
The Splicing of the Mitochondrial Calcium Uniporter Genuine Activator MICU1 Is Driven by RBFOX2 Splicing Factor during Myogenic Differentiation

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
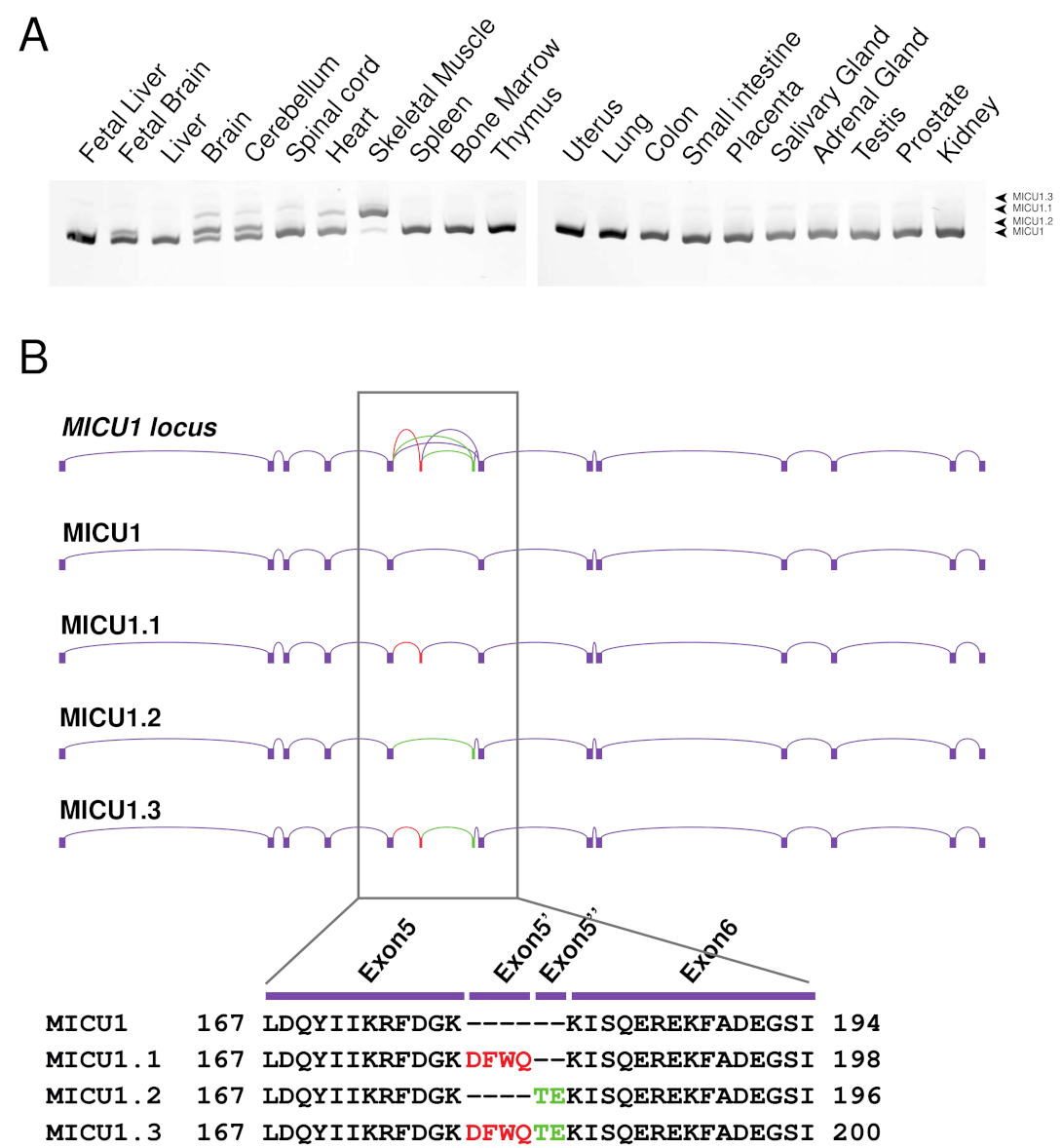
2.1. Characterization of the Human MICU1 Splice Variants
2.2. Human MICU1 Splice Variants Display Different Effects on Mitochondrial Ca2+ Uptake
2.3. MICU1 Introns Flanking 5′ Extra-Exon Present Multiple Splicing Factors Binding Sites
2.4. MICU1 Alternative Splicing Events Do Not Require the Splicing Factor MBNL1
2.5. MICU1 Alterative Splicing Is Regulated during Embryonic Development by the Splicing Factor RBFOX2
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cell Culture and Transfection
4.3. Aequorin Ca2+ Measurements
4.4. RNA Extraction, Reverse Transcription, PCR and qPCR
4.5. siRNAs and Constructs
4.6. Western Blotting
4.7. Prediction of Splicing Factors Binding Sites
4.8. Statistical Analysis of Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brett, D.; Pospisil, H.; Valcárcel, J.; Reich, J.; Bork, P. Alternative splicing and genome complexity. Nat. Genet. 2002, 30, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Mariano, A.G.B.; Andrew, P.B.; Erika, L.L. Alternative splicing in disease and therapy. Nat. Biotechnol. 2004, 22, 535–546. [Google Scholar]
- Modrek, B.; Lee, C. A genomic view of alternative splicing. Nat. Genet. 2002, 30, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. Rna 2008, 14, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and mechanisms of alternative splicing in cancer—Implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Lipscombe, D.; Lopez Soto, E.J. Alternative splicing of neuronal genes: New mechanisms and new therapies. Curr. Opin. Neurobiol. 2019, 57, 26–31. [Google Scholar] [CrossRef]
- Merkin, J.; Russell, C.; Chen, P.; Burge, C.B. Evolutionary Dynamics of Gene and Isoform Regulation in Mammalian Tissues. Science 2012, 338, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Castle, J.C.; Zhang, C.; Shah, J.K.; Kulkarni, A.V.; Kalsotra, A.; Cooper, T.A.; Johnson, J.M. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008, 40, 1416–1425. [Google Scholar] [CrossRef] [Green Version]
- Pascual, M.; Vicente, M.; Monferrer, L.; Artero, R. The Muscleblind family of proteins: An emerging class of regulators of developmentally programmed alternative splicing. Differentiation 2006, 74, 65–80. [Google Scholar] [CrossRef]
- Kuroyanagi, H.; Ohno, G.; Mitani, S.; Hagiwara, M. The Fox-1 Family and SUP-12 Coordinately Regulate Tissue-Specific Alternative Splicing In Vivo. Mol. Cell. Biol. 2007, 27, 8612–8621. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorian, M.; Smith, C.W.J. Decoding muscle alternative splicing. Curr. Opin. Genet. Dev. 2011, 21, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Toyota, N.; Shimada, Y. Differentiation of troponin in cardiac and skeletal muscles in chicken embryos as studied by immunofluorescence microscopy. J. Cell Biol. 1981, 91, 497–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.W.J.; Nadal-Ginard, B. Mutually exclusive splicing of α-tropomyosin exons enforced by an unusual lariat branch point location: Implications for constitutive splicing. Cell 1989, 56, 749–758. [Google Scholar] [CrossRef]
