
The Complex Mechanisms by Which Neurons Die Following DNA Damage in Neurodegenerative Diseases


Abstract
:1. Introduction
2. The DNA Damage Response
2.1. DNA Damage Signalling in Neurons
2.2. Detecting the Presence of DNA Damage
2.3. DNA Repair Mechanisms in Neurons
3. Neuronal Senescence
4. Re-Entry of Neurons into the Cell Cycle
5. Mitochondria and DNA Damage in Neurons
6. Cell Death Mechanisms Relevant to DNA Damage
6.1. Cell Death in Neurons
6.2. Apoptosis
6.3. Autophagy-Dependent Cell Death
6.4. Necrosis: Necroptosis and MPTP-Dependent Regulated Necrosis
6.4.1. Necroptosis
6.4.2. MPTP-Dependent Regulated Necrosis
6.5. Parthanatos
6.6. Ferroptosis
6.7. Pyroptosis
6.8. Lysosome Dependent Cell Death
7. Mechanisms of Cell Death Induced by DNA Damage in Neurodegenerative Diseases
7.1. Alzheimer’s Disease (AD)
7.1.1. DNA Damage and Apoptosis in AD
7.1.2. DNA Damage and Parthanatos in AD
7.1.3. DNA Damage, p53 and Neuronal Death in AD
7.2. Parkinson’s Disease
7.2.1. DNA Damage and Apoptosis in PD
7.2.2. DNA Damage and Parthanatos in PD
7.2.3. DNA Damage, p53, and Neuronal Death in PD
7.3. Amyotrophic Lateral Sclerosis (ALS)
7.3.1. DNA Damage and Apoptosis in ALS
7.3.2. DNA Damage and Parthanatos in ALS
7.3.3. DNA Damage, p53, and Neuronal Death in ALS
7.4. Frontotemporal Dementia (FTD)
7.5. Huntington’s Disease (HD)
8. Therapeutic Potential of DNA Damage in Preventing Cell Death in Neurodegenerative Diseases
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Commun. 2020, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Lindahl, T. Maintenance of genome stability. Genom. Proteom. Bioinform. 2016, 14, 119–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyama, T.; Wilson, D.M., 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [Green Version]
- Polo, L.M.; Xu, Y.; Hornyak, P.; Garces, F.; Zeng, Z.; Hailstone, R.; Matthews, S.J.; Caldecott, K.W.; Oliver, A.W.; Pearl, L.H. Efficient Single-Strand Break Repair Requires Binding to Both Poly(ADP-Ribose) and DNA by the Central BRCT Domain of XRCC1. Cell Rep. 2019, 26, 573–581.e575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target. 2020, 5, 60. [Google Scholar] [CrossRef]
- Madabhushi, R.; Pan, L.; Tsai, L.-H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Hill, S.E.; Nathan, W.J.; Paiano, J.; Callen, E.; Wang, D.; Shinoda, K.; van Wietmarschen, N.; Colon-Mercado, J.M.; Zong, D.; et al. Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 2021, 593, 440–444. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, J.J.; Stewart, G.S. A single strand that links multiple neuropathologies in human disease. Brain 2013, 136, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Ko, H.L.; Ren, E.C. Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules 2012, 2, 524–548. [Google Scholar] [CrossRef] [Green Version]
- Langelier, M.-F.; Billur, R.; Sverzhinsky, A.; Black, B.E.; Pascal, J.M. HPF1 dynamically controls the PARP1/2 balance between initiating and elongating ADP-ribose modifications. Nat. Commun. 2021, 12, 6675. [Google Scholar] [CrossRef] [PubMed]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharm. 2012, 84, 137–146. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoch, N.C.; Hanzlikova, H.; Rulten, S.L.; Tétreault, M.; Komulainen, E.; Ju, L.; Hornyak, P.; Zeng, Z.; Gittens, W.; Rey, S.A.; et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 2017, 541, 87–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Bürkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharm. 2018, 175, 192–222. [Google Scholar] [CrossRef] [PubMed]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.C.; Corbett, A.H.; Doetsch, P.W. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015, 43, 10083–10101. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef]
- Gottlieb, T.M.; Jackson, S.P. The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell 1993, 72, 131–142. [Google Scholar] [CrossRef]
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X.; et al. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol. Cell 2015, 58, 172–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Herrero, F.; de Jager, M.; Dekker, N.H.; Kanaar, R.; Wyman, C.; Dekker, C. Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature 2005, 437, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Sun, Y.; Jiang, X.; Ayrapetov, M.K.; Moskwa, P.; Yang, S.; Weinstock, D.M.; Price, B.D. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J. Cell Biol. 2010, 191, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Kusch, T.; Florens, L.; Macdonald, W.H.; Swanson, S.K.; Glaser, R.L.; Yates, J.R., 3rd; Abmayr, S.M.; Washburn, M.P.; Workman, J.L. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 2004, 306, 2084–2087. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Lukas, C.; Melander, F.; Bartek, J.; Lukas, J. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J. Cell. Biol. 2005, 170, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Difilippantonio, S.; Gapud, E.; Wong, N.; Huang, C.-Y.; Mahowald, G.; Chen, H.T.; Kruhlak, M.J.; Callen, E.; Livak, F.; Nussenzweig, M.C.; et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature 2008, 456, 529–533. [Google Scholar] [CrossRef]
- Chapman, J.R.; Jackson, S.P. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Aziz, K.; Aziz, A.; Deng, M.; Qin, B.; Luo, K.; Jeganathan, K.B.; Zhang, H.; Liu, T.; Yu, J.; et al. L3MBTL2 orchestrates ubiquitin signalling by dictating the sequential recruitment of RNF8 and RNF168 after DNA damage. Nat. Cell Biol. 2018, 20, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Fortini, P.; Ferretti, C.; Dogliotti, E. The response to DNA damage during differentiation: Pathways and consequences. Mutat Res. 2013, 743–744, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Bartkova, J.; Latella, L.; Falck, J.; Mailand, N.; Schroeder, T.; Sehested, M.; Lukas, J.; Bartek, J. DNA damage-activated kinase Chk2 is independent of proliferation or differentiation yet correlates with tissue biology. Cancer Res. 2001, 61, 4990–4993. [Google Scholar]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef]
- Jones, G.G.; Reaper, P.M.; Pettitt, A.R.; Sherrington, P.D. The ATR-p53 pathway is suppressed in noncycling normal and malignant lymphocytes. Oncogene 2004, 23, 1911–1921. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Fishel, M.L.; Vasko, M.R.; Kelley, M.R. DNA repair in neurons: So if they don’t divide what’s to repair? Mutat Res. 2007, 614, 24–36. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. Human nucleotide excision repair syndromes: Molecular clues to unexpected intricacies. Eur. J. Cancer 1994, 30A, 1912–1921. [Google Scholar] [CrossRef] [Green Version]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitomi, K.; Iwai, S.; Tainer, J.A. The intricate structural chemistry of base excision repair machinery: Implications for DNA damage recognition, removal, and repair. DNA Repair 2007, 6, 410–428. [Google Scholar] [CrossRef]
- Kanaar, R.; Hoeijmakers, J.H.; van Gent, D.C. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998, 8, 483–489. [Google Scholar] [CrossRef]
- Tsukamoto, Y.; Ikeda, H. Double-strand break repair mediated by DNA end-joining. Genes Cells 1998, 3, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Env. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Dharmalingam, P.; Vasquez, V.; Mitra, J.; Boldogh, I.; Rao, K.S.; Kent, T.A.; Mitra, S.; Hegde, M.L. Chronic oxidative damage together with genome repair deficiency in the neurons is a double whammy for neurodegeneration: Is damage response signaling a potential therapeutic target? Mech. Ageing Dev. 2017, 161, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef]
- Von Zglinicki, T.; Wan, T.; Miwa, S. Senescence in Post-Mitotic Cells: A Driver of Aging? Antioxid. Redox Signal. 2021, 34, 308–323. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Ishikawa, S.; Ishikawa, F. Proteostasis failure and cellular senescence in long-term cultured postmitotic rat neurons. Aging Cell 2020, 19, e13071. [Google Scholar] [CrossRef]
- Sah, E.; Krishnamurthy, S.; Ahmidouch, M.Y.; Gillispie, G.J.; Milligan, C.; Orr, M.E. The Cellular Senescence Stress Response in Post-Mitotic Brain Cells: Cell Survival at the Expense of Tissue Degeneration. Life 2021, 11, 229. [Google Scholar] [CrossRef] [PubMed]
- De Zio, D.; Cianfanelli, V.; Cecconi, F. New insights into the link between DNA damage and apoptosis. Antioxid. Redox Signal. 2013, 19, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Herrup, K.; Busser, J.C. The induction of multiple cell cycle events precedes target-related neuronal death. Development 1995, 121, 2385–2395. [Google Scholar] [CrossRef]
