.A.; Kosumi, K.; Masugi, Y.; Twombly, T.S.; Cao, Y.; et al. Fusobacterium nucleatum in colorectal cancer relates to immune response differentially by tumor microsatellite instability status. Cancer Immunol. Res. 2018, 6, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Simoni, Y.; Becht, E.; Loh, C.Y.; Li, N.; Lachance, D.; Koo, S.L.; Lim, T.P.; Tan, E.K.W.; Mathew, R.; et al. Human Tumor-Infiltrating MAIT Cells Display Hallmarks of Bacterial Antigen Recognition in Colorectal Cancer. Cell Rep. Med. 2020, 1, 100039. [Google Scholar] [CrossRef] [PubMed]
- Cremonesi, E.; Governa, V.; Garzon, J.F.G.; Mele, V.; Amicarella, F.; Muraro, M.G.; Trella, E.; Galati-Fournier, V.; Oertli, D.; Däster, S.R.; et al. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut 2018, 67, 1984–1994. [Google Scholar] [CrossRef]
- Roberti, M.P.; Yonekura, S.; Duong, C.P.M.; Picard, M.; Ferrere, G.; Tidjani Alou, M.; Rauber, C.; Iebba, V.; Lehmann, C.H.K.; Amon, L.; et al. Chemotherapy-induced ileal crypt apoptosis and the ileal microbiome shape immunosurveillance and prognosis of proximal colon cancer. Nat. Med. 2020, 26, 919–931. [Google Scholar] [CrossRef]
- Toussi, D.N.; Liu, X.; Massari, P. The FomA porin from Fusobacterium nucleatum is a toll-like receptor 2 agonist with immune adjuvant activity. Clin. Vaccine Immunol. 2012, 19, 1093–1101. [Google Scholar] [CrossRef] [Green Version]
- Park, S.R.; Kim, D.J.; Han, S.H.; Kang, M.J.; Lee, J.Y.; Jeong, Y.J.; Lee, S.J.; Kim, T.H.; Ahn, S.G.; Yoon, J.H.; et al. Diverse toll-like receptors mediate cytokine production by fusobacterium nucleatum and aggregatibacter actinomycetemcomitans in macrophages. Infect. Immun. 2014, 82, 1914–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisonneuve, C.; Tsang, D.K.L.; Foerster, E.G.; Robert, L.M.; Mukherjee, T.; Prescott, D.; Tattoli, I.; Lemire, P.; Winer, D.A.; Winer, S.; et al. Nod1 promotes colorectal carcinogenesis by regulating the immunosuppressive functions of tumor-infiltrating myeloid cells. Cell Rep. 2021, 34, 108677. [Google Scholar] [CrossRef]
- Lamprinaki, D.; Garcia-Vello, P.; Marchetti, R.; Hellmich, C.; McCord, K.A.; Bowles, K.M.; Macauley, M.S.; Silipo, A.; De Castro, C.; Crocker, P.R.; et al. Siglec-7 Mediates Immunomodulation by Colorectal Cancer-Associated Fusobacterium nucleatum ssp. animalis. Front. Immunol. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gallausiaux, C.; Malabirade, A.; Habier, J.; Wilmes, P. Fusobacterium nucleatum Extracellular Vesicles Modulate Gut Epithelial Cell Innate Immunity via FomA and TLR2. Front. Immunol. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.Y.C.; Cosgrove, L.; Lockett, T.; Head, R.; Topping, D.L. A review of the potential mechanisms for the lowering of colorectal oncogenesis by butyrate. Br. J. Nutr. 2012, 108, 820–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, S.; Konishi, Y.; Narukawa, M.; Sugiura, Y.; Yoshimoto, S.; Arai, Y.; Sato, S.; Yoshida, Y.; Tsuji, S.; Uemura, K.; et al. Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Nakkarach, A.; Foo, H.L.; Song, A.A.L.; Mutalib, N.E.A.; Nitisinprasert, S.; Withayagiat, U. Anti-cancer and anti-inflammatory effects elicited by short chain fatty acids produced by Escherichia coli isolated from healthy human gut microbiota. Microb. Cell Fact. 2021, 20, 1–17. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Fu, L.; Li, Y.; Wang, W.; Gong, M.; Zhang, J.; Dong, X.; Huang, J.; Wang, Q.; Mackay, C.R.; et al. Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8+ T cell immunity. Cell Metab. 2021, 33, 988–1000.e7. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C.; et al. The bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin. Infect. Dis. 2015, 60, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Dalmasso, G.; Cougnoux, A.; Delmas, J.; Darfeuille-Michaud, A.; Bonnet, R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2015, 5, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Proença, M.A.; Biselli, J.M.; Succi, M.; Severino, F.E.; Berardinelli, G.N.; Caetano, A.; Reis, R.M.; Hughes, D.J.; Silva, A.E. Relationship between fusobacterium nucleatum, inflammatory mediators and microRNAs in colorectal carcinogenesis. World J. Gastroenterol. 2018, 24, 5351–5365. [Google Scholar] [CrossRef] [PubMed]
