
Cell Type-Specific Induction of Inflammation-Associated Genes in Crohn’s Disease and Colorectal Cancer

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
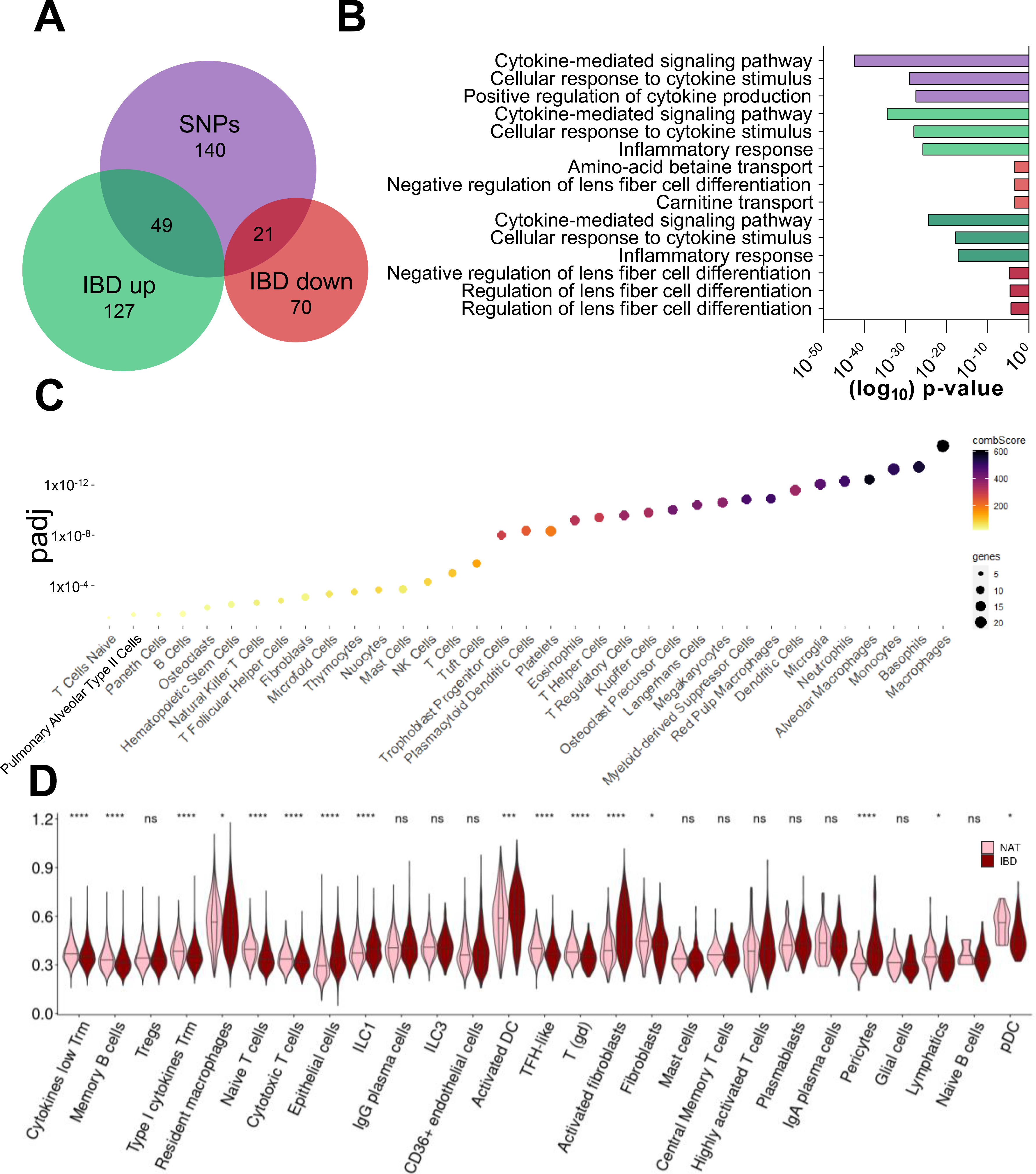
2.1. Only Distinct Cell Populations Contribute to the Upregulation of Inflammation-Induced Genes in CD Lesions