- Zot, A.S.; Potter, J.D. Structural Aspects of Troponin-Tropomyosin Regulation of Skeletal Muscle Contraction. Annu. Rev. Biophys. Biophys. Chem. 1987, 16, 535–559. [Google Scholar] [CrossRef]
- Farah, C.S.; Reinach, F.C. The troponin complex and regulation of muscle contraction. FASEB J. 1995, 9, 755–767. [Google Scholar] [CrossRef]
- Hinkle, E.R.; Wiedner, H.J.; Black, A.J.; Giudice, J. RNA processing in skeletal muscle biology and disease. Transcription 2019, 10, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Imbriano, C.; Molinari, S. Alternative splicing of transcription factors genes in muscle physiology and pathology. Genes 2018, 9, 107. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Kolonin, A.M.; Fiorotto, M.L.; Cooper, T.A. Rbfox-Splicing Factors Maintain Skeletal Muscle Mass by Regulating Calpain3 and Proteostasis. Cell Rep. 2018, 24, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Nakka, K.; Ghigna, C.; Gabellini, D.; Dilworth, F.J. Diversification of the muscle proteome through alternative splicing. Skelet. Muscle 2018, 8, 1–18. [Google Scholar] [CrossRef]
- Lin, X.; Miller, J.W.; Mankodi, A.; Kanadia, R.N.; Yuan, Y.; Moxley, R.T.; Swanson, M.S.; Thornton, C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006, 15, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Fu, Y.H.; Pizzuti, A.; Fenwick, R.G.; King, J.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; Nasser, G.A.; Ashizawa, T.; de Jong, P.; et al. An Unstable Triplet Repeat in a Gene Related to Myotonic Muscular Dystrophy. Science 1992, 255, 1256–1258. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barceló, J.; O’Hoy, K.; et al. Myotonic Dystrophy Mutation: An Unstable CTG Repeat in the 3′ Untranslated region of the Gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P.W. Myotonic Dystrophy Type 2 Caused by a CCTG Expansion in Intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Narasimhan, A.; Greiner, R.; Bathe, O.F.; Baracos, V.; Damaraju, S. Differentially expressed alternatively spliced genes in skeletal muscle from cancer patients with cachexia. J. Cachexia. Sarcopenia Muscle 2018, 9, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Orengo, J.P.; Ward, A.J.; Cooper, T.A. Alternative splicing dysregulation secondary to skeletal muscle regeneration. Ann. Neurol. 2011, 69, 681–690. [Google Scholar] [CrossRef] [Green Version]