- Prorok-Hamon, M.; Friswell, M.K.; Alswied, A.; Roberts, C.L.; Song, F.; Flanagan, P.K.; Knight, P.; Codling, C.; Marchesi, J.R.; Winstanley, C.; et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 2014, 63, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immune Target | NTC Number | Phase | Title | Start Year |
|---|---|---|---|---|
| Chemokines | ||||
| CXCR1/2 inhibition | NCT04599140 | 1/2 | SX-682 and Nivolumab for the Treatment of RAS-Mutated, MSS Unresectable or Metastatic Colorectal Cancer, the STOPTRAFFIC-1 Trial | 2020 |
| CSFs | ||||
| G-CSF | NCT00541125 | 2 | Vaccine Therapy With or Without Sargramostim in Treating Patients With Advanced or Metastatic Cancer | 2007 |
| GM-CSF | NCT00028496 | 1 | Vaccine Therapy in Treating Patients With Cancer of the Gastrointestinal Tract | 2001 |
| NCT00012246 | 2 | Vaccine Therapy in Treating Patients With Stage IIB, Stage III, or Stage IV Colorectal Cancer | 2002 | |
| NCT00091286 | 1 | Cellular Immune Augmentation in Colon and Rectal Cancer | 2003 | |
| NCT00257322 | 2 | Vaccine Therapy and Radiation to Liver Metastasis in Patients With CEA-Positive Solid Tumors | 2003 | |
| NCT00081848 | 1 | Vaccine Therapy and Sargramostim With or Without Docetaxel in Treating Patients With Metastatic Lung Cancer or Metastatic Colorectal Cancer | 2004 | |
| NCT00088933 | 1 | GM-CSF and Combination Chemotherapy in Treating Patients Who Are Undergoing Surgery for Stage II or Stage III Colon Cancer | 2004 | |
| NCT00262808 | 2 | Vaccine Therapy in Treating Patients With Liver or Lung Metastases From Colorectal Cancer | 2004 | |
| NCT00103142 | 2 | Study of Colon GVAX and Cyclophosphamide in Patients With Metastatic Colorectal Cancer | 2005 | |
| NCT00656123 | 1 | Neoadjuvant Study of Recombinant Vaccinia Virus to Treat Metastatic Colorectal Carcinoma in Patients Undergoing Complete Resection of Liver Tumors | 2008 | |
| NCT01329809 | 2 | Safety Study of Recombinant Vaccinia Virus Administered Intravenously in Patients With Metastatic, Refractory Colorectal Carcinoma | 2011 | |
| NCT01380600 | 1 | SGI-110 in Combination With an Allogeneic Colon Cancer Cell Vaccine (GVAX) and Cyclophosphamide (CY) in Metastatic Colorectal Cancer (mCRC) | 2011 | |
| NCT01966289 | 1 | RhGM-CSF as Adjuvant Immunotherapy in Treating Stage III Colon Cancer | 2014 | |
| NCT02466906 | 2 | Study of GVAX (With CY) and Pembrolizumab in MMR-p Advanced Colorectal Cancer | 2015 | |
| NCT02981524 | 2 | Vaccine Therapy With or Without Sargramostim in Treating Patients With Advanced or Metastatic Cancer | 2017 | |
| IFNs | ||||
| IFN | NCT00309530 | 3 | Randomized Study on Adjuvant Chemotherapy and Adjuvant Chemo-Immunotherapy in Colon Carcinoma Dukes C | 1990 |
| IFNα | NCT00003063 | 3 | Biological Therapy With Combination Chemotherapy in Patients With Colorectal Cancer | 1991 |
| NCT01060501 | 3 | Modulation of Adjuvant 5-FU by Folinic Acid and Interferon-alpha in Colon Cancer | 1992 | |
| NCT02387307 | 1 | A Study of rSIFN-co in Subjects With Advanced Solid Tumors | 2013 | |
| NCT04798612 | 2 | Effect of Low-dose Interferon-alfa2a on Peri-operative Immune Suppression | 2021 | |
| IFNα GM-CSF | NCT00016042 | 1 | Fluorouracil and Biological Therapy in Treating Patients With Metastatic Kidney or Colorectal Cancer | 2001 |
| IFNα GM-CSF | NCT00030342 | 1/2 | Biological Therapy and Chemotherapy in Treating Patients With Metastatic Kidney Cancer or Colorectal Cancer | 2001 |