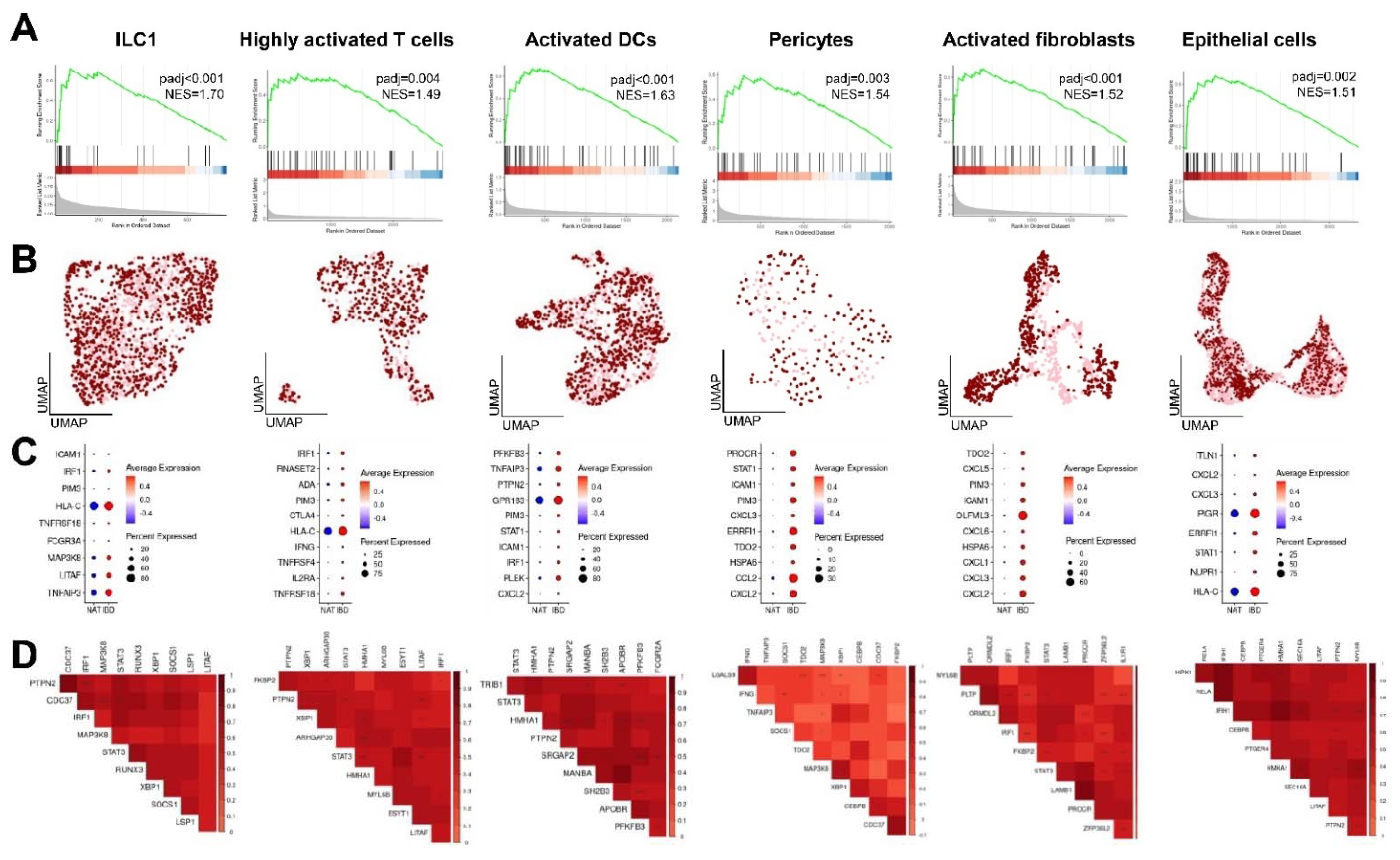
2.2. Cell Type-Specific Enrichment and Co-Expression Patterns of CD-Induced Genes
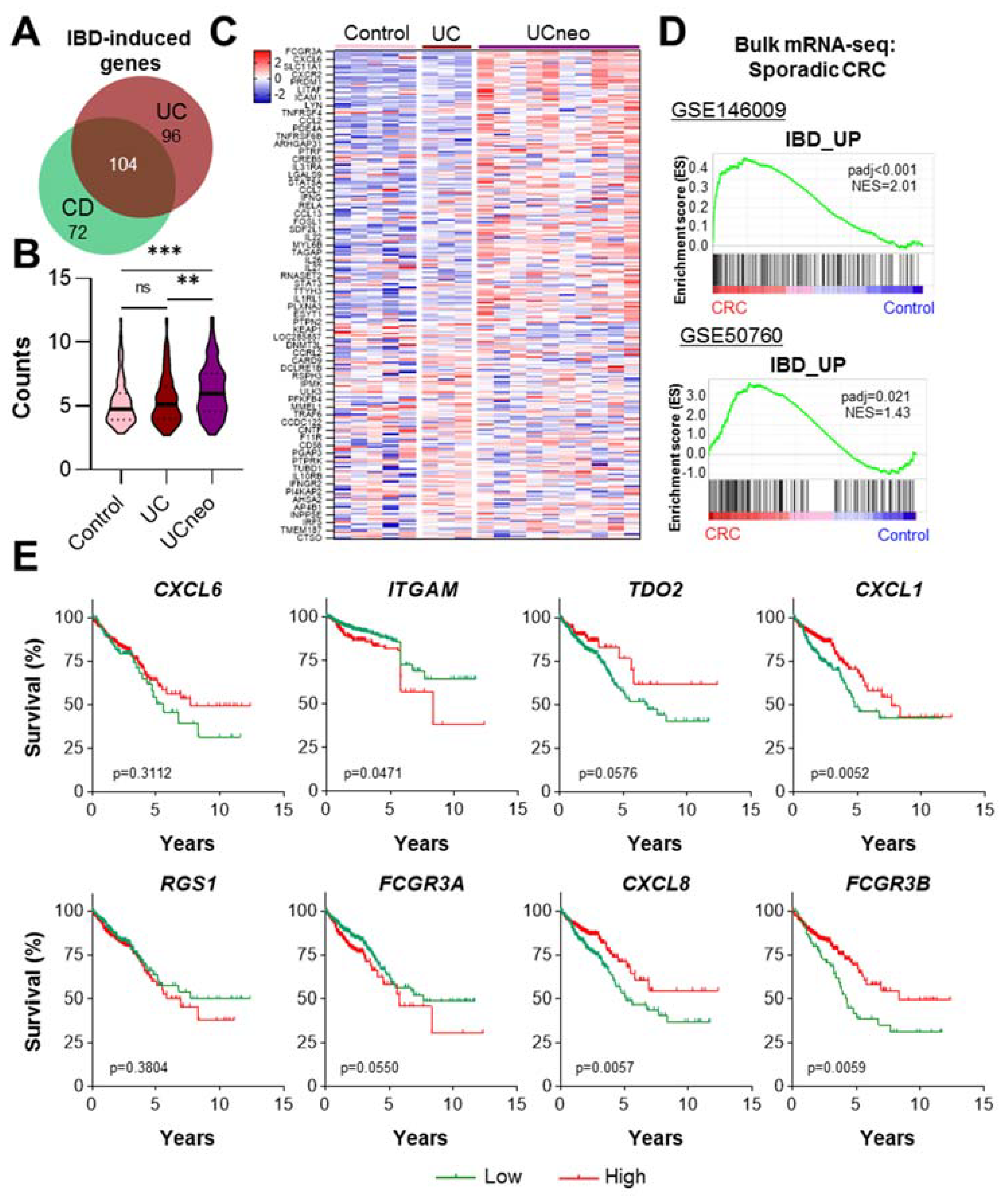
2.3. CD-Induced Genes Are Upregulated in UC-Associated Neoplasia and Sporadic CRC
