
The Anxiolytic Drug Buspirone Prevents Rotenone-Induced Toxicity in a Mouse Model of Parkinson’s Disease



Abstract
:1. Introduction
2. Results
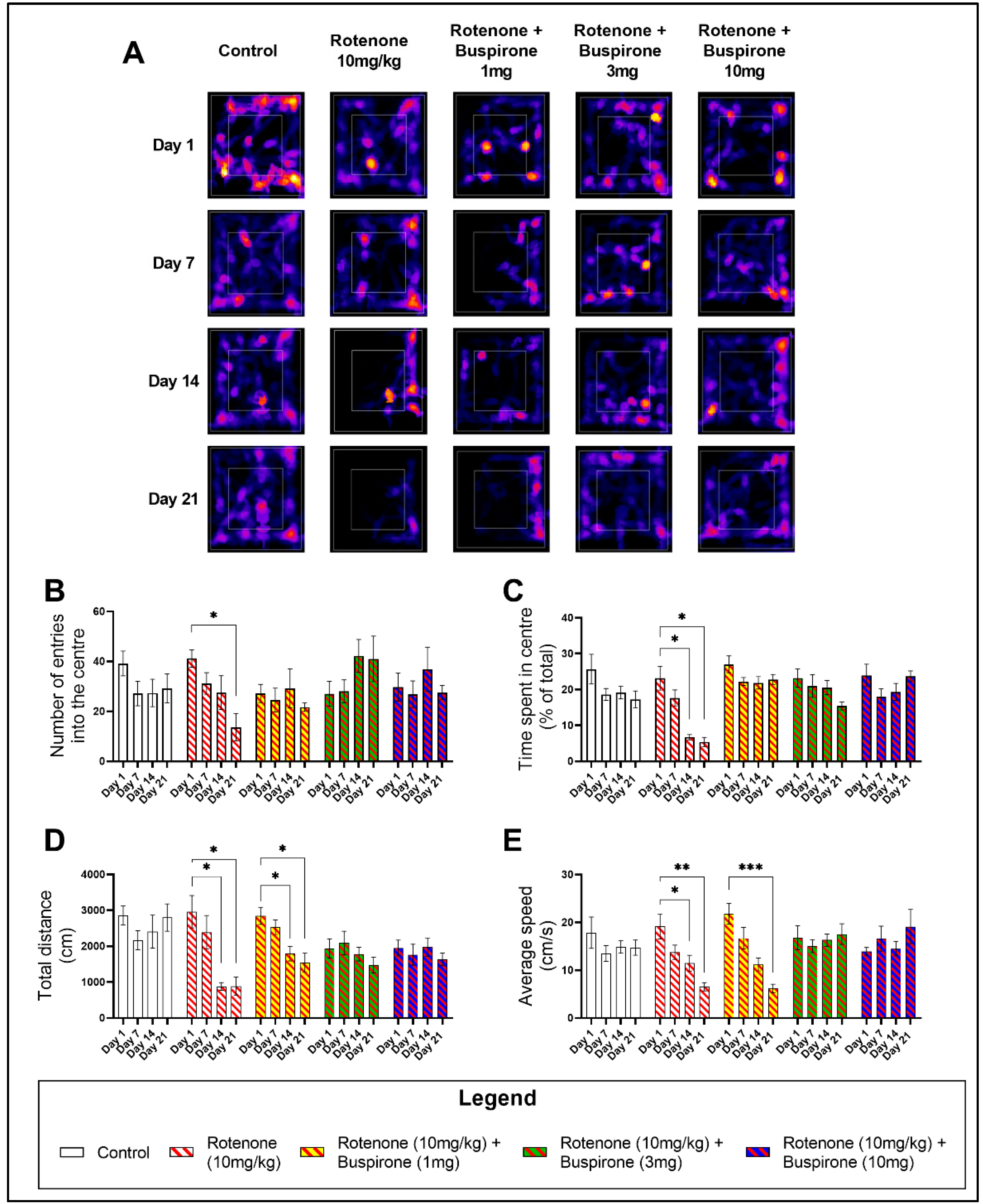
2.1. Buspirone Prevents Rotenone-Induced Deficits in Locomotor and Exploratory Behaviour
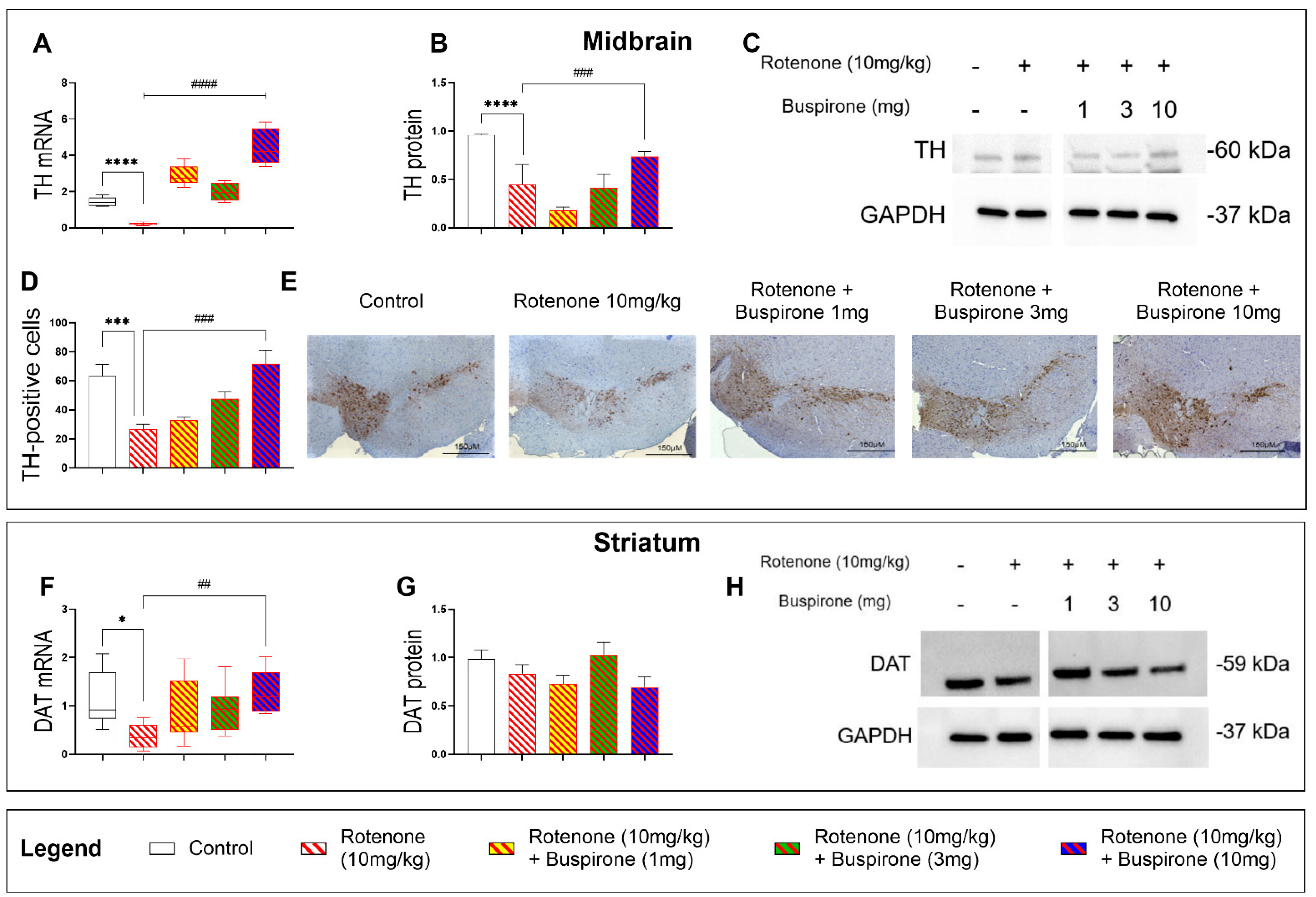