- Schwartz, E.I.; Smilenov, L.B.; Price, M.A.; Osredkar, T.; Baker, R.A.; Ghosh, S.; Shi, F.D.; Vollmer, T.L.; Lencinas, A.; Stearns, D.M.; et al. Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell Cycle 2007, 6, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Casafont, I.; Palanca, A.; Lafarga, V.; Berciano, M.T.; Lafarga, M. Effect of ionizing radiation in sensory ganglion neurons: Organization and dynamics of nuclear compartments of DNA damage/repair and their relationship with transcription and cell cycle. Acta Neuropathol. 2011, 122, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Tomashevski, A.; Webster, D.R.; Grammas, P.; Gorospe, M.; Kruman, I.I. Cyclin-C-dependent cell-cycle entry is required for activation of non-homologous end joining DNA repair in postmitotic neurons. Cell Death Differ. 2010, 17, 1189–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrup, K.; Neve, R.; Ackerman, S.L.; Copani, A. Divide and die: Cell cycle events as triggers of nerve cell death. J. Neurosci. 2004, 24, 9232–9239. [Google Scholar] [CrossRef]
- Ippati, S.; Deng, Y.; van der Hoven, J.; Heu, C.; van Hummel, A.; Chua, S.W.; Paric, E.; Chan, G.; Feiten, A.; Fath, T.; et al. Rapid initiation of cell cycle reentry processes protects neurons from amyloid-beta toxicity. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Mandal, A.; Drerup, C.M. Axonal Transport and Mitochondrial Function in Neurons. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, G.; Pachouri, U.C.; Khaidem, D.C.; Kundu, A.; Chopra, C.; Singh, P. Mitochondrial DNA Damage and Diseases. F1000Research 2015, 4, 176. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Grootjans, S.; Goossens, V.; Dondelinger, Y.; Krysko, D.V.; Takahashi, N.; Vandenabeele, P. Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods 2013, 61, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal cell death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Clarke, G.; Lumsden, C.J. Scale-free neurodegeneration: Cellular heterogeneity and the stretched exponential kinetics of cell death. J. Biol. 2005, 233, 515–525. [Google Scholar] [CrossRef]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Horvitz, H.R. Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 1999, 59, 1701s–1706s. [Google Scholar]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Hyman, B.T.; Yuan, J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat. Rev. Neurosci. 2012, 13, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Nagase, H.; Kawane, K.; Mukae, N.; Fukuyama, H. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 2003, 10, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, B.D.; Rampalli, S.; Burns, L.E.; Brunette, S.; Dilworth, F.J.; Megeney, L.A. Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proc. Natl. Acad. Sci. USA 2010, 107, 4230–4235. [Google Scholar] [CrossRef] [Green Version]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar]
- Miller, F.D.; Pozniak, C.D.; Walsh, G.S. Neuronal life and death: An essential role for the p53 family. Cell Death Differ. 2000, 7, 880–888. [Google Scholar] [CrossRef]
- Schuler, M.; Green, D.R. Transcription, apoptosis and p53: Catch-22. Trends Genet. 2005, 21, 182–187. [Google Scholar] [CrossRef]
- Fortini, P.; Dogliotti, E. Mechanisms of dealing with DNA damage in terminally differentiated cells. Mutat. Res. 2010, 685, 38–44. [Google Scholar] [CrossRef]
- Vaughn, A.E.; Deshmukh, M. Essential postmitochondrial function of p53 uncovered in DNA damage-induced apoptosis in neurons. Cell Death Differ. 2007, 14, 973–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, K.M.; Linhoff, M.W.; Potts, P.R.; Deshmukh, M. Decreased apoptosome activity with neuronal differentiation sets the threshold for strict IAP regulation of apoptosis. J. Cell Biol. 2004, 167, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by Self-Digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Son, J.H.; Shim, J.H.; Kim, K.H.; Ha, J.Y.; Han, J.Y. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 2012, 44, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Ichim, G.; Green, D.R. Die another way--non-apoptotic mechanisms of cell death. J. Cell Sci. 2014, 127, 2135–2144. [Google Scholar] [CrossRef] [Green Version]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 2019, 35, 477–500. [Google Scholar] [CrossRef]
- Hewitt, G.; Korolchuk, V.I. Repair, reuse, recycle: The expanding role of autophagy in genome maintenance. Trends Cell Biol. 2017, 27, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA damage response and autophagy: A meaningful partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [Green Version]
- Czarny, P.; Pawlowska, E.; Bialkowska-Warzecha, J.; Kaarniranta, K.; Blasiak, J. Autophagy in DNA damage response. Int. J. Mol. Sci. 2015, 16, 2641–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Orlotti, N.I.; Cimino-Reale, G.; Borghini, E.; Pennati, M.; Sissi, C.; Perrone, F.; Palumbo, M.; Daidone, M.G.; Folini, M.; Zaffaroni, N. Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated DNA damage. Autophagy 2012, 8, 1185–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galati, S.; Boni, C.; Gerra, M.C.; Lazzaretti, M.; Buschini, A. Autophagy: A player in response to oxidative stress and DNA damage. Oxid. Med. Cell Longev. 2019, 2019, 5692958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.R.; Menck, C.F.M.; Leandro, G.S. Autophagy roles in the modulation of DNA repair pathways. Int. J. Mol. Sci. 2017, 18, 2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Abrams, J. p53: The janus of autophagy? Nat. Cell Biol. 2008, 10, 637–639. [Google Scholar] [CrossRef]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, J.M.; Tanzer, M.C.; Lucet, I.S.; Young, S.N.; Spall, S.K.; Sharma, P.; Pierotti, C.; Garnier, J.M.; Dobson, R.C.; Webb, A.I.; et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 15072–15077. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricker, M.; Vilalta, A.; Tolkovsky, A.M.; Brown, G.C. Caspase inhibitors protect neurons by enabling selective necroptosis of inflamed microglia. J. Biol. Chem. 2013, 288, 9145–9152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Li, J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013, 4, e716. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Wu, W.; Li, H.; Li, S.; Huang, L.T.; Yang, Y.Q.; Sun, Q.; Wang, C.X.; Yu, Z.; Hang, C.H. Necroptosis, a novel type of programmed cell death, contributes to early neural cells damage after spinal cord injury in adult mice. J. Spinal Cord Med. 2015, 38, 745–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Qiu, J.; Liang, M.; Golinski, J.; van Leyen, K.; Jung, J.E.; You, Z.; Lo, E.H.; Degterev, A.; Whalen, M.J. Akt and mTOR mediate programmed necrosis in neurons. Cell Death Dis. 2014, 5, e1084. [Google Scholar] [CrossRef] [Green Version]
- Biton, S.; Ashkenazi, A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell 2011, 145, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 2014, 1842, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Canta, A.; Pozzi, E.; Carozzi, V.A. Mitochondrial dysfunction in chemotherapy-induced peripheral neuropathy (CIPN). Toxics 2015, 3, 198–223. [Google Scholar] [CrossRef] [Green Version]
- Dawson, T.M.; Dawson, V.L. Mitochondrial mechanisms of neuronal cell death: Potential therapeutics. Annu. Rev. Pharm. Toxicol. 2017, 57, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, S.W.; Koh, D.W.; Lew, J.; Coombs, C.; Bowers, W.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J. Neurosci. 2004, 24, 10963–10973. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef] [Green Version]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharm. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Kam, T.I.; Dawson, T.M.; Dawson, V.L. Poly (ADP-ribose) (PAR)-dependent cell death in neurodegenerative diseases. Int. Rev. Cell Mol. Biol. 2020, 353, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.A.; Kim, N.S.; Yu, S.W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kim, N.S.; Haince, J.F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef] [Green Version]
- Mashimo, M.; Kato, J.; Moss, J. ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 18964–18969. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Bacher, M.; Meinhardt, A.; Lan, H.Y.; Dhabhar, F.S.; Mu, W.; Metz, C.N.; Chesney, J.A.; Gemsa, D.; Donnelly, T.; Atkins, R.C.; et al. MIF expression in the rat brain: Implications for neuronal function. Mol. Med. 1998, 4, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Chai, X.; Zhang, W.; Li, L.; Wu, Y.; Zhu, X.; Zhao, S. Profile of MIF in developing hippocampus: Association with cell proliferation and neurite outgrowth. Front. Mol. Neurosci. 2020, 13, 147. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.H.; Tseng, W.H.; Chi, J.T. The Intersection of DNA damage response and ferroptosis-a rationale for combination therapeutics. Biology 2020, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox. Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Adachi-Tominari, K.; Sano, O.; Kamei, T.; Nogami, M.; Ogi, K.; Okano, H.; Yano, M. Involvement of ferroptosis in human motor neuron cell death. Biochem. Biophys. Res. Commun. 2021, 566, 24–29. [Google Scholar] [CrossRef]