- van der Wal, E.; Bergsma, A.J.; Pijnenburg, J.M.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Antisense Oligonucleotides Promote Exon Inclusion and Correct the Common c.-32-13T>G GAA Splicing Variant in Pompe Disease. Mol. Ther. Nucleic Acids 2017, 7, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Ng, B.; Yang, F.; Huston, D.P.; Yan, Y.; Yang, Y.; Xiong, Z.; Peterson, L.E.; Wang, H.; Yang, X.F. Increased noncanonical splicing of autoantigen transcripts provides the structural basis for expression of untolerized epitopes. J. Allergy Clin. Immunol. 2004, 114, 1463–1470. [Google Scholar] [CrossRef] [Green Version]
- Young, C.S.; Pyle, A.D. Exon Skipping Therapy. Cell 2016, 167, 1144. [Google Scholar] [CrossRef]
- Liao, P.; Yu, D.; Li, G.; Tan, F.Y.; Jia, L.S.; Yeow, L.C.; Tuck, W.S. A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. J. Biol. Chem. 2007, 282, 35133–35142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, P.; Yu, D.; Lu, S.; Tang, Z.; Mui, C.L.; Zeng, S.; Lin, W.; Tuck, W.S. Smooth muscle-selective alternatively spliced exon generates functional variation in Cav1.2 calcium channels. J. Biol. Chem. 2004, 279, 50329–50335. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Nakamori, M.; Lueck, J.D.; Pouliquin, P.; Aoike, F.; Fujimura, H.; Dirksen, R.T.; Takahashi, M.P.; Dulhunty, A.F.; Sakoda, S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet. 2005, 14, 2189–2200. [Google Scholar] [CrossRef] [Green Version]
- Futatsugi, A.; Kuwajima, G.; Mikoshiba, K. Tissue-specific and developmentally regulated alternative splicing in mouse skeletal muscle ryanodine receptor mRNA. Biochem. J. 1995, 305, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Brandl, C.J.; deLeon, S.; Martin, D.R.; MacLennan, D.H. Adult forms of the Ca2+ATPase of sarcoplasmic reticulum. Expression in developing skeletal muscle. J. Biol. Chem. 1987, 262, 3768–3774. [Google Scholar] [CrossRef]
- Tang, Z.Z.; Yarotskyy, V.; Wei, L.; Sobczak, K.; Nakamori, M.; Eichinger, K.; Moxley, R.T.; Dirksen, R.T.; Thornton, C.A. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca v1.1 calcium channel. Hum. Mol. Genet. 2012, 21, 1312–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feno, S.; Rizzuto, R.; Raffaello, A.; Vecellio Reane, D. The molecular complexity of the Mitochondrial Calcium Uniporter. Cell Calcium 2021, 93, 102322. [Google Scholar] [CrossRef]
- Vecellio Reane, D.; Vallese, F.; Checchetto, V.; Acquasaliente, L.; Butera, G.; De Filippis, V.; Szabò, I.; Zanotti, G.; Rizzuto, R.; Raffaello, A. A MICU1 Splice Variant Confers High Sensitivity to the Mitochondrial Ca2+ Uptake Machinery of Skeletal Muscle. Mol. Cell 2016, 64, 760–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabò, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 Finely Tune the Mitochondrial Ca2+ Uniporter by Exerting Opposite Effects on MCU Activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [Green Version]
- Paz, I.; Kosti, I.; Ares, M.; Cline, M.; Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014, 42, W361–W367. [Google Scholar] [CrossRef]