2.2. Buspirone Prevents the Loss of Dopaminergic Neurons in Rotenone-Treated Mice
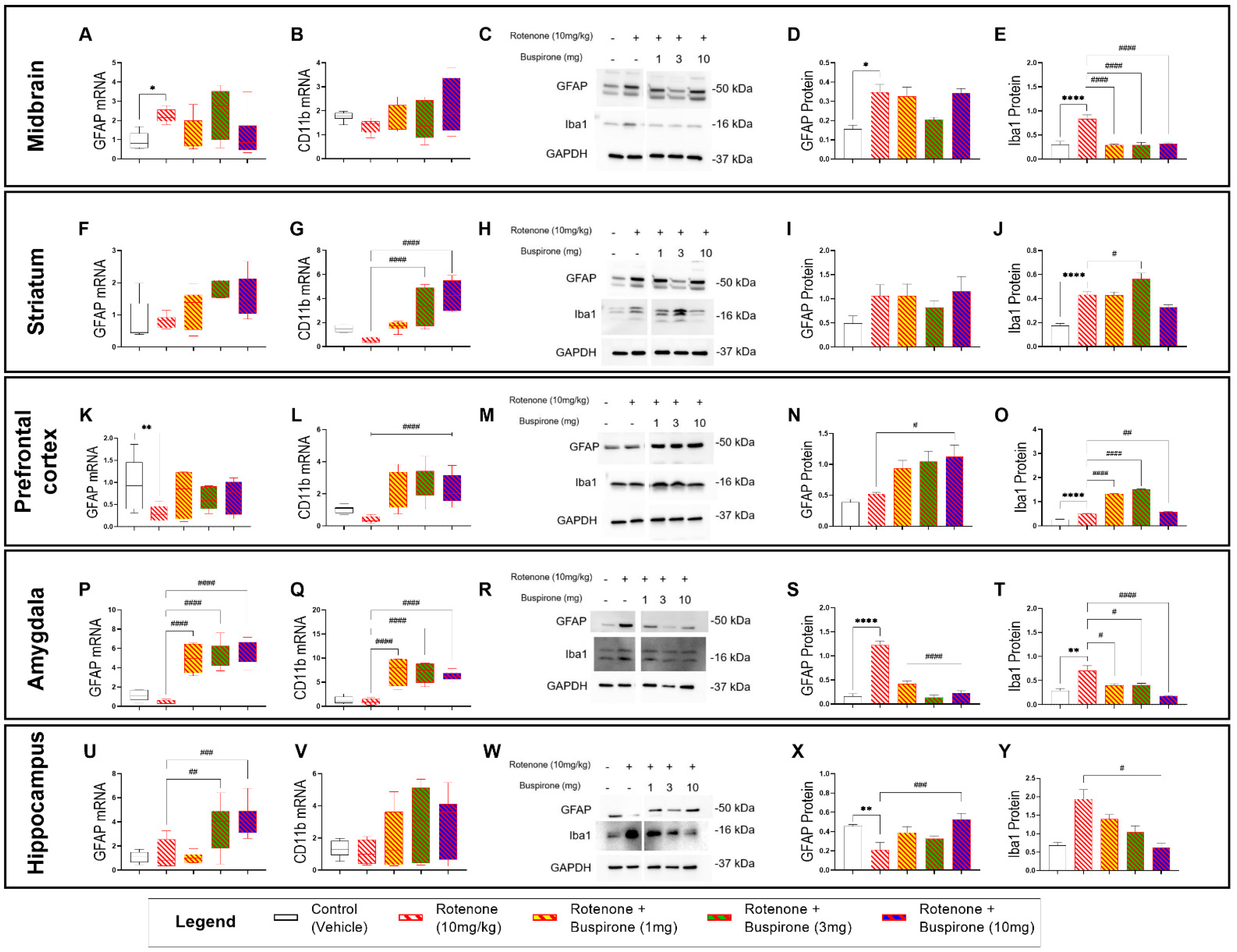
2.3. Buspirone Triggers Brain-Region-Specific Changes