- Poltorack, C.D.; Dixon, S.J. Understanding the role of cysteine in ferroptosis: Progress & paradoxes. FEBS J. 2021. [Google Scholar] [CrossRef]
- Cozzi, A.; Orellana, D.I.; Santambrogio, P.; Rubio, A.; Cancellieri, C.; Giannelli, S.; Ripamonti, M.; Taverna, S.; Di Lullo, G.; Rovida, E.; et al. Stem cell modeling of neuroferritinopathy reveals iron as a determinant of senescence and ferroptosis during neuronal aging. Stem Cell Rep. 2019, 13, 832–846. [Google Scholar] [CrossRef] [Green Version]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Chen, H.; Xu, H.; Wu, Y.; Wu, C.; Jia, C.; Li, Y.; Sheng, S.; Xu, C.; Xu, H.; et al. Role of pyroptosis in traumatic brain and spinal cord injuries. Int. J. Biol. Sci. 2020, 16, 2042–2050. [Google Scholar] [CrossRef]
- Lu, F.; Lan, Z.; Xin, Z.; He, C.; Guo, Z.; Xia, X.; Hu, T. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. J. Cell Physiol. 2020, 235, 3207–3221. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C.R.; Frost, E.L.; Bellinger, C.E.; Bolte, A.C.; McKee, C.A.; Hurt, M.E.; Paysour, M.J.; Ennerfelt, H.E.; Lukens, J.R. AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature 2020, 580, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Aits, S.; Jaattela, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogarty, M.P.; McCormack, R.M.; Noonan, J.; Murphy, D.; Gowran, A.; Campbell, V.A. A role for p53 in the beta-amyloid-mediated regulation of the lysosomal system. Neurobiol. Aging 2010, 31, 1774–1786. [Google Scholar] [CrossRef] [PubMed]
- Gowran, A.; Campbell, V.A. A role for p53 in the regulation of lysosomal permeability by delta 9-tetrahydrocannabinol in rat cortical neurones: Implications for neurodegeneration. J. Neurochem. 2008, 105, 1513–1524. [Google Scholar] [CrossRef]
- Windelborn, J.A.; Lipton, P. Lysosomal release of cathepsins causes ischemic damage in the rat hippocampal slice and depends on NMDA-mediated calcium influx, arachidonic acid metabolism, and free radical production. J. Neurochem. 2008, 106, 56–69. [Google Scholar] [CrossRef]
- Bove, J.; Martinez-Vicente, M.; Dehay, B.; Perier, C.; Recasens, A.; Bombrun, A.; Antonsson, B.; Vila, M. BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy 2014, 10, 889–900. [Google Scholar] [CrossRef] [Green Version]
- Villalpando Rodriguez, G.E.; Torriglia, A. Calpain 1 induce lysosomal permeabilization by cleavage of lysosomal associated membrane protein 2. Biochim. Biophys. Acta 2013, 1833, 2244–2253. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.J.; Zhang, X.D.; Sun, W.; Qi, L.; Wu, J.C.; Qin, Z.H. DRAM1 regulates apoptosis through increasing protein levels and lysosomal localization of BAX. Cell Death Dis. 2015, 6, e1624. [Google Scholar] [CrossRef]
- Darios, F.; Stevanin, G. Impairment of lysosome function and autophagy in rare neurodegenerative diseases. J. Mol. Biol. 2020, 432, 2714–2734. [Google Scholar] [CrossRef]
- Vaquer-Alicea, J.; Diamond, M.I. Propagation of protein aggregation in neurodegenerative diseases. Annu. Rev. Biochem. 2019, 88, 785–810. [Google Scholar] [CrossRef] [PubMed]
- Sepe, S.; Milanese, C.; Gabriels, S.; Derks, K.W.; Payan-Gomez, C.; van Ijcken, J.W.F.; Rijksen, Y.M.; Nigg, A.L.; Moreno, S.; Cerri, S.; et al. Inefficient DNA repair is an aging-related modifier of Parkinson’s disease. Cell Rep. 2016, 15, 1866–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiwaku, H.; Okazawa, H. Impaired DNA damage repair as a common feature of neurodegenerative diseases and psychiatric disorders. Curr. Mol. Med. 2015, 15, 119–128. [Google Scholar] [CrossRef]
- Abugable, A.A.; Morris, J.L.M.; Palminha, N.M.; Zaksauskaite, R.; Ray, S.; El-Khamisy, S.F. DNA repair and neurological disease: From molecular understanding to the development of diagnostics and model organisms. DNA Repair 2019, 81, 102669. [Google Scholar] [CrossRef]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Ageing as a primary risk factor for Parkinson’s disease: Evidence from studies of non-human primates. Nat. Rev. Neurosci. 2011, 12, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Milanese, C.; Cerri, S.; Ulusoy, A.; Gornati, S.V.; Plat, A.; Gabriels, S.; Blandini, F.; Di Monte, D.A.; Hoeijmakers, J.H.; Mastroberardino, P.G. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis. 2018, 9, 818. [Google Scholar] [CrossRef] [Green Version]
- Coppedè, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2015, 776, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Kok, J.R.; Palminha, N.M.; Dos Santos Souza, C.; El-Khamisy, S.F.; Ferraiuolo, L. DNA damage as a mechanism of neurodegeneration in ALS and a contributor to astrocyte toxicity. Cell. Mol. Life Sci. 2021, 78, 5707–5729. [Google Scholar] [CrossRef]
- Shadfar, S.; Hwang, C.J.; Lim, M.-S.; Choi, D.-Y.; Hong, J.T. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch. Pharmacal. Res. 2015, 38, 2106–2119. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- d’Errico, P.; Meyer-Luehmann, M. Mechanisms of Pathogenic Tau and Aβ protein spreading in Alzheimer’s disease. Front. Aging Neurosci. 2020, 12, 265. [Google Scholar] [CrossRef]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef]
- Tian, J.; Guo, L.; Sui, S.; Driskill, C.; Phensy, A.; Wang, Q.; Gauba, E.; Zigman, J.M.; Swerdlow, R.H.; Kroener, S.; et al. Disrupted hippocampal growth hormone secretagogue receptor 1α interaction with dopamine receptor D1 plays a role in Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaav6278. [Google Scholar] [CrossRef]
- Lin, X.; Kapoor, A.; Gu, Y.; Chow, M.J.; Peng, J.; Zhao, K.; Tang, D. Contributions of DNA damage to Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 1666. [Google Scholar] [CrossRef] [Green Version]
- Guennewig, B.; Lim, J.; Marshall, L.; McCorkindale, A.N.; Paasila, P.J.; Patrick, E.; Kril, J.J.; Halliday, G.M.; Cooper, A.A.; Sutherland, G.T. Defining early changes in Alzheimer’s disease from RNA sequencing of brain regions differentially affected by pathology. Sci. Rep. 2021, 11, 4865. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharm. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS. J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [Green Version]
- Martínez, G.; Duran-Aniotz, C.; Cabral-Miranda, F.; Vivar, J.P.; Hetz, C. Endoplasmic reticulum proteostasis impairment in aging. Aging Cell 2017, 16, 615–623. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Dang, S.; Wu, S.; Wang, J.; Li, H.; Huang, M.; He, W.; Li, Y.M.; Wong, C.C.; Shi, Y. Cleavage of amyloid precursor protein by an archaeal presenilin homologue PSH. Proc. Natl. Acad. Sci. USA 2015, 112, 3344–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welikovitch, L.A.; Do Carmo, S.; Maglóczky, Z.; Szocsics, P.; Lőke, J.; Freund, T.; Cuello, A.C. Evidence of intraneuronal Aβ accumulation preceding tau pathology in the entorhinal cortex. Acta Neuropathol. 2018, 136, 901–917. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanbhag, N.M.; Evans, M.D.; Mao, W.; Nana, A.L.; Seeley, W.W.; Adame, A.; Rissman, R.A.; Masliah, E.; Mucke, L. Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, P.-C.; Patnaik, D.; Watson, L.A.; Gao, F.; Pan, L.; Wang, J.; Adaikkan, C.; Penney, J.; Cam, H.P.; Huang, W.-C.; et al. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease. Nat. Commun. 2020, 11, 2484. [Google Scholar] [CrossRef]
- Jacobsen, E.; Beach, T.; Shen, Y.; Li, R.; Chang, Y. Deficiency of the Mre11 DNA repair complex in Alzheimer’s disease brains. Mol. Brain Res. 2004, 128, 1–7. [Google Scholar] [CrossRef]
- Shackelford, D.A. DNA end joining activity is reduced in Alzheimer’s disease. Neurobiol. Aging 2006, 27, 596–605. [Google Scholar] [CrossRef]
- Dobbin, M.M.; Madabhushi, R.; Pan, L.; Chen, Y.; Kim, D.; Gao, J.; Ahanonu, B.; Pao, P.-C.; Qiu, Y.; Zhao, Y.; et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat. Neurosci 2013, 16, 1008–1015. [Google Scholar] [CrossRef]
- Cruz, J.C.; Tseng, H.C.; Goldman, J.A.; Shih, H.; Tsai, L.H. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 2003, 40, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Misiak, M.; Vergara Greeno, R.; Baptiste, B.A.; Sykora, P.; Liu, D.; Cordonnier, S.; Fang, E.F.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. DNA polymerase β decrement triggers death of olfactory bulb cells and impairs olfaction in a mouse model of Alzheimer’s disease. Aging Cell 2017, 16, 162–172. [Google Scholar] [CrossRef]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Bradley-Whitman, M.A.; Timmons, M.D.; Beckett, T.L.; Murphy, M.P.; Lynn, B.C.; Lovell, M.A. Nucleic acid oxidation: An early feature of Alzheimer’s disease. J. Neurochem. 2014, 128, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Song, H.; Croteau, D.L.; Akbari, M.; Bohr, V.A. Genome instability in Alzheimer disease. Mech. Ageing Dev. 2017, 161, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-H.; Siddoway, B.; Kaeser, G.E.; Segota, I.; Rivera, R.; Romanow, W.J.; Liu, C.S.; Park, C.; Kennedy, G.; Long, T.; et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 2018, 563, 639–645. [Google Scholar] [CrossRef]