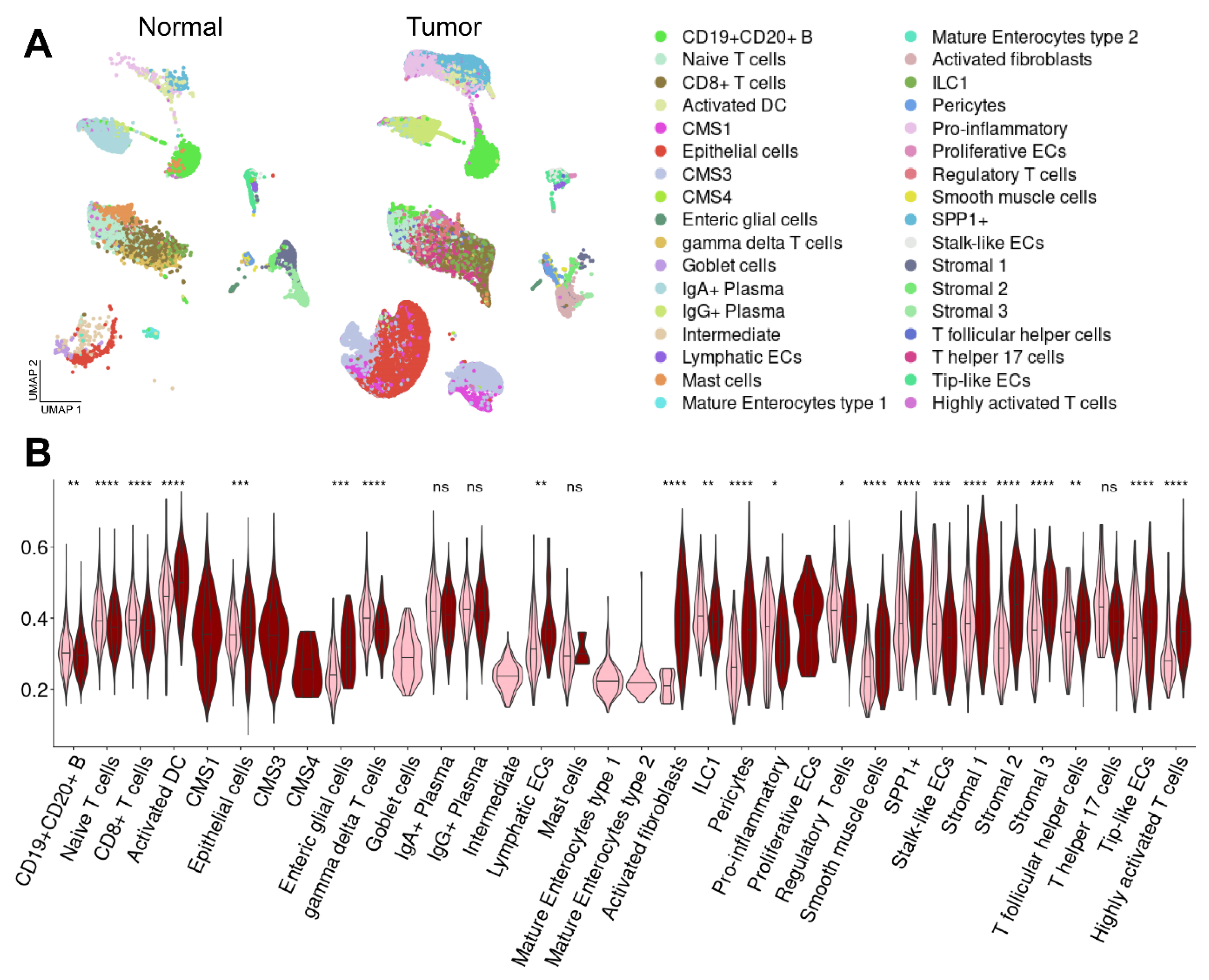
2.4. Enrichment of CD Risk Genes in Sporadic CRC on a Single-Cell Level
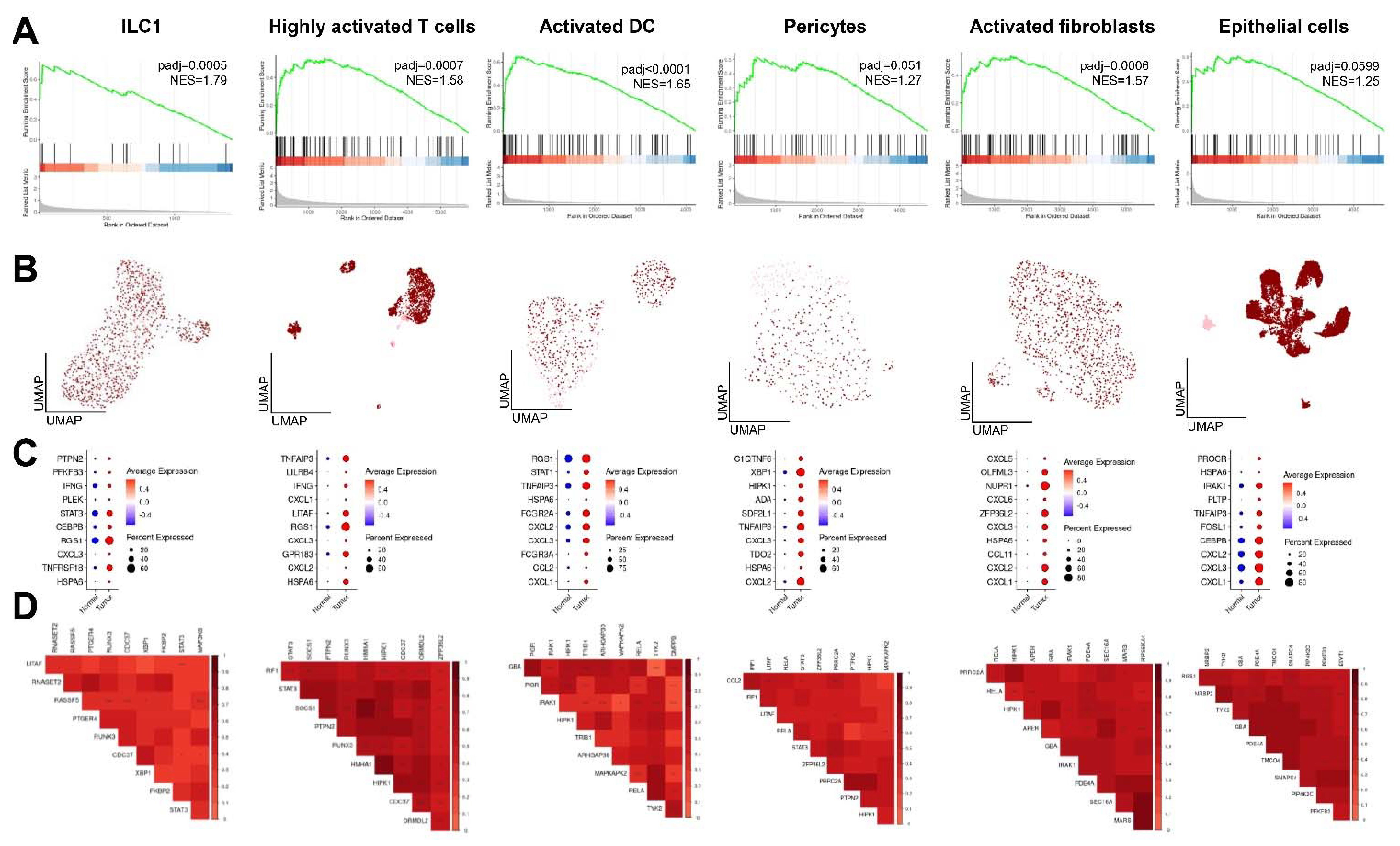
2.5. Enrichment and Co-Expression Patterns of CD-Induced Genes in Distinct Cell Populations in CRC
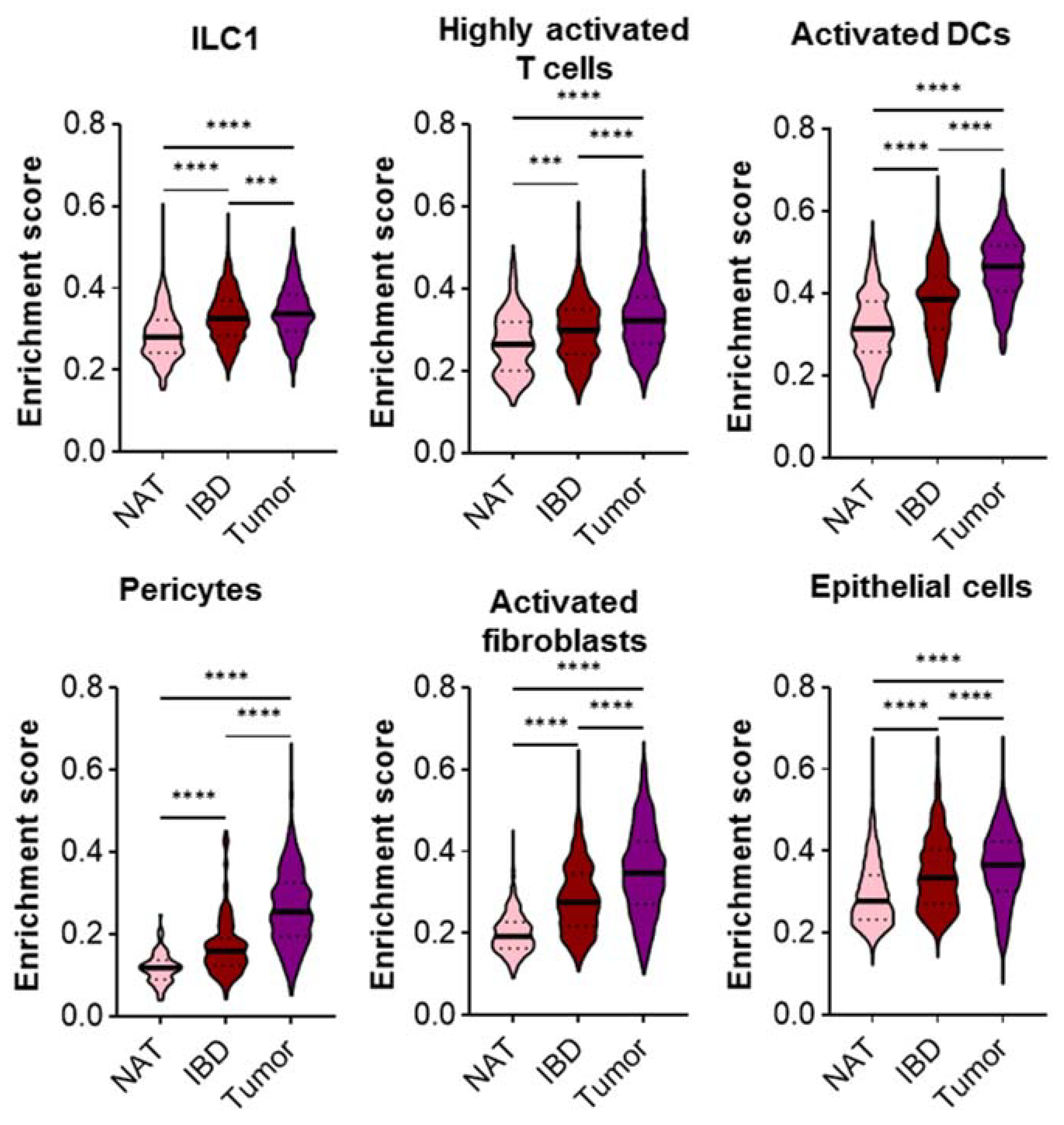
2.6. The Expression of CD Risk Genes Gradually Increases from Control NAT, CD to CRC Cells
3. Discussion
4. Materials and Methods
4.1. CD Signature
4.2. Analysis of Microarray Sequencing Data
4.3. Analysis of CRC Bulk mRNA Sequencing Data
4.4. Single-Cell RNA-seq (scRNA-seq) Analysis
4.5. Statistics and Graphs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- CCFA. The Facts about Inflammatory Bowel Diseases. Available online: http://www.crohnscolitisfoundation.org/assets/pdfs/updatedibdfactbook.pdf (accessed on 30 December 2021).