in the Expression of Glial Activation Markers
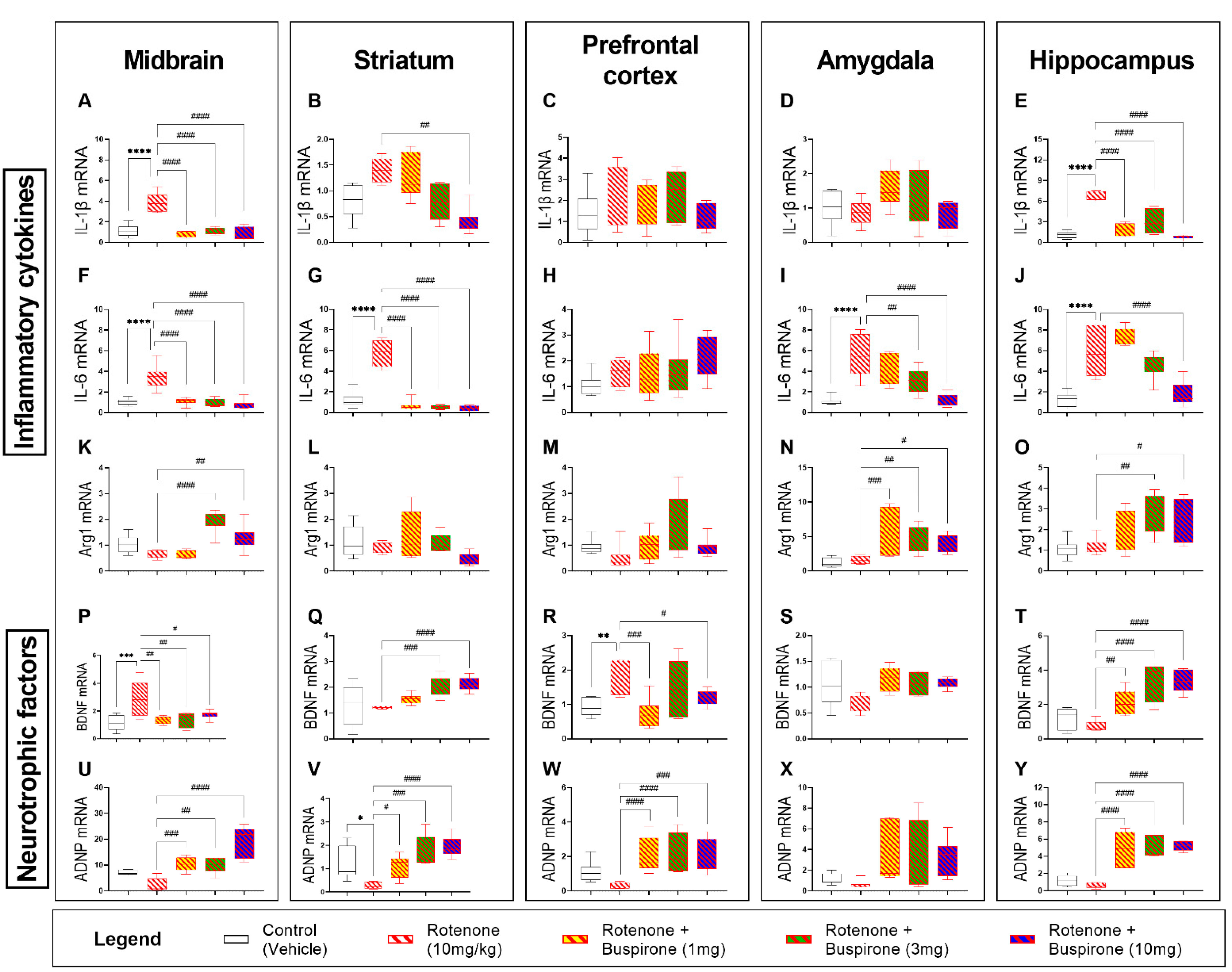
2.4. Buspirone Reduces Neuroinflammation and Promotes the Expression of Neurotrophic Factors