- Anderson, A.J.; Su, J.H.; Cotman, C.W. DNA damage and apoptosis in Alzheimer’s disease: Colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J. Neurosci. 1996, 16, 1710–1719. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Nguyen, T.V.; Pike, C.J. Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci. 2005, 25, 1149–1158. [Google Scholar] [CrossRef]
- Forloni, G.; Chiesa, R.; Smiroldo, S.; Verga, L.; Salmona, M.; Tagliavini, F.; Angeretti, N. Apoptosis mediated neurotoxicity induced by chronic application of beta amyloid fragment 25-35. Neuroreport 1993, 4, 523–526. [Google Scholar] [CrossRef]
- Das, H.; Sarkar, S.; Paidi, R.K.; Biswas, S.C. Subtle genomic DNA damage induces intraneuronal production of amyloid-β (1-42) by increasing β-secretase activity. Faseb. J. 2021, 35, e21569. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Res. 2000, 855, 116–123. [Google Scholar] [CrossRef]
- Weissman, L.; Jo, D.G.; Sørensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007, 35, 5545–5555. [Google Scholar] [CrossRef]
- Sugo, N.; Aratani, Y.; Nagashima, Y.; Kubota, Y.; Koyama, H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J. 2000, 19, 1397–1404. [Google Scholar] [CrossRef] [Green Version]
- Canugovi, C.; Shamanna, R.A.; Croteau, D.L.; Bohr, V.A. Base excision DNA repair levels in mitochondrial lysates of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1293–1300. [Google Scholar] [CrossRef] [Green Version]
- Mao, G.; Pan, X.; Zhu, B.B.; Zhang, Y.; Yuan, F.; Huang, J.; Lovell, M.A.; Lee, M.P.; Markesbery, W.R.; Li, G.M.; et al. Identification and characterization of OGG1 mutations in patients with Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 2759–2766. [Google Scholar] [CrossRef] [Green Version]
- Demars, M.; Hu, Y.-S.; Gadadhar, A.; Lazarov, O. Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J. Neurosci. Res. 2010, 88, 2103–2117. [Google Scholar] [CrossRef] [Green Version]
- Kieroń, M.; Żekanowski, C.; Falk, A.; Wężyk, M. Oxidative DNA Damage Signalling in Neural Stem Cells in Alzheimer’s Disease. Oxid. Med. Cell Longev. 2019, 2019, 2149812. [Google Scholar] [CrossRef] [Green Version]
- Okun, E.; Marton, D.; Cohen, D.; Griffioen, K.; Kanfi, Y.; Illouz, T.; Madar, R.; Cohen, H.Y. Sirt6 alters adult hippocampal neurogenesis. PLoS ONE 2017, 12, e0179681. [Google Scholar] [CrossRef] [Green Version]
- Kaluski, S.; Portillo, M.; Besnard, A.; Stein, D.; Einav, M.; Zhong, L.; Ueberham, U.; Arendt, T.; Mostoslavsky, R.; Sahay, A.; et al. Neuroprotective Functions for the Histone Deacetylase SIRT6. Cell Rep. 2017, 18, 3052–3062. [Google Scholar] [CrossRef] [Green Version]
- López-Arribillaga, E.; Rodilla, V.; Pellegrinet, L.; Guiu, J.; Iglesias, M.; Roman, A.C.; Gutarra, S.; González, S.; Muñoz-Cánoves, P.; Fernández-Salguero, P.; et al. Bmi1 regulates murine intestinal stem cell proliferation and self-renewal downstream of Notch. Development 2015, 142, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.D.; Jones, P.A. Tissue-specific alternative splicing in the human INK4a/ARF cell cycle regulatory locus. Oncogene 1999, 18, 3810–3820. [Google Scholar] [CrossRef] [Green Version]
- Ismail, I.H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010, 191, 45–60. [Google Scholar] [CrossRef]
- Lin, X.; Ojo, D.; Wei, F.; Wong, N.; Gu, Y.; Tang, D. A Novel Aspect of Tumorigenesis-BMI1 Functions in Regulating DNA Damage Response. Biomolecules 2015, 5, 3396–3415. [Google Scholar] [CrossRef] [Green Version]
- El Hajjar, J.; Chatoo, W.; Hanna, R.; Nkanza, P.; Tétreault, N.; Tse, Y.C.; Wong, T.P.; Abdouh, M.; Bernier, G. Heterochromatic genome instability and neurodegeneration sharing similarities with Alzheimer’s disease in old Bmi1+/− mice. Sci Rep. 2019, 9, 594. [Google Scholar] [CrossRef]
- Flamier, A.; El Hajjar, J.; Adjaye, J.; Fernandes, K.J.; Abdouh, M.; Bernier, G. Modeling Late-Onset Sporadic Alzheimer’s Disease through BMI1 Deficiency. Cell Rep. 2018, 23, 2653–2666. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, T.; Baer, R.; Gautier, J. DNA double-strand break repair pathway choice and cancer. DNA Repair 2014, 19, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Wezyk, M.; Szybinska, A.; Wojsiat, J.; Szczerba, M.; Day, K.; Ronnholm, H.; Kele, M.; Berdynski, M.; Peplonska, B.; Fichna, J.P.; et al. Overactive BRCA1 affects presenilin 1 in induced pluripotent stem cell-derived neurons in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 175–202. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Do TUNEL and other apoptosis assays detect cell death in preclinical studies? Int. J. Mol. Sci. 2020, 21, 9090. [Google Scholar] [CrossRef]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Strosznajder, J.B.; Jeśko, H.; Strosznajder, R.P. Effect of amyloid beta peptide on poly(ADP-ribose) polymerase activity in adult and aged rat hippocampus. Acta Biochim. Pol. 2000, 47, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Love, S.; Barber, R.; Wilcock, G.K. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain 1999, 122, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Strosznajder, J.B.; Czapski, G.A.; Adamczyk, A.; Strosznajder, R.P. Poly(ADP-ribose) polymerase-1 in amyloid beta toxicity and Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 78–84. [Google Scholar] [CrossRef]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Libien, J.; Shaik, F.; Wolk, J.; Hernández, A.I. Nucleolar PARP-1 expression is decreased in Alzheimer’s disease: Consequences for epigenetic regulation of rDNA and cognition. Neural. Plast. 2016, 2016, 8987928. [Google Scholar] [CrossRef]
- Wu, M.F.; Yin, J.H.; Hwang, C.S.; Tang, C.M.; Yang, D.I. NAD attenuates oxidative DNA damages induced by amyloid beta-peptide in primary rat cortical neurons. Free Radic. Res. 2014, 48, 794–805. [Google Scholar] [CrossRef]
- Narne, P.; Pandey, V.; Simhadri, P.K.; Phanithi, P.B. Poly(ADP-ribose)polymerase-1 hyperactivation in neurodegenerative diseases: The death knell tolls for neurons. Semin. Cell Dev. Biol. 2017, 63, 154–166. [Google Scholar] [CrossRef]
- Martire, S.; Mosca, L.; d’Erme, M. PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev. 2015, 146-148, 53–64. [Google Scholar] [CrossRef]
- Nho, K.; Corneveaux, J.J.; Kim, S.; Lin, H.; Risacher, S.L.; Shen, L.; Swaminathan, S.; Ramanan, V.K.; Liu, Y.; Foroud, T.; et al. Whole-exome sequencing and imaging genetics identify functional variants for rate of change in hippocampal volume in mild cognitive impairment. Mol. Psychiatry 2013, 18, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Regier, M.; Liang, J.; Choi, A.; Verma, K.; Libien, J.; Hernández, A.I. Evidence for decreased nucleolar PARP-1 as an early marker of cognitive impairment. Neural. Plast. 2019, 2019, 4383258. [Google Scholar] [CrossRef] [Green Version]
- Nair, V.D.; McNaught, K.S.; González-Maeso, J.; Sealfon, S.C.; Olanow, C.W. p53 mediates nontranscriptional cell death in dopaminergic cells in response to proteasome inhibition. J. Biol. Chem. 2006, 281, 39550–39560. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, Y.; Shimohama, S.; Kamoshima, W.; Matsuoka, Y.; Nomura, Y.; Taniguchi, T. Changes of p53 in the brains of patients with Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1997, 232, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Shin, C.; Kim, M.J.; Lee, B.; Park, K.H.; Cha, C.I. Immunocytochemical study on the distribution of p53 in the hippocampus and cerebellum of the aged rat. Brain Res. 2000, 885, 137–141. [Google Scholar] [CrossRef]
- Davenport, C.M.; Sevastou, I.G.; Hooper, C.; Pocock, J.M. Inhibiting p53 pathways in microglia attenuates microglial-evoked neurotoxicity following exposure to Alzheimer peptides. J. Neurochem. 2010, 112, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Farmer, K.M.; Ghag, G.; Puangmalai, N.; Montalbano, M.; Bhatt, N.; Kayed, R. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 132. [Google Scholar] [CrossRef]
- Zhang, Y.; McLaughlin, R.; Goodyer, C.; LeBlanc, A. Selective cytotoxicity of intracellular amyloid beta peptide1-42 through p53 and Bax in cultured primary human neurons. J. Cell Biol. 2002, 156, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Shadfar, S.; Kim, Y.-G.; Katila, N.; Neupane, S.; Ojha, U.; Bhurtel, S.; Srivastav, S.; Jeong, G.-S.; Park, P.-H.; Hong, J.T.; et al. Neuroprotective effects of antidepressants via upregulation of neurotrophic factors in the MPTP model of Parkinson’s disease. Mol. Neurobiol. 2018, 55, 554–566. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s disease: Dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef]