- Goodwin, M.; Mohan, A.; Batra, R.; Lee, K.Y.; Charizanis, K.; Fernández Gómez, F.J.; Eddarkaoui, S.; Sergeant, N.; Buée, L.; Kimura, T.; et al. MBNL Sequestration by Toxic RNAs and RNA Misprocessing in the Myotonic Dystrophy Brain. Cell Rep. 2015, 12, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.W.; Urbinati, C.R.; Teng-Umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef] [Green Version]
- Mankodi, A.; Teng-Umnuay, P.; Krym, M.; Henderson, D.; Swanson, M.; Thornton, C.A. Ribonuclear Inclusions in Skeletal Muscle in Myotonic Dystrophy Types 1 and 2. Ann. Neurol. 2003, 54, 760–768. [Google Scholar] [CrossRef]
- Botta, A.; Malena, A.; Loro, E.; Del Moro, G.; Suman, M.; Pantic, B.; Szabadkai, G.; Vergani, L. Altered Ca2+ Homeostasis and Endoplasmic Reticulum Stress in Myotonic Dystrophy Type 1 Muscle Cells. Genes 2013, 4, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Martínez, A.; Soblechero-Martín, P.; De-la-Puente-Ovejero, L.; Nogales-Gadea, G.; Arechavala-Gomeza, V. An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I. Genes 2020, 11, 1109. [Google Scholar] [CrossRef]
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A Muscleblind Knockout Model for Myotonic Dystrophy. Science 2003, 302, 1978–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bargiela, A.; Sabater-Arcis, M.; Espinosa-Espinosa, J.; Zulaica, M.; Lopez de Munain, A.; Artero, R. Increased Muscleblind levels by chloroquine treatment improve myotonic dystrophy type 1 phenotypes in in vitro and in vivo models. Proc. Natl. Acad. Sci. USA 2019, 116, 25203–25213. [Google Scholar] [CrossRef]
- Hino, S.I.; Kondo, S.; Sekiya, H.; Saito, A.; Kanemoto, S.; Murakami, T.; Chihara, K.; Aoki, Y.; Nakamori, M.; Takahashi, M.P.; et al. Molecular mechanisms responsible for aberrant splicing of SERCA1 in myotonic dystrophy type 1. Hum. Mol. Genet. 2007, 16, 2834–2843. [Google Scholar] [CrossRef]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic Dystrophy in Transgenic Mice Expressing an Expanded CUG Repeat. Science 2000, 289, 1769–1772. [Google Scholar] [CrossRef] [Green Version]
- Schätzl, T.; Kaiser, L.; Deigner, H.P. Facioscapulohumeral muscular dystrophy: Genetics, gene activation and downstream signalling with regard to recent therapeutic approaches: An update. Orphanet J. Rare Dis. 2021, 16, 129. [Google Scholar] [CrossRef]
- Hall, M.P.; Nagel, R.J.; Fagg, W.S.; Shiue, L.; Cline, M.S.; Perriman, R.J.; Donohue, J.P.; Ares, M. Quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation. RNA 2013, 19, 627–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bland, C.S.; Wang, E.T.; Vu, A.; David, M.P.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res. 2010, 38, 7651–7664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar] [CrossRef]
- Sebbag-Sznajder, N.; Raitskin, O.; Angenitzki, M.; Sato, T.A.; Sperling, J.; Sperling, R. Regulation of alternative splicing within the supraspliceosome. J. Struct. Biol. 2012, 177, 152–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Jacewicz, A.; Delgado, B.D.; Baradaran, R.; Long, S.B. Structures reveal gatekeeping of the mitochondrial Ca2+ uniporter by MICU1-MICU2. Elife 2020, 9, 1–30. [Google Scholar] [CrossRef]