- Ben-Yosef, N.; Frampton, M.; Schiff, E.R.; Daher, S.; Abu Baker, F.; Safadi, R.; Israeli, E.; Segal, A.W.; Levine, A.P. Genetic analysis of four consanguineous multiplex families with inflammatory bowel disease. Gastroenterol. Rep. 2021, 9, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Morsy, Y.; Brillant, N.; Franc, Y.; Scharl, M.; Wawrzyniak, M.; On behalf of the Swiss Ibd Cohort Study Group. Unravelling the Impact of the Genetic Variant rs1042058 within the TPL2 Risk Gene Locus on Molecular and Clinical Disease Course Patients with Inflammatory Bowel Disease. Cells 2021, 10, 3589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, X.; Jiang, X.; Yang, X.; Wen, C.; Zhi, M.; Gao, X.; Hu, P.; Liu, H. Polymorphisms of the TNF Gene and Three Susceptibility Loci Are Associated with Crohn’s Disease and Perianal Fistula Crohn’s Disease: A Study among the Han Population from South China. Med. Sci. Monit. 2019, 25, 9637–9650. [Google Scholar] [CrossRef]
- Cleynen, I.; Boucher, G.; Jostins, L.; Schumm, L.P.; Zeissig, S.; Ahmad, T.; Andersen, V.; Andrews, J.M.; Annese, V.; Brand, S.; et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: A genetic association study. Lancet 2016, 387, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Kakuta, Y.; Iwaki, H.; Umeno, J.; Kawai, Y.; Kawahara, M.; Takagawa, T.; Shimoyama, Y.; Naito, T.; Moroi, R.; Kuroha, M.; et al. Crohn’s disease and early exposure to thiopurines are independent risk factors for mosaic chromosomal alterations in patients with inflammatory bowel diseases. J. Crohns Colitis 2021. [Google Scholar] [CrossRef] [PubMed]
- Serra, E.G.; Schwerd, T.; Moutsianas, L.; Cavounidis, A.; Fachal, L.; Pandey, S.; Kammermeier, J.; Croft, N.M.; Posovszky, C.; Rodrigues, A.; et al. Somatic mosaicism and common genetic variation contribute to the risk of very-early-onset inflammatory bowel disease. Nat. Commun. 2020, 11, 995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liang, Y.; Lu, J.; Tan, Y. An alternative splicing signature in human Crohn’s disease. BMC Gastroenterol. 2021, 21, 420. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tan, Y. Dysregulation of alternative splicing is associated with the pathogenesis of ulcerative colitis. Biomed. Eng. Online 2021, 20, 121. [Google Scholar] [CrossRef] [PubMed]
- Peloquin, J.M.; Goel, G.; Kong, L.; Huang, H.; Haritunians, T.; Sartor, R.B.; Daly, M.J.; Newberry, R.D.; McGovern, D.P.; Yajnik, V.; et al. Characterization of candidate genes in inflammatory bowel disease-associated risk loci. JCI Insight 2016, 1, e87899. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.A.R.; Rodrigues, B.L.; Ayrizono, M.d.L.S.; Leal, R.F. The Immunological Basis of Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2016, 2016, 2097274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, K.; Antanaviciute, A.; Fawkner-Corbett, D.; Jagielowicz, M.; Aulicino, A.; Lagerholm, C.; Davis, S.; Kinchen, J.; Chen, H.H.; Alham, N.K.; et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 2019, 567, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Borcherding, N.; Andrews, J. Escape: Easy Single Cell Analysis Platform for Enrichment: R Package, Version 1.2.0; Bioconductor: Seattle, WA, USA, 2021; Available online: https://bioconductor.org/packages/release/bioc/html/escape.html (accessed on 14 February 2022).