- DeMaagd, G.; Philip, A. Parkinson’s disease and its management: Part 1: Disease Entity, Risk Factors, Pathophysiology, Clinical Presentation, and Diagnosis. Pharm. Ther. 2015, 40, 504–532. [Google Scholar]
- Katila, N.; Bhurtel, S.; Park, P.H.; Choi, D.Y. Metformin attenuates rotenone-induced oxidative stress and mitochondrial damage via the AKT/Nrf2 pathway. Neurochem. Int. 2021, 148, 105120. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.Y.; Katila, N.; Neupane, S.; Shadfar, S.; Ojha, U.; Bhurtel, S.; Srivastav, S.; Son, D.J.; Park, P.-H.; Yoon, D.Y.; et al. Enhanced dopaminergic neurotoxicity mediated by MPTP in IL-32β transgenic mice. Neurochem. Int. 2017, 102, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting mitochondrial impairment in Parkinson’s disease: Challenges and opportunities. Front. Cell Dev. Biol. 2021, 8, 1704. [Google Scholar] [CrossRef]
- Neupane, S.; Srivastav, S.; Bhurtel, S.; Katila, N.; Shadfar, S.; Park, P.H.; Hong, J.T.; Choi, D.Y. Enhanced neuroinflammatory responses after systemic LPS injection in IL-32β transgenic mice. J. Chem. Neuroanat. 2018, 94, 173–182. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Power, J.H.; Barnes, O.L.; Chegini, F. Lewy bodies and the mechanisms of neuronal cell death in Parkinson’s Disease and dementia with lewy bodies. Brain Pathol. 2017, 27, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Fu, Y.; Halliday, G.M.; Sue, C.M. PARK genes link mitochondrial dysfunction and alpha-synuclein pathology in sporadic Parkinson’s disease. Front. Cell Dev. Biol. 2021, 9, 612476. [Google Scholar] [CrossRef]
- Gonzalez-Hunt, C.P.; Sanders, L.H. DNA damage and repair in Parkinson’s disease: Recent advances and new opportunities. J. Neurosci. Res. 2021, 99, 180–189. [Google Scholar] [CrossRef]
- Martín-Jiménez, R.; Lurette, O.; Hebert-Chatelain, E. Damage in mitochondrial DNA associated with Parkinson’s disease. DNA Cell Biol. 2020, 39, 1421–1430. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Yu, T.; Liu, Y.; Yan, J.; Guo, Y.; Jing, Y.; Yang, X.; Song, Y.; Tian, Y. DNA damage preceding dopamine neuron degeneration in A53T human α-synuclein transgenic mice. Biochem. Biophys. Res. Commun. 2016, 481, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P.; Olanow, C.W. Understanding cell death in Parkinson’s disease. Ann. Neurol. 1998, 44, S72–S84. [Google Scholar] [CrossRef] [PubMed]
- Danielson, S.R.; Andersen, J.K. Oxidative and nitrative protein modifications in Parkinson’s disease. Free Radic. Biol. Med. 2008, 44, 1787–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weil, R.S.; Lashley, T.L.; Bras, J.; Schrag, A.E.; Schott, J.M. Current concepts and controversies in the pathogenesis of Parkinson’s disease dementia and Dementia with Lewy Bodies. F1000Research 2017, 6, 1604. [Google Scholar] [CrossRef] [Green Version]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Gary, D.S.; Mattson, M.P. Integrin signaling via the PI3-kinase-Akt pathway increases neuronal resistance to glutamate-induced apoptosis. J. Neurochem. 2001, 76, 1485–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [Green Version]
- Béraud, D.; Twomey, M.; Bloom, B.; Mittereder, A.; Ton, V.; Neitzke, K.; Chasovskikh, S.; Mhyre, T.R.; Maguire-Zeiss, K.A. α-Synuclein Alters Toll-Like Receptor Expression. Front. Neurosci. 2011, 5, 80. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.K. Does neuronal loss in Parkinson’s disease involve programmed cell death? Bioessays 2001, 23, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Hunot, S.; Michel, P.P.; Muriel, M.P.; Vyas, S.; Faucheux, B.A.; Mouatt-Prigent, A.; Turmel, H.; Srinivasan, A.; Ruberg, M.; et al. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2875–2880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanath, V.; Wu, Y.; Boonplueang, R.; Chen, S.; Stevenson, F.F.; Yantiri, F.; Yang, L.; Beal, M.F.; Andersen, J.K. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease. J. Neurosci. 2001, 21, 9519–9528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatton, W.G.; Chalmers-Redman, R.M.; Elstner, M.; Leesch, W.; Jagodzinski, F.B.; Stupak, D.P.; Sugrue, M.M.; Tatton, N.A. Glyceraldehyde-3-phosphate dehydrogenase in neurodegeneration and apoptosis signaling. J. Neural. Transm. Suppl. 2000, 77–100. [Google Scholar] [CrossRef]
- Tarze, A.; Deniaud, A.; Le Bras, M.; Maillier, E.; Molle, D.; Larochette, N.; Zamzami, N.; Jan, G.; Kroemer, G.; Brenner, C. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene 2007, 26, 2606–2620. [Google Scholar] [CrossRef] [Green Version]
- El-Agnaf, O.M.; Jakes, R.; Curran, M.D.; Middleton, D.; Ingenito, R.; Bianchi, E.; Pessi, A.; Neill, D.; Wallace, A. Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments. FEBS Lett. 1998, 440, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 10919. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Rong, Z.; Tu, P.; Xu, P.; Sun, Y.; Yu, F.; Tu, N.; Guo, L.; Yang, Y. The Mitochondrial Response to DNA Damage. Front. Cell Dev. Biol. 2021, 9, 669379. [Google Scholar] [CrossRef]
- Dagda, R.K.; Cherra, S.J., 3rd; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H.; Parks, J.K.; Miller, S.W.; Tuttle, J.B.; Trimmer, P.A.; Sheehan, J.P.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann. Neurol. 1996, 40, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Tatton, N.A.; Kish, S.J. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange staining. Neuroscience 1997, 77, 1037–1048. [Google Scholar] [CrossRef]
- Zhang, J.; Pieper, A.; Snyder, S.H. Poly(ADP-ribose) synthetase activation: An early indicator of neurotoxic DNA damage. J. Neurochem. 1995, 65, 1411–1414. [Google Scholar] [CrossRef]
- Mandir, A.S.; Przedborski, S.; Jackson-Lewis, V.; Wang, Z.Q.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Hoffman, B.E.; Guastella, D.B.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc. Natl. Acad. Sci. USA 1999, 96, 5774–5779. [Google Scholar] [CrossRef] [Green Version]
- Cosi, C.; Marien, M. Implication of poly (ADP-ribose) polymerase (PARP) in neurodegeneration and brain energy metabolism. Decreases in mouse brain NAD+ and ATP caused by MPTP are prevented by the PARP inhibitor benzamide. Ann. N. Y. Acad. Sci. 1999, 890, 227–239. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Grammatopoulos, T.N.; Altmann, S.; Amore, A.; Standaert, D.G.; Hyman, B.T.; Kazantsev, A.G. Pharmacological inhibition of PARP-1 reduces alpha-synuclein- and MPP+-induced cytotoxicity in Parkinson’s disease in vitro models. Biochem. Biophys. Res. Commun. 2007, 357, 596–602. [Google Scholar] [CrossRef]
- Cosi, C.; Colpaert, F.; Koek, W.; Degryse, A.; Marien, M. Poly(ADP-ribose) polymerase inhibitors protect against MPTP-induced depletions of striatal dopamine and cortical noradrenaline in C57B1/6 mice. Brain Res. 1996, 729, 264–269. [Google Scholar] [CrossRef]
- Lee, Y.; Karuppagounder, S.S.; Shin, J.H.; Lee, Y.I.; Ko, H.S.; Swing, D.; Jiang, H.; Kang, S.U.; Lee, B.D.; Kang, H.C.; et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci. 2013, 16, 1392–1400. [Google Scholar] [CrossRef]
- Ham, S.; Yun, S.P.; Kim, H.; Kim, D.; Seo, B.A.; Kim, H.; Shin, J.Y.; Dar, M.A.; Lee, G.H.; Lee, Y.I.; et al. Amyloid-like oligomerization of AIMP2 contributes to α-synuclein interaction and Lewy-like inclusion. Sci. Transl. Med. 2020, 12, eaax0091. [Google Scholar] [CrossRef] [PubMed]
- Sidorkina, O.; Espey, M.G.; Miranda, K.M.; Wink, D.A.; Laval, J. Inhibition of poly(ADP-RIBOSE) polymerase (PARP) by nitric oxide and reactive nitrogen oxide species. Free Radic. Biol. Med. 2003, 35, 1431–1438. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Hare, D.J.; Duce, J.A.; George, J.L.; Adlard, P.A.; McLean, C.; Rogers, J.T.; Cherny, R.A.; Finkelstein, D.I.; et al. Parkinson’s disease iron deposition caused by nitric oxide-induced loss of β-amyloid precursor protein. J. Neurosci. 2015, 35, 3591–3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunot, S.; Boissière, F.; Faucheux, B.; Brugg, B.; Mouatt-Prigent, A.; Agid, Y.; Hirsch, E.C. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 1996, 72, 355–363. [Google Scholar] [CrossRef]
- Mogi, M.; Kondo, T.; Mizuno, Y.; Nagatsu, T. p53 protein, interferon-gamma, and NF-kappaB levels are elevated in the parkinsonian brain. Neurosci. Lett. 2007, 414, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, D.C.; Choi, B.H.; Ha, H.; Kim, K.T. Regulation of p53 by activated protein kinase C-delta during nitric oxide-induced dopaminergic cell death. J. Biol. Chem. 2006, 281, 2215–2224. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, C.A.; Sunyach, C.; Giaime, E.; West, A.; Corti, O.; Brice, A.; Safe, S.; Abou-Sleiman, P.M.; Wood, N.W.; Takahashi, H.; et al. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nat. Cell Biol. 2009, 11, 1370–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, A.I.; Garrison, S.P.; Zambetti, G.P.; O’Malley, K.L. 6-OHDA generated ROS induces DNA damage and p53- and PUMA-dependent cell death. Mol. Neurodegener. 2011, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Yee, K.S.; Wilkinson, S.; James, J.; Ryan, K.M.; Vousden, K.H. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2009, 16, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.R.; Ghafouri, M.; Mukerjee, R.; Bagashev, A.; Chabrashvili, T.; Sawaya, B.E. Role of p53 in neurodegenerative diseases. Neurodegener Dis. 2012, 9, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor neuron susceptibility in ALS/FTD. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef] [Green Version]