- Wang, Y.; Han, Y.; She, J.; Nguyen, N.X.; Mootha, V.K.; Bai, X.C.; Jiang, Y. Structural insights into the ca2+-dependent gating of the human mitochondrial calcium uniporter. Elife 2020, 9, 1–19. [Google Scholar] [CrossRef]
- Wu, W.; Zheng, J.; Jia, Z. Structural characterization of the mitochondrial Ca2+ uniporter provides insights into Ca2+ uptake and regulation. iScience 2021, 24, 102895. [Google Scholar] [CrossRef]
- Park, J.; Lee, Y.; Park, T.; Kang, J.Y.; Mun, S.A.; Jin, M.; Yang, J.; Eom, S.H. Structure of the MICU1–MICU2 heterodimer provides insights into the gatekeeping threshold shift. IUCrJ 2020, 7, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Shen, Q.; Zhang, R.; Qiu, Z.; Wang, Y.; Zheng, J.; Jia, Z. The structure of the MICU1-MICU2 complex unveils the regulation of the mitochondrial calcium uniporter. EMBO J. 2020, 39, e104285. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Jiang, W.; Kaushik, V.K.; Mootha, V.K.; Grabarek, Z. Crystal structure of MICU2 and comparison with MICU1 reveal insights into the uniporter gating mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 3546–3555. [Google Scholar] [CrossRef] [Green Version]
- Fan, M.; Zhang, J.; Tsai, C.W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.F.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; Li, S.; Wang, Z.; Liu, Y.; Feng, J.; Zhu, Y.; Shen, Y. Structural and mechanistic insights into MICU1 regulation of mitochondrial calcium uptake. EMBO J. 2014, 33, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Wang, M.; Wang, J.; Nie, Z.; Wu, G.; Yang, X.; Shen, Y. Dimerization of MICU Proteins Controls Ca2+ Influx through the Mitochondrial Ca2+ Uniporter. Cell Rep. 2019, 26, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; de Juan-Sanz, J.; Farrell, R.J.; Ryan, T.A. Molecular Tuning of the Axonal Mitochondrial Ca2+ Uniporter Ensures Metabolic Flexibility of Neurotransmission. Neuron 2020, 105, 678–687. [Google Scholar] [CrossRef]
- Tufi, R.; Gleeson, T.P.; von Stockum, S.; Hewitt, V.L.; Lee, J.J.; Terriente-Felix, A.; Sanchez-Martinez, A.; Ziviani, E.; Whitworth, A.J. Comprehensive Genetic Characterization of Mitochondrial Ca2+ Uniporter Components Reveals Their Different Physiological Requirements In Vivo. Cell Rep. 2019, 27, 1541–1550. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.F.; Yang, W.; Liao, Z.Y.; Wu, Y.X.; Fan, Z.; Guo, A.; Yu, J.; Chen, Q.N.; Wu, J.H.; Zhou, J.; et al. MICU3 regulates mitochondrial Ca2+-dependent antioxidant response in skeletal muscle aging. Cell Death Dis. 2021, 12, 1115. [Google Scholar] [CrossRef]
- Bandman, E. Contractile protein isoforms in muscle development. Dev. Biol. 1992, 154, 273–283. [Google Scholar] [CrossRef]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and Disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [Green Version]