- Pekow, J.; Dougherty, U.; Huang, Y.; Gometz, E.; Nathanson, J.; Cohen, G.; Levy, S.; Kocherginsky, M.; Venu, N.; Westerhoff, M.; et al. Gene signature distinguishes patients with chronic ulcerative colitis harboring remote neoplastic lesions. Inflamm. Bowel Dis. 2013, 19, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paredes, J.; Zabaleta, J.; Garai, J.; Ji, P.; Imtiaz, S.; Spagnardi, M.; Alvarado, J.; Li, L.; Akadri, M.; Barrera, K.; et al. Immune-Related Gene Expression and Cytokine Secretion Is Reduced among African American Colon Cancer Patients. Front. Oncol. 2020, 10, 1498. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Kim, S.-Y.; Kim, J.-H.; Roh, S.A.; Cho, D.-H.; Kim, Y.S.; Kim, J.C. A nineteen gene-based risk score classifier predicts prognosis of colorectal cancer patients. Mol. Oncol. 2014, 8, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Saul, D.; Kosinsky, R.L. Single-Cell Transcriptomics Reveals the Expression of Aging- and Senescence-Associated Genes in Distinct Cancer Cell Populations. Cells 2021, 10, 3126. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.C.; Chang, C.; Boschetti, G.; Ungaro, R.; Giri, M.; Grout, J.A.; Gettler, K.; Chuang, L.-S.; Nayar, S.; Greenstein, A.J.; et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019, 178, 1493–1508.e20. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-O.; Hong, Y.; Etlioglu, H.E.; Cho, Y.B.; Pomella, V.; van den Bosch, B.; Vanhecke, J.; Verbandt, S.; Hong, H.; Min, J.-W.; et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 2020, 52, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Saul, D.; Monroe, D.G.; Rowsey, J.L.; Kosinsky, R.L.; Vos, S.J.; Doolittle, M.L.; Farr, J.N.; Khosla, S. Modulation of fracture healing by the transient accumulation of senescent cells. eLife 2021, 10, e69958. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M.; et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.-R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Iacono, G.; Massoni-Badosa, R.; Heyn, H. Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol. 2019, 20, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillen, C.D.; Walmsley, R.S.; Prior, P.; Andrews, H.A.; Allan, R.N. Ulcerative colitis and Crohn’s disease: A comparison of the colorectal cancer risk in extensive colitis. Gut 1994, 35, 1590–1592. [Google Scholar] [CrossRef] [PubMed]
- Lutgens, M.W.M.D.; van Oijen, M.G.H.; van der Heijden, G.J.M.G.; Vleggaar, F.P.; Siersema, P.D.; Oldenburg, B. Declining risk of colorectal cancer in inflammatory bowel disease: An updated meta-analysis of population-based cohort studies. Inflamm. Bowel Dis. 2013, 19, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Weiser, M.; Simon, J.M.; Kochar, B.; Tovar, A.; Israel, J.W.; Robinson, A.; Gipson, G.R.; Schaner, M.S.; Herfarth, H.H.; Sartor, R.B.; et al. Molecular classification of Crohn’s disease reveals two clinically relevant subtypes. Gut 2018, 67, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Vitali, F.; Wein, A.; Rath, T.; Eckstein, M.; Neufert, C.; Siebler, J.; Atreya, R.; Hartmann, A.; Hohenberger, W.; Weber, K.; et al. The outcome of patients with inflammatory bowel disease-associated colorectal cancer is not worse than that of patients with sporadic colorectal cancer-a matched-pair analysis of survival. Int. J. Colorectal Dis. 2022, 37, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, Y.; Golan, M.A.; Annunziata, M.L.; Du, J.; Dougherty, U.; Kong, J.; Musch, M.; Huang, Y.; Pekow, J.; et al. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J. Clin. Investig. 2013, 123, 3983–3996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Yu, J.; Liu, K.; Liu, Y.; Mao, C.; Gao, W. The Pathogenic Roles of IL-22 in Colitis: Its Transcription Regulation by Musculin in T Helper Subsets and Innate Lymphoid Cells. Front. Immunol. 2021, 12, 758730. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Crinier, A.; Escalière, B.; Ye, Y.; Wang, Z.; Zhang, T.; Batista, L.; Liu, H.; Hong, L.; Wu, N.; et al. Single-cell transcriptomic landscape reveals tumor specific innate lymphoid cells associated with colorectal cancer progression. Cell Rep. Med. 2021, 2, 100353. [Google Scholar] [CrossRef] [PubMed]
- Kappeler, A.; Mueller, C. The role of activated cytotoxic T cells in inflammatory bowel disease. Histol. Histopathol. 2000, 15, 167–172. [Google Scholar] [CrossRef]