| IFNα, IFNγ, GM-CSF | NCT00002475 | 2 | Cyclophosphamide Plus Vaccine Therapy in Treating Patients With Advanced Cancer | 1991 |
| IFNγ | NCT00002796 | 1/2 | Phase I-II Study of Fluorouracil in Combination With Phenylbutyrate in Advanced Colorectal Cancer | 1997 |
| ILs | ||||
| IL-2 | NCT00019591 | 1/2 | Vaccine Therapy With or Without Interleukin-2 in Treating Patients With Locally Advanced or Metastatic Colorectal Cancer | 1999 |
| NCT00020267 | 1 | Vaccine Therapy in Treating Patients With Metastatic Cancer | 2000 | |
| NCT03190941 | 1/2 | Administering Peripheral Blood Lymphocytes Transduced With a Murine T-Cell Receptor Recognizing the G12V Variant of Mutated RAS in HLA-A*11:01 Patients | 2017 | |
| NCT03745326 | 1/2 | Administering Peripheral Blood Lymphocytes Transduced With a Murine T-Cell Receptor Recognizing the G12D Variant of Mutated RAS in HLA-A*11:01 Patients | 2019 | |
| NCT04426669 | 1/2 | A Study of Metastatic Gastrointestinal Cancers Treated With Tumor Infiltrating Lymphocytes in Which the Gene Encoding the Intracellular Immune Checkpoint CISH Is Inhibited Using CRISPR Genetic Engineering | 2020 | |
| IL-2, GM-CSF | NCT00019331 | 2 | Vaccine Therapy Plus Biological Therapy in Treating Adults With Metastatic Solid Tumors | 1997 |
| IL-2 fusion | NCT00128622 | 1 | Denileukin Diftitox Followed by Vaccine Therapy in Treating Patients With Metastatic Cancer | 2005 |
| IL-2, GM-CSF | NCT00019084 | 2 | Vaccine Therapy and Biological Therapy in Treating Patients With Advanced Cancer | 1996 |
| IL-7 | NCT01339000 | 2 | Improving the Immune System With Human IL-7 Vaccine in Older Subjects Who Have Had Chemotherapy | 2011 |
| IL-12 | NCT00003046 | 1 | Interleukin-12 in Treating Patients With Cancer in the Abdomen | 1997 |
| NCT00003439 | 1 | Interleukin-12 in Treating Patients With Refractory Advanced-Stage Ovarian Cancer or Abdominal Cancer | 1998 | |
| NCT00004074 | 1 | Interleukin-12 and Trastuzumab in Treating Patients With Cancer That Has High Levels of HER2/Neu | 1999 | |
| NCT00072098 | 1 | Interleukin-12 Gene in Treating Patients With Liver Metastases Secondary to Colorectal Cancer | 2003 | |
| IL-15 super-agonist | NCT03127098 | 1/2 | QUILT-3.040: ETBX-011 (Ad5 [E1-, E2b-]-CEA(6D)) Vaccine in Combination With ALT-803 (Super-agonist IL-15) in Subjects Having CEA-Expressing Cancer | 2017 |
| TGF-β | ||||
| TGF-β trap | NCT03436563 | 1/2 | M7824 in Patients With Metastatic Colorectal Cancer or With Advanced Solid Tumors With Microsatellite Instability | 2018 |
| TGF-β trap IL-12 fusion | NCT04708470 | 1/2 | Phase I/II Trial of the Combination of Bintrafusp Alfa (M7824), Entinostat and NHS-IL12 (M9241) in Patients With Advanced Cancer | 2021 |
| TNFs | ||||
| TNF | NCT00436410 | 1 | Tumor Necrosis Factor in Patients Undergoing Surgery for Primary Cancer or Metastatic Cancer | 2006 |
| TNFα conjugate | NCT00098943 | 1 | NGR-TNF in Treating Patients With Advanced Solid Tumors | 2004 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plundrich, D.; Chikhladze, S.; Fichtner-Feigl, S.; Feuerstein, R.; Briquez, P.S. Molecular Mechanisms of Tumor Immunomodulation in the Microenvironment of Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 2782. https://doi.org/10.3390/ijms23052782
Plundrich D, Chikhladze S, Fichtner-Feigl S, Feuerstein R, Briquez PS. Molecular Mechanisms of Tumor Immunomodulation in the Microenvironment of Colorectal Cancer. International Journal of Molecular Sciences. 2022; 23(5):2782. https://doi.org/10.3390/ijms23052782
Chicago/Turabian StylePlundrich, Dorothea, Sophia Chikhladze, Stefan Fichtner-Feigl, Reinhild Feuerstein, and Priscilla S. Briquez. 2022. "Molecular Mechanisms of Tumor Immunomodulation in the Microenvironment of Colorectal Cancer" International Journal of Molecular Sciences 23, no. 5: 2782. https://doi.org/10.3390/ijms23052782