- Pedrotti, S.; Giudice, J.; Dagnino-Acosta, A.; Knoblauch, M.; Singh, R.K.; Hanna, A.; Mo, Q.; Hicks, J.; Hamilton, S.; Cooper, T.A. The RNA-binding protein Rbfox1 regulates splicing required for skeletal muscle structure and function. Hum. Mol. Genet. 2015, 24, 2360–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.K.; Xia, Z.; Bland, C.S.; Kalsotra, A.; Scavuzzo, M.A.; Curk, T.; Ule, J.; Li, W.; Cooper, T.A. Rbfox2-Coordinated Alternative Splicing of Mef2d and Rock2 Controls Myoblast Fusion during Myogenesis. Mol. Cell 2014, 55, 592–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Zhang, Z.; Castle, J.; Sun, S.; Johnson, J.; Krainer, A.R.; Zhang, M.Q. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2 (Genes and Development (2008) 22 (2550–2563)). Genes Dev. 2008, 22, 2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runfola, V.; Sebastian, S.; Dilworth, F.J.; Gabellini, D. Rbfox proteins regulate tissue-specific alternative splicing of Mef2D required for muscle differentiation. J. Cell Sci. 2015, 128, 631–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvinge, H. Regulation of alternative mRNA splicing: Old players and new perspectives. FEBS Lett. 2018, 592, 2987–3006. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef]
- Mammucari, C.; Gherardi, G.; Zamparo, I.; Raffaello, A.; Boncompagni, S.; Chemello, F.; Cagnin, S.; Braga, A.; Zanin, S.; Pallafacchina, G.; et al. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 2015, 10, 1269–1279. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabó, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [Green Version]
- Pinton, P.; Rimessi, A.; Romagnoli, A.; Prandini, A.; Rizzuto, R. Biosensors for the detection of calcium and pH. Methods Cell Biol. 2007, 80, 297–325. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 2013, 3, 71–85. [Google Scholar] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vecellio Reane, D.; Cerqua, C.; Sacconi, S.; Salviati, L.; Trevisson, E.; Raffaello, A. The Splicing of the Mitochondrial Calcium Uniporter Genuine Activator MICU1 Is Driven by RBFOX2 Splicing Factor during Myogenic Differentiation. Int. J. Mol. Sci. 2022, 23, 2517. https://doi.org/10.3390/ijms23052517
Vecellio Reane D, Cerqua C, Sacconi S, Salviati L, Trevisson E, Raffaello A. The Splicing of the Mitochondrial Calcium Uniporter Genuine Activator MICU1 Is Driven by RBFOX2 Splicing Factor during Myogenic Differentiation. International Journal of Molecular Sciences. 2022; 23(5):2517. https://doi.org/10.3390/ijms23052517
Chicago/Turabian StyleVecellio Reane, Denis, Cristina Cerqua, Sabrina Sacconi, Leonardo Salviati, Eva Trevisson, and Anna Raffaello. 2022. "The Splicing of the Mitochondrial Calcium Uniporter Genuine Activator MICU1 Is Driven by RBFOX2 Splicing Factor during Myogenic Differentiation" International Journal of Molecular Sciences 23, no. 5: 2517. https://doi.org/10.3390/ijms23052517