- Medinas, D.B.; Rozas, P.; Martínez Traub, F.; Woehlbier, U.; Brown, R.H.; Bosco, D.A.; Hetz, C. Endoplasmic reticulum stress leads to accumulation of wild-type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 8209–8214. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.; Jiao, Y.; Ferrando, L.M.; Yablonska, S.; Li, F.; Horoszko, E.C.; Lacomis, D.; Friedlander, R.M.; Carlisle, D.L. Neuronal mitochondrial dysfunction in sporadic amyotrophic lateral sclerosis is developmentally regulated. Sci. Rep. 2021, 11, 18916. [Google Scholar] [CrossRef]
- Hemerková, P.; Vališ, M. Role of oxidative stress in the pathogenesis of amyotrophic lateral sclerosis: Antioxidant metalloenzymes and therapeutic strategies. Biomolecules 2021, 11, 437. [Google Scholar] [CrossRef]
- Burk, K.; Pasterkamp, R.J. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 2019, 137, 859–877. [Google Scholar] [CrossRef] [Green Version]
- Verheijen, B.M.; Morimoto, S.; Sasaki, R.; Oyanagi, K.; Kokubo, Y.; Kuzuhara, S.; van Leeuwen, F.W. Expression of mutant ubiquitin and proteostasis impairment in kii amyotrophic lateral sclerosis/parkinsonism-dementia complex brains. J. Neuropathol. Exp. Neurol. 2020, 79, 902–907. [Google Scholar] [CrossRef]
- King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.; Vickers, J.C. Excitotoxicity in ALS: Overstimulation, or overreaction? Exp. Neurol. 2016, 275, 162–171. [Google Scholar] [CrossRef]
- Konopka, A.; Whelan, D.R.; Jamali, M.S.; Perri, E.; Shahheydari, H.; Toth, R.P.; Parakh, S.; Robinson, T.; Cheong, A.; Mehta, P.; et al. Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol. Neurodegener. 2020, 15, 51. [Google Scholar] [CrossRef]
- Tank, E.M.; Figueroa-Romero, C.; Hinder, L.M.; Bedi, K.; Archbold, H.C.; Li, X.; Weskamp, K.; Safren, N.; Paez-Colasante, X.; Pacut, C.; et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 2018, 9, 2845. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Marrone, L.; Drexler, H.C.A.; Wang, J.; Tripathi, P.; Distler, T.; Heisterkamp, P.; Anderson, E.N.; Kour, S.; Moraiti, A.; Maharana, S.; et al. FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol. 2019, 138, 67–84. [Google Scholar] [CrossRef] [Green Version]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Andrade, N.S.; Ramic, M.; Esanov, R.; Liu, W.; Rybin, M.J.; Gaidosh, G.; Abdallah, A.; Del’Olio, S.; Huff, T.C.; Chee, N.T.; et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 2020, 15, 13. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Curle, A.J.; Haider, A.M.; Balmus, G. The role of DNA damage response in amyotrophic lateral sclerosis. Essays Biochem. 2020, 64, 847–861. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, E.N.; Mitra, J.; Wang, H.; Rangaswamy, S.; Hegde, P.M.; Basu, P.; Rao, K.S.; Hegde, M.L. Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Hum. Mol. Genet. 2019, 28, 2459–2476. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, S.; Oztürk, A.; Hicks, G.G. FUS-regulated RNA metabolism and DNA damage repair: Implications for amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis. Rare Dis. 2014, 2, e29515. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef]
- Deng, Q.; Holler, C.J.; Taylor, G.; Hudson, K.F.; Watkins, W.; Gearing, M.; Ito, D.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T.; et al. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, M.; Toth, R.; Vandermoere, F.; Morrice, N.A.; Rouse, J. Identification and characterization of FUS/TLS as a new target of ATM. Biochem. J. 2008, 415, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Konopka, A.; Atkin, J.D. The Emerging Role of DNA Damage in the pathogenesis of the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2018, 19, 3137. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.; Herranz-Martin, S.; Karyka, E.; Liao, C.; Lewis, K.; Elsayed, W.; Lukashchuk, V.; Chiang, S.C.; Ray, S.; Mulcahy, P.J.; et al. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat. Neurosci. 2017, 20, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Konopka, A.; Soo, K.Y.; Ito, D.; Atkin, J.D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, 2882–2896. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA Damage in iPSC-derived motor neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Kisby, G.E.; Milne, J.; Sweatt, C. Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. Neuroreport 1997, 8, 1337–1340. [Google Scholar] [CrossRef]
- Kikuchi, H.; Furuta, A.; Nishioka, K.; Suzuki, S.O.; Nakabeppu, Y.; Iwaki, T. Impairment of mitochondrial DNA repair enzymes against accumulation of 8-oxo-guanine in the spinal motor neurons of amyotrophic lateral sclerosis. Acta Neuropathol. 2002, 103, 408–414. [Google Scholar] [CrossRef]
- Shaikh, A.Y.; Martin, L.J. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromolecular. Med. 2002, 2, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olkowski, Z.L. Mutant AP endonuclease in patients with amyotrophic lateral sclerosis. Neuroreport 1998, 9, 239–242. [Google Scholar] [CrossRef]
- Penndorf, D.; Tadić, V.; Witte, O.W.; Grosskreutz, J.; Kretz, A. DNA strand breaks and TDP-43 mislocation are absent in the murine hSOD1G93A model of amyotrophic lateral sclerosis in vivo and in vitro. PLoS ONE 2017, 12, e0183684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higelin, J.; Catanese, A.; Semelink-Sedlacek, L.L.; Oeztuerk, S.; Lutz, A.K.; Bausinger, J.; Barbi, G.; Speit, G.; Andersen, P.M.; Ludolph, A.C.; et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 2018, 30, 150–162. [Google Scholar] [CrossRef]
- Watanabe, Y.; Nakagawa, T.; Akiyama, T.; Nakagawa, M.; Suzuki, N.; Warita, H.; Aoki, M.; Nakayama, K. An Amyotrophic Lateral Sclerosis-associated mutant of C21ORF2 Is stabilized by NEK1-mediated Hyperphosphorylation and the inability to bind FBXO3. iScience 2020, 23, 101491. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.F.; Riley, D.J.; Chen, P.L. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 2011, 10, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, P.L.; Chen, C.F.; Jiang, X.; Riley, D.J. Never-in-mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 2008, 7, 3194–3201. [Google Scholar] [CrossRef]
- Chen, L.; Na, R.; Danae McLane, K.; Thompson, C.S.; Gao, J.; Wang, X.; Ran, Q. Overexpression of ferroptosis defense enzyme Gpx4 retards motor neuron disease of SOD1G93A mice. Sci. Rep. 2021, 11, 12890. [Google Scholar] [CrossRef]
- Morrice, J.R.; Gregory-Evans, C.Y.; Shaw, C.A. Necroptosis in amyotrophic lateral sclerosis and other neurological disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 347–353. [Google Scholar] [CrossRef]
- Murakami, T.; Nagai, M.; Miyazaki, K.; Morimoto, N.; Ohta, Y.; Kurata, T.; Takehisa, Y.; Kamiya, T.; Abe, K. Early decrease of mitochondrial DNA repair enzymes in spinal motor neurons of presymptomatic transgenic mice carrying a mutant SOD1 gene. Brain Res. 2007, 1150, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Liu, Z.; Chen, K.; Price, A.C.; Pan, Y.; Swaby, J.A.; Golden, W.C. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: Mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 2007, 500, 20–46. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F. An overview of DNA repair in amyotrophic lateral sclerosis. Sci. World J. 2011, 11, 1679–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Waard, M.C.; van der Pluijm, I.; Zuiderveen Borgesius, N.; Comley, L.H.; Haasdijk, E.D.; Rijksen, Y.; Ridwan, Y.; Zondag, G.; Hoeijmakers, J.H.; Elgersma, Y.; et al. Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathol. 2010, 120, 461–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.K.; Spiller, K.J.; Ge, G.; Zheng, A.; Xu, Y.; Zhou, M.; Tripathy, K.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015, 130, 643–660. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Lee, K.; Matsuoka, M. TDP-43-induced death is associated with altered regulation of BIM and Bcl-xL and attenuated by caspase-mediated TDP-43 cleavage. J. Biol. Chem. 2011, 286, 13171–13183. [Google Scholar] [CrossRef] [Green Version]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Investig. 2011, 121, 726–738. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.D.; Zhu, M.W.; Shan, D.; Wang, S.Y.; Yin, X.; Yang, Y.Q.; Wang, T.H.; Zhang, C.T.; Wang, Y.; Liang, W.W.; et al. Spy1, a unique cell cycle regulator, alters viability in ALS motor neurons and cell lines in response to mutant SOD1-induced DNA damage. DNA Repair 2019, 74, 51–62. [Google Scholar] [CrossRef]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- De Conti, L.; Akinyi, M.V.; Mendoza-Maldonado, R.; Romano, M.; Baralle, M.; Buratti, E. TDP-43 affects splicing profiles and isoform production of genes involved in the apoptotic and mitotic cellular pathways. Nucleic. Acids Res. 2015, 43, 8990–9005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Box, J.K.; Paquet, N.; Adams, M.N.; Boucher, D.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Nucleophosmin: From structure and function to disease development. BMC Mol. Biol. 2016, 17, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abo-Rady, M.; Kalmbach, N.; Pal, A.; Schludi, C.; Janosch, A.; Richter, T.; Freitag, P.; Bickle, M.; Kahlert, A.-K.; Petri, S.; et al. Knocking out C9ORF72 exacerbates axonal trafficking defects associated with hexanucleotide repeat expansion and reduces levels of heat shock proteins. Stem Cell Rep. 2020, 14, 390–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, L.F.; Cerqueira, F.M.; Macedo, A.F.; Garcia, C.C.; Angeli, J.P.; Schumacher, R.I.; Sogayar, M.C.; Augusto, O.; Carrì, M.T.; Di Mascio, P.; et al. Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS. Biochim. Biophys. Acta 2010, 1802, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Transgenic mice with human mutant genes causing Parkinson’s disease and amyotrophic lateral sclerosis provide common insight into mechanisms of motor neuron selective vulnerability to degeneration. Rev. Neurosci. 2007, 18, 115–136. [Google Scholar] [CrossRef]
- Schmidt, R.H.; Nickerson, J.M.; Boatright, J.H. Exercise as gene therapy: BDNF and DNA damage repair. Asia Pac. J. Ophthalmol. 2016, 5, 309–311. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.D.; Wu, C.L.; Hwang, W.C.; Yang, D.I. More Insight into BDNF against Neurodegeneration: Anti-apoptosis, anti-oxidation, and suppression of autophagy. Int. J. Mol. Sci. 2017, 18, 545. [Google Scholar] [CrossRef] [Green Version]
- Estévez, A.G.; Kamaid, A.; Thompson, J.A.; Cornwell, T.L.; Radi, R.; Barbeito, L.; Beckman, J.S. Cyclic guanosine 5’ monophosphate (GMP) prevents expression of neuronal nitric oxide synthase and apoptosis in motor neurons deprived of trophic factors in rats. Neurosci. Lett. 2002, 326, 201–205. [Google Scholar] [CrossRef]
- Estévez, A.G.; Spear, N.; Thompson, J.A.; Cornwell, T.L.; Radi, R.; Barbeito, L.; Beckman, J.S. Nitric oxide-dependent production of cGMP supports the survival of rat embryonic motor neurons cultured with brain-derived neurotrophic factor. J. Neurosci. 1998, 18, 3708–3714. [Google Scholar] [CrossRef] [Green Version]
- Noon, A.T.; Shibata, A.; Rief, N.; Löbrich, M.; Stewart, G.S.; Jeggo, P.A.; Goodarzi, A.A. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 2010, 12, 177–184. [Google Scholar] [CrossRef]
- Alagoz, M.; Chiang, S.C.; Sharma, A.; El-Khamisy, S.F. ATM deficiency results in accumulation of DNA-topoisomerase I covalent intermediates in neural cells. PLoS ONE 2013, 8, e58239. [Google Scholar] [CrossRef] [PubMed]
- Boehrs, J.K.; He, J.; Halaby, M.J.; Yang, D.Q. Constitutive expression and cytoplasmic compartmentalization of ATM protein in differentiated human neuron-like SH-SY5Y cells. J. Neurochem. 2007, 100, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Engelhardt, J.I.; Henkel, J.S.; Siklós, L.; Soós, J.; Goodman, C.; Appel, S.H. Widespread increased expression of the DNA repair enzyme PARP in brain in ALS. Neurology 2004, 62, 319–322. [Google Scholar] [CrossRef] [PubMed]
- McGurk, L.; Lee, V.M.; Trojanowksi, J.Q.; Van Deerlin, V.M.; Lee, E.B.; Bonini, N.M. Poly-A binding protein-1 localization to a subset of TDP-43 inclusions in amyotrophic lateral sclerosis occurs more frequently in patients harboring an expansion in C9orf72. J. Neuropathol. Exp. Neurol. 2014, 73, 837–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGurk, L.; Mojsilovic-Petrovic, J.; van Deerlin, V.M.; Shorter, J.; Kalb, R.G.; Lee, V.M.; Trojanowski, J.Q.; Lee, E.B.; Bonini, N.M. Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2018, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.K.; Shin, K.S.; Kang, S.J. AIF translocates to the nucleus in the spinal motor neurons in a mouse model of ALS. Neurosci. Lett. 2006, 406, 205–210. [Google Scholar] [CrossRef]
- Kim, S.H.; Henkel, J.S.; Beers, D.R.; Sengun, I.S.; Simpson, E.P.; Goodman, J.C.; Engelhardt, J.I.; Siklós, L.; Appel, S.H. PARP expression is increased in astrocytes but decreased in motor neurons in the spinal cord of sporadic ALS patients. J. Neuropathol. Exp. Neurol. 2003, 62, 88–103. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Matsuoka, M. Overexpression of nuclear FUS induces neuronal cell death. Neuroscience 2015, 287, 113–124. [Google Scholar] [CrossRef]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol. Dis. 2000, 7, 613–622. [Google Scholar] [CrossRef]
- Maor-Nof, M.; Shipony, Z.; Lopez-Gonzalez, R.; Nakayama, L.; Zhang, Y.J.; Couthouis, J.; Blum, J.A.; Castruita, P.A.; Linares, G.R.; Ruan, K.; et al. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 2021, 184, 689–708.e620. [Google Scholar] [CrossRef] [PubMed]
- González de Aguilar, J.L.; Gordon, J.W.; René, F.; de Tapia, M.; Lutz-Bucher, B.; Gaiddon, C.; Loeffler, J.P. Alteration of the Bcl-x/Bax ratio in a transgenic mouse model of amyotrophic lateral sclerosis: Evidence for the implication of the p53 signaling pathway. Neurobiol. Dis. 2000, 7, 406–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.J.; Lavakumar, M.; Tampi, D.; Balachandran, S.; Tampi, R.R. Frontotemporal dementia: Latest evidence and clinical implications. Adv. Psychopharmacol. 2018, 8, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Broce, I.J.; Castruita, P.A.; Yokoyama, J.S. Moving toward patient-tailored treatment in ALS and FTD: The potential of genomic assessment as a tool for biological discovery and trial recruitment. Front. Neurosci. 2021, 15, 639078. [Google Scholar] [CrossRef]
- Lau, D.H.W.; Hartopp, N.; Welsh, N.J.; Mueller, S.; Glennon, E.B.; Mórotz, G.M.; Annibali, A.; Gomez-Suaga, P.; Stoica, R.; Paillusson, S.; et al. Disruption of ER−mitochondria signalling in fronto-temporal dementia and related amyotrophic lateral sclerosis. Cell Death Dis. 2018, 9, 327. [Google Scholar] [CrossRef] [Green Version]
- Anoar, S.; Woodling, N.S.; Niccoli, T. Mitochondria dysfunction in Frontotemporal Dementia/Amyotrophic Lateral Sclerosis: Lessons from drosophila models. Front. Neurosci. 2021, 15, 1602. [Google Scholar] [CrossRef]
- Palluzzi, F.; Ferrari, R.; Graziano, F.; Novelli, V.; Rossi, G.; Galimberti, D.; Rainero, I.; Benussi, L.; Nacmias, B.; Bruni, A.C.; et al. A novel network analysis approach reveals DNA damage, oxidative stress and calcium/cAMP homeostasis-associated biomarkers in frontotemporal dementia. PLoS ONE 2017, 12, e0185797. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Xu, J. C9orf72 Dipeptide repeats cause selective neurodegeneration and Cell-autonomous excitotoxicity in drosophila glutamatergic neurons. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 7741–7752. [Google Scholar] [CrossRef] [Green Version]
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front. Neurosci. 2020, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Su, J.H.; Nichol, K.E.; Sitch, T.; Sheu, P.; Chubb, C.; Miller, B.L.; Tomaselli, K.J.; Kim, R.C.; Cotman, C.W. DNA Damage and activated caspase-3 expression in neurons and astrocytes: Evidence for apoptosis in frontotemporal dementia. Exp. Neurol. 2000, 163, 9–19. [Google Scholar] [CrossRef]
- Ashraf, A.; So, P.-W. Spotlight on ferroptosis: Iron-dependent cell death in Alzheimer’s disease. Front. Aging Neurosci. 2020, 12, 196. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.-J.; et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Ghidoni, R.; Benussi, L.; Glionna, M.; Franzoni, M.; Binetti, G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 2008, 71, 1235–1239. [Google Scholar] [CrossRef]
- Bandey, I.; Chiou, S.H.; Huang, A.P.; Tsai, J.C.; Tu, P.H. Progranulin promotes Temozolomide resistance of glioblastoma by orchestrating DNA repair and tumor stemness. Oncogene 2015, 34, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Kao, A.W.; Eisenhut, R.J.; Martens, L.H.; Nakamura, A.; Huang, A.; Bagley, J.A.; Zhou, P.; de Luis, A.; Neukomm, L.J.; Cabello, J.; et al. A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4441–4446. [Google Scholar] [CrossRef] [Green Version]