- Kosinsky, R.L.; Zerche, M.; Kutschat, A.P.; Nair, A.; Ye, Z.; Saul, D.; von Heesen, M.; Friton, J.J.; Schwarzer, A.C.; Paglilla, N.; et al. RNF20 and RNF40 regulate vitamin D receptor-dependent signaling in inflammatory bowel disease. Cell Death Differ. 2021, 28, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.M.; Bamidele, A.O.; Svingen, P.A.; Sagstetter, M.R.; Smyrk, T.C.; Gaballa, J.M.; Hamdan, F.H.; Kosinsky, R.L.; Gibbons, H.R.; Sun, Z.; et al. BMI1 maintains the Treg epigenomic landscape to prevent inflammatory bowel disease. J. Clin. Investig. 2021, 131, e140755. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Wang, H.; Hong, Y.; Ren, A.; Wang, H.; Liu, L.; Zhao, Q. Identification of an at-risk subpopulation with high immune infiltration based on the peroxisome pathway and TIM3 in colorectal cancer. BMC Cancer 2022, 22, 44. [Google Scholar] [CrossRef]
- Kanauchi, Y.; Yamamoto, T.; Yoshida, M.; Zhang, Y.; Lee, J.; Hayashi, S.; Kadowaki, M. Cholinergic anti-inflammatory pathway ameliorates murine experimental Th2-type colitis by suppressing the migration of plasmacytoid dendritic cells. Sci. Rep. 2022, 12, 54. [Google Scholar] [CrossRef]
- Chandra, R.; Karalis, J.D.; Liu, C.; Murimwa, G.Z.; Voth Park, J.; Heid, C.A.; Reznik, S.I.; Huang, E.; Minna, J.D.; Brekken, R.A. The Colorectal Cancer Tumor Microenvironment and Its Impact on Liver and Lung Metastasis. Cancers 2021, 13, 6206. [Google Scholar] [CrossRef] [PubMed]
- Pryczynicz, A.; Cepowicz, D.; Zaręba, K.; Gryko, M.; Hołody-Zaręba, J.; Kędra, B.; Kemona, A.; Guzińska-Ustymowicz, K. Dysfunctions in the Mature Dendritic Cells Are Associated with the Presence of Metastases of Colorectal Cancer in the Surrounding Lymph Nodes. Gastroenterol. Res. Pract. 2016, 2016, 2405437. [Google Scholar] [CrossRef] [PubMed]
- Yarilina, A.; Park-Min, K.-H.; Antoniv, T.; Hu, X.; Ivashkiv, L.B. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol. 2008, 9, 378–387. [Google Scholar] [CrossRef]
- Pfizenmaier, K.; Scheurich, P.; Schlüter, C.; Krönke, M. Tumor necrosis factor enhances HLA-A,B,C and HLA-DR gene expression in human tumor cells. J. Immunol. 1987, 138, 975–980. [Google Scholar] [PubMed]
- Pereira, R.; Lago, P.; Faria, R.; Torres, T. Safety of Anti-TNF Therapies in Immune-Mediated Inflammatory Diseases: Focus on Infections and Malignancy. Drug Dev. Res. 2015, 76, 419–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vulliemoz, M.; Brand, S.; Juillerat, P.; Mottet, C.; Ben-Horin, S.; Michetti, P. TNF-Alpha Blockers in Inflammatory Bowel Diseases: Practical Recommendations and a User’s Guide: An Update. Digestion 2020, 101 (Suppl. 1), 16–26. [Google Scholar] [CrossRef] [PubMed]
- Hamdeh, S.; Aziz, M.; Altayar, O.; Olyaee, M.; Murad, M.H.; Hanauer, S.B. Early vs. Late Use of Anti-TNFa Therapy in Adult Patients with Crohn Disease: A Systematic Review and Meta-Analysis. Inflamm. Bowel Dis. 2020, 26, 1808–1818. [Google Scholar] [CrossRef]
- Rieder, F.; Fiocchi, C. Intestinal fibrosis in inflammatory bowel disease-Current knowledge and future perspectives. J. Crohns Colitis 2008, 2, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Garza Treviño, E.N.; González, P.D.; Valencia Salgado, C.I.; Martinez Garza, A. Effects of pericytes and colon cancer stem cells in the tumor microenvironment. Cancer Cell Int. 2019, 19, 173. [Google Scholar] [CrossRef]
- Boccarelli, A.; Del Buono, N.; Esposito, F. Colorectal cancer in Crohn’s disease evaluated with genes belonging to fibroblasts of the intestinal mucosa selected by NMF. Pathol. Res. Pract. 2022, 229, 153728. [Google Scholar] [CrossRef]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Boesch, M.; Spizzo, G.; Seeber, A. Concise Review: Aggressive Colorectal Cancer: Role of Epithelial Cell Adhesion Molecule in Cancer Stem Cells and Epithelial-to-Mesenchymal Transition. Stem Cells Transl. Med. 2018, 7, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.C.; Banks, R.E.; Haidar, A.; Gearing, A.J.; Hemingway, I.K.; Ibbotson, S.H.; Dixon, M.F.; Axon, A.T. Adhesion molecules in inflammatory bowel disease. Gut 1995, 36, 724–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saul, D.; Leite Barros, L.; Wixom, A.Q.; Gellhaus, B.; Gibbons, H.R.; Faubion, W.A.; Kosinsky, R.L. Cell Type-Specific Induction of Inflammation-Associated Genes in Crohn’s Disease and Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 3082. https://doi.org/10.3390/ijms23063082
Saul D, Leite Barros L, Wixom AQ, Gellhaus B, Gibbons HR, Faubion WA, Kosinsky RL. Cell Type-Specific Induction of Inflammation-Associated Genes in Crohn’s Disease and Colorectal Cancer. International Journal of Molecular Sciences. 2022; 23(6):3082. https://doi.org/10.3390/ijms23063082
Chicago/Turabian StyleSaul, Dominik, Luísa Leite Barros, Alexander Q. Wixom, Benjamin Gellhaus, Hunter R. Gibbons, William A. Faubion, and Robyn Laura Kosinsky. 2022. "Cell Type-Specific Induction of Inflammation-Associated Genes in Crohn’s Disease and Colorectal Cancer" International Journal of Molecular Sciences 23, no. 6: 3082. https://doi.org/10.3390/ijms23063082