- Chitramuthu, B.P.; Baranowski, D.C.; Kay, D.G.; Bateman, A.; Bennett, H.P.J. Progranulin modulates zebrafish motoneuron development in vivoand rescues truncation defects associated with knockdown of Survival motor neuron 1. Mol. Neurodegener. 2010, 5, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of cell survival by secreted proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Wheeler, V.C.; Gutekunst, C.A.; Vrbanac, V.; Lebel, L.A.; Schilling, G.; Hersch, S.; Friedlander, R.M.; Gusella, J.F.; Vonsattel, J.P.; Borchelt, D.R.; et al. Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum. Mol. Genet. 2002, 11, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Semaka, A.; Creighton, S.; Warby, S.; Hayden, M.R. Predictive testing for Huntington disease: Interpretation and significance of intermediate alleles. Clin. Genet. 2006, 70, 283–294. [Google Scholar] [CrossRef]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein misfolding and ER stress in Huntington’s Disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ratan, R.R. Oxidative stress and Huntington’s Disease: The good, the bad, and the ugly. J. Huntingt. Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef] [Green Version]
- Caviston, J.P.; Holzbaur, E.L.F. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009, 19, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanov, M.B.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Beal, M.F. Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2001, 79, 1246–1249. [Google Scholar] [CrossRef]
- Maiuri, T.; Suart, C.E.; Hung, C.L.K.; Graham, K.J.; Barba Bazan, C.A.; Truant, R. DNA Damage repair in Huntington’s Disease and other neurodegenerative diseases. Neurother. J. Am. Soc. Exp. Neurotherapeutics 2019, 16, 948–956. [Google Scholar] [CrossRef]
- Maiuri, T.; Hung, C.L.K.; Suart, C.; Begeja, N.; Barba-Bazan, C.; Peng, Y.; Savic, N.; Wong, T.; Truant, R. DNA Repair in Huntington’s disease and spinocerebellar ataxias: Somatic instability and alternative hypotheses. J. Huntingt. Dis. 2021, 10, 165–173. [Google Scholar] [CrossRef]
- Synofzik, M.; Schüle, R. Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways. Mov. Disord. 2017, 32, 332–345. [Google Scholar] [CrossRef]
- Møllersen, L.; Rowe, A.D.; Illuzzi, J.L.; Hildrestrand, G.A.; Gerhold, K.J.; Tveterås, L.; Bjølgerud, A.; Wilson, D.M., 3rd; Bjørås, M.; Klungland, A. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum. Mol. Genet. 2012, 21, 4939–4947. [Google Scholar] [CrossRef]
- Manley, K.; Shirley, T.L.; Flaherty, L.; Messer, A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat. Genet. 1999, 23, 471–473. [Google Scholar] [CrossRef]
- Massey, T.H.; Jones, L. The central role of DNA damage and repair in CAG repeat diseases. Dis Model. Mech 2018, 11, dmm031930. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Wang, C.-E.; Zhao, B.; Li, W.; Ouyang, Z.; Liu, Z.; Yang, H.; Fan, P.; O’Neill, A.; Gu, W.; et al. Expression of Huntington’s disease protein results in apoptotic neurons in the brains of cloned transgenic pigs. Hum. Mol. Genet. 2010, 19, 3983–3994. [Google Scholar] [CrossRef] [PubMed]
- Bano, D.; Zanetti, F.; Mende, Y.; Nicotera, P. Neurodegenerative processes in Huntington’s disease. Cell Death Dis. 2011, 2, e228. [Google Scholar] [CrossRef]
- Gelman, A.; Rawet-Slobodkin, M.; Elazar, Z. Huntingtin facilitates selective autophagy. Nat. Cell Biol. 2015, 17, 214–215. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, E.; Hasegawa, K.; Fujita, K.; Ichinose, S.; Yagishita, S.; Murata, M.; Tagawa, K.; Akashi, T.; Eishi, Y.; Okazawa, H. A novel form of necrosis, TRIAD, occurs in human Huntington’s disease. Acta Neuropathol. Commun. 2017, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, B.I.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 2005, 47, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.Q.; Wang, X.X.; Wang, Y.; Chuang, D.M.; DiFiglia, M.; Chase, T.N.; Qin, Z.H. Susceptibility of striatal neurons to excitotoxic injury correlates with basal levels of Bcl-2 and the induction of P53 and c-Myc immunoreactivity. Neurobiol. Dis. 2005, 20, 562–573. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [Green Version]
- Illuzzi, J.; Yerkes, S.; Parekh-Olmedo, H.; Kmiec, E.B. DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J. Neurosci. Res. 2009, 87, 733–747. [Google Scholar] [CrossRef]
- Shen, X.; Chen, J.; Li, J.; Kofler, J.; Herrup, K. Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. eNeuro 2016, 3, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N.; et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015, 6, 8897. [Google Scholar] [CrossRef] [PubMed]
- Eilam, R.; Peter, Y.; Groner, Y.; Segal, M. Late degeneration of nigro-striatal neurons in ATM-/- mice. Neuroscience 2003, 121, 83–98. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Yang, D.; Pribadi, M.; Kim, T.S.; Krishnan, G.; Choi, S.Y.; Lee, S.; Coppola, G.; Gao, F.-B. Partial inhibition of the overactivated Ku80-dependent DNA repair pathway rescues neurodegeneration in C9ORF72-ALS/FTD. Proc. Natl. Acad. Sci. USA 2019, 116, 9628–9633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanungo, J. DNA-dependent protein kinase and DNA repair: Relevance to Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, J.P.Y.; Ma, H.T.; Poon, R.Y.C. Synergism between ATM and PARP1 inhibition involves DNA damage and abrogating the G(2) DNA damage checkpoint. Mol. Cancer 2020, 19, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.S.; Nowsheen, S.; Wang, T.; Thotala, D.K.; Xia, F. Glycogen synthase kinase 3β inhibition enhances repair of DNA double-strand breaks in irradiated hippocampal neurons. Neuro-Oncol. 2011, 13, 459–470. [Google Scholar] [CrossRef] [Green Version]
- Fielder, E.; von Zglinicki, T.; Jurk, D. The DNA Damage response in neurons: Die by apoptosis or survive in a senescence-like state? J. Alzheimer’s Dis. 2017, 60, S107–S131. [Google Scholar] [CrossRef]
- Surova, O.; Zhivotovsky, B. Various modes of cell death induced by DNA damage. Oncogene 2013, 32, 3789–3797. [Google Scholar] [CrossRef] [Green Version]




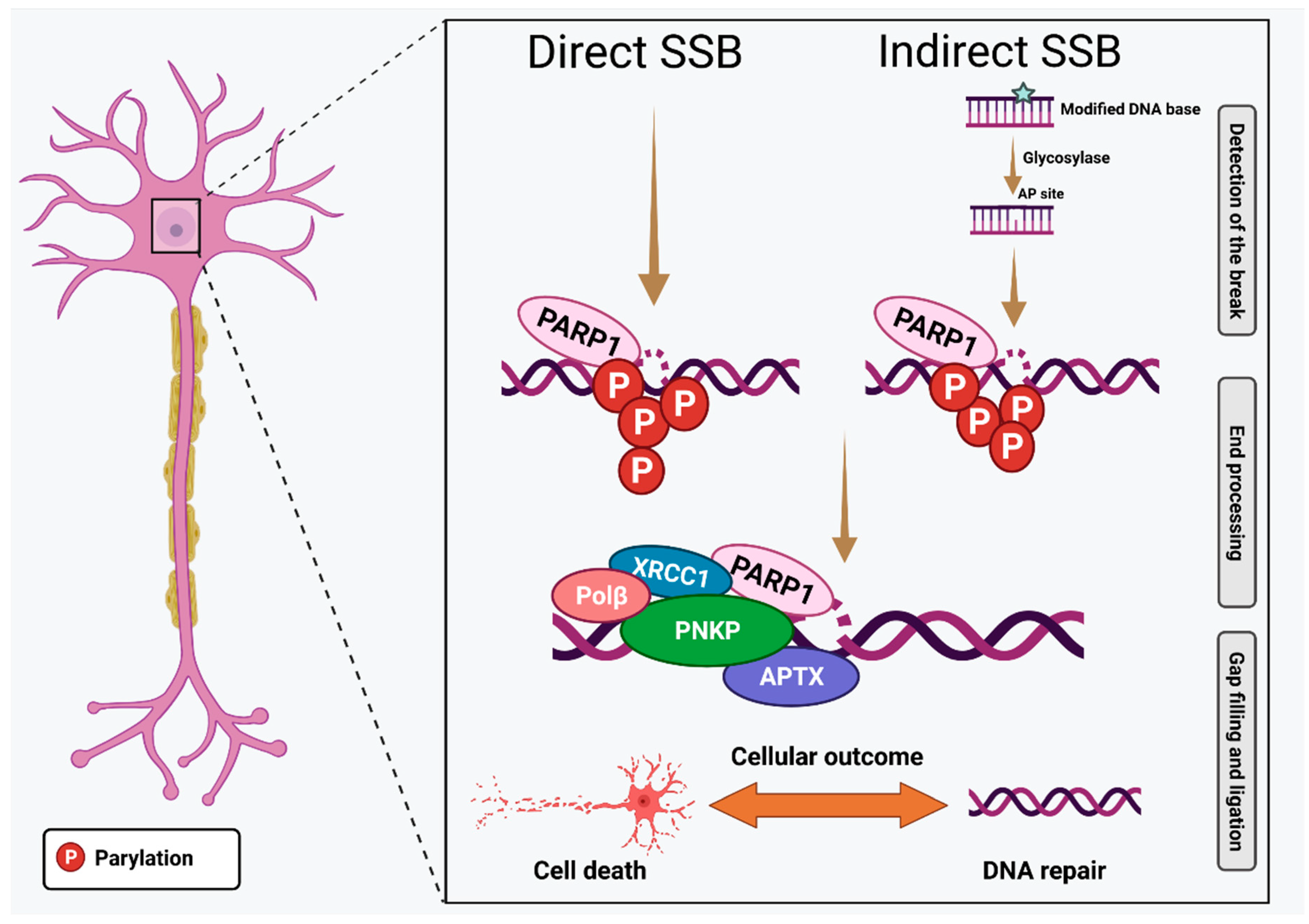{kind=link}
{kind=link}
{kind=link}
| Pathway | Form of Apoptosis? | NCDD Definition | Intra- or Extracellular Trigger? | Described in Neurons? | Related to DNA Damage in Neurons? |
|---|---|---|---|---|---|
| Apoptosis Intrinsic | Yes | Demarcated by MOMP (both) or CASP8 (extrinsic) and induced by executioner caspases, mainly CASP3 | Both | Yes | Yes |
| Apoptosis Extrinsic | Yes | Demarcated by MOMP (both) or CASP8 (extrinsic) and induced by executioner caspases, mainly CASP3 | Extracellular | Yes | No |
| Anoikis | Yes | Form of intrinsic apoptosis elicited by loss of integrin-dependent attachment to extracellular matrix | Both | No | No |
| MPT-driven necrosis | Non-apoptotic | Initiated by oxidative stress or Ca2+ overload, necrotic phenotype, dependent on CYPD | Intracellular | Yes | Neurotoxic |
| Necroptosis | Non-apoptotic | Depends on MLKL, RIPK3, and sometimes RIPK1 activity | Both | Yes | No |
| TRIAD | Non-apoptotic | Transcriptional repression-induced atypical cell death | - | Yes | No |
| Ferroptosis | Non-apoptotic | Initiated by oxidative stress, depends on GPX4 | Intracellular | Yes | Yes |
| Pyroptosis | Non-apoptotic | Depends on plasma membrane pores by gasdermin proteins, inflammatory caspases | Both | Yes | Yes |
| Parthanatos | Non-apoptotic | Initiated by PARP1 hyperactivation, coupled to AIF and MIF DNA degradation | Intracellular? | Yes | Yes |
| Entosis | Non-apoptotic | Initiated from actomyosin-dependent internalisation executed by lysosomes | Intracellular? | No | No |
| NETotic cell death | Nonapoptotic | ROS-dependent restricted to hematopoietic cells, associated with NET extrusion | No | No | |
| Lysosome-dependent cell death | Non-apoptotic | Demarcated by primary LMP and triggered by cathepsins or MOMP and executioner caspases | Intracellular? | Yes | No |
| Autophagy-dependent cell death | Non-apoptotic | Depends on the autophagy machinery | Intracellular? | Yes | No |
| Immunogenic cell death | Non-apoptotic | Activates an adaptive immune response in immunocompetent hosts | No | No | |
| Oxeiptosis * | Non-apoptotic | Activated by ROS, dependent on KEAP1, mediated by PGAM5 and AIFM | Intracellular? | No | No |
| Alkaliptosis * | Non-apoptotic | Activated by pH changes, an alkalinisation-dependent form of regulated necrosis. | Intracellular? | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shadfar, S.; Brocardo, M.; Atkin, J.D. The Complex Mechanisms by Which Neurons Die Following DNA Damage in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 2484. https://doi.org/10.3390/ijms23052484
Shadfar S, Brocardo M, Atkin JD. The Complex Mechanisms by Which Neurons Die Following DNA Damage in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2022; 23(5):2484. https://doi.org/10.3390/ijms23052484
Chicago/Turabian StyleShadfar, Sina, Mariana Brocardo, and Julie D. Atkin. 2022. "The Complex Mechanisms by Which Neurons Die Following DNA Damage in Neurodegenerative Diseases" International Journal of Molecular Sciences 23, no. 5: 2484. https://doi.org/10.3390/ijms23052484