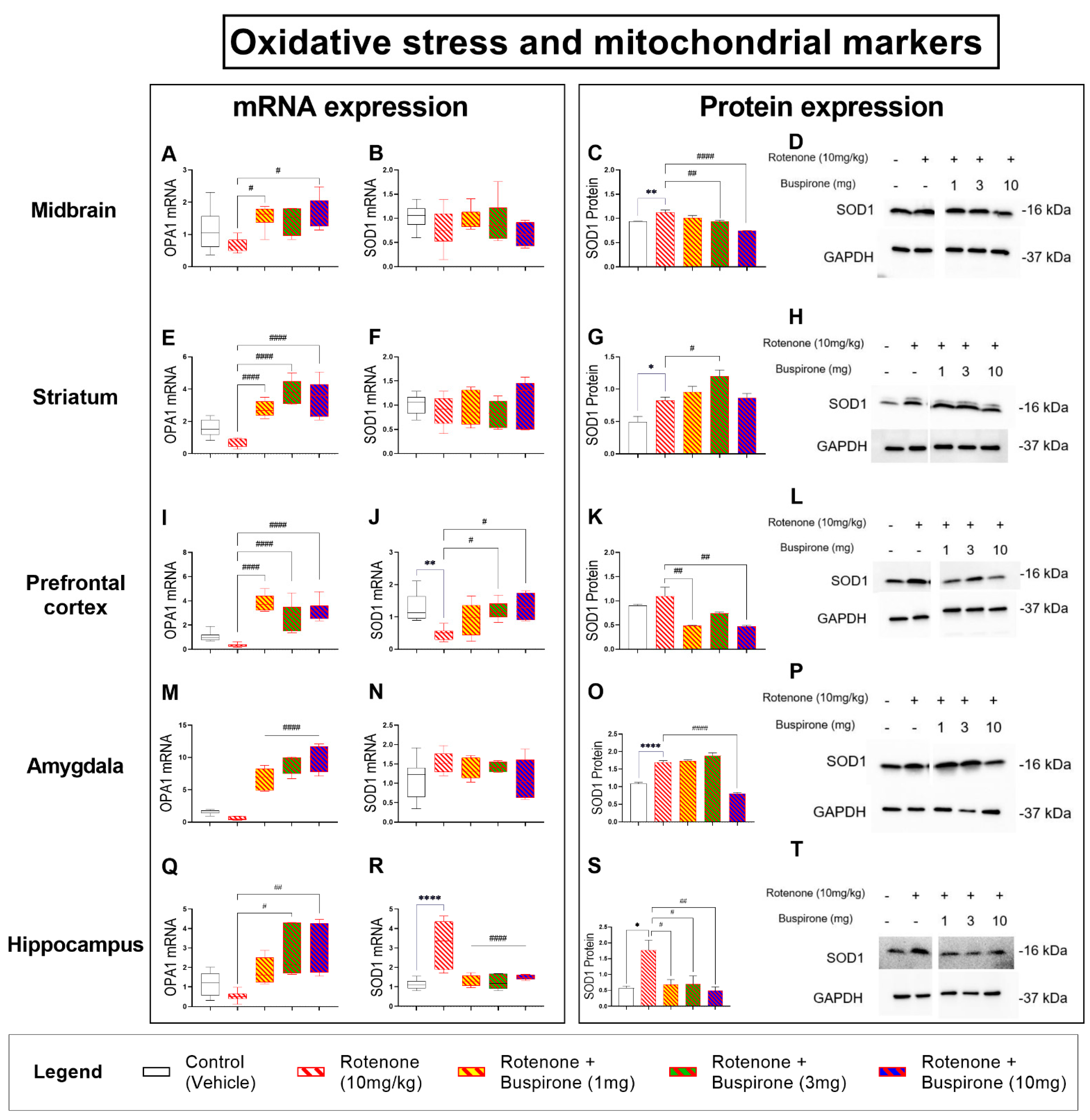
2.5. Buspirone Dampens Rotenone-Induced Oxidative Stress in Distinct Brain Regions
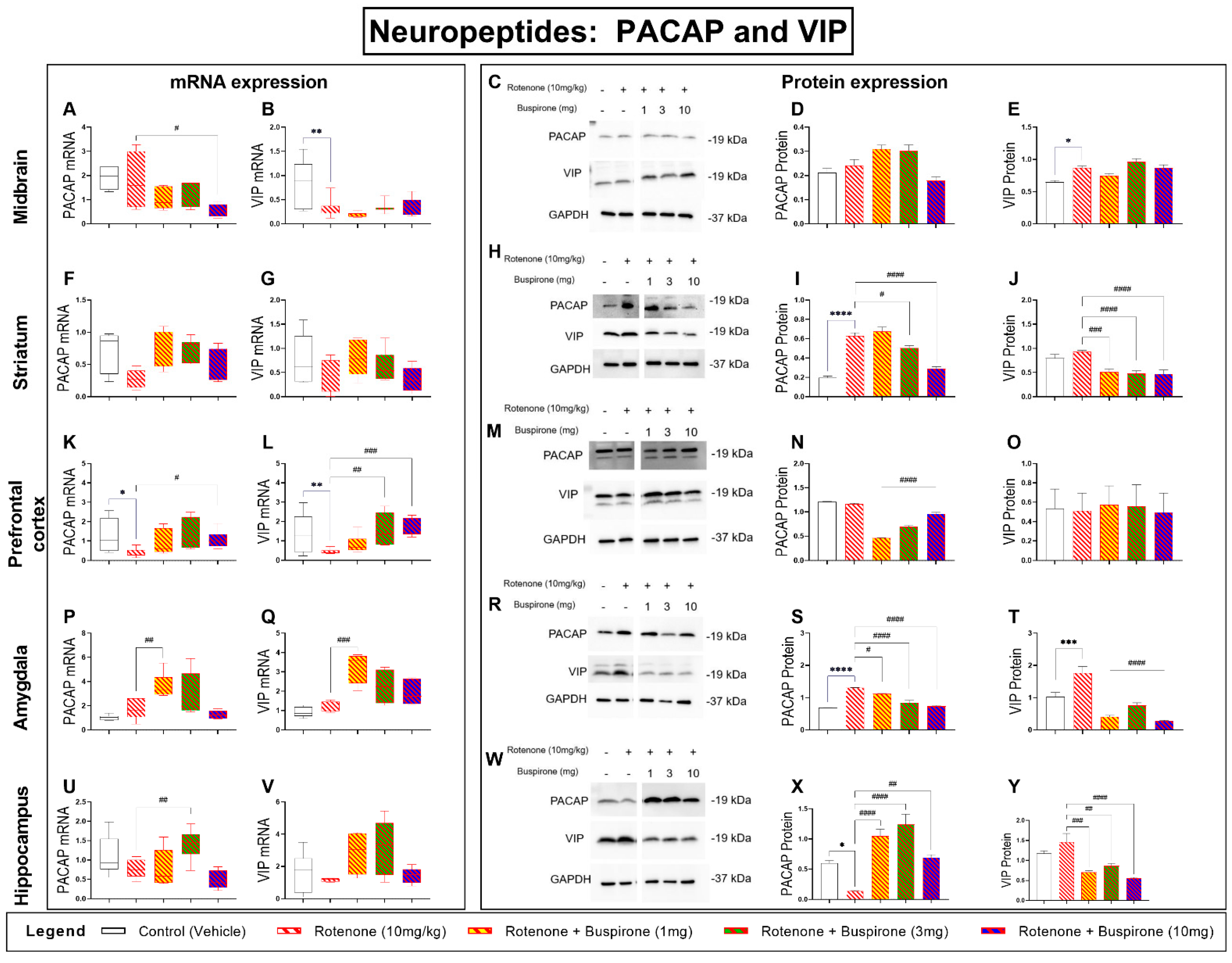
2.6. Buspirone Prevents Dysregulations of the PACAP and VIP Neuropeptides in the Brains of Rotenone-Treated Mice
3. Discussion
4. Materials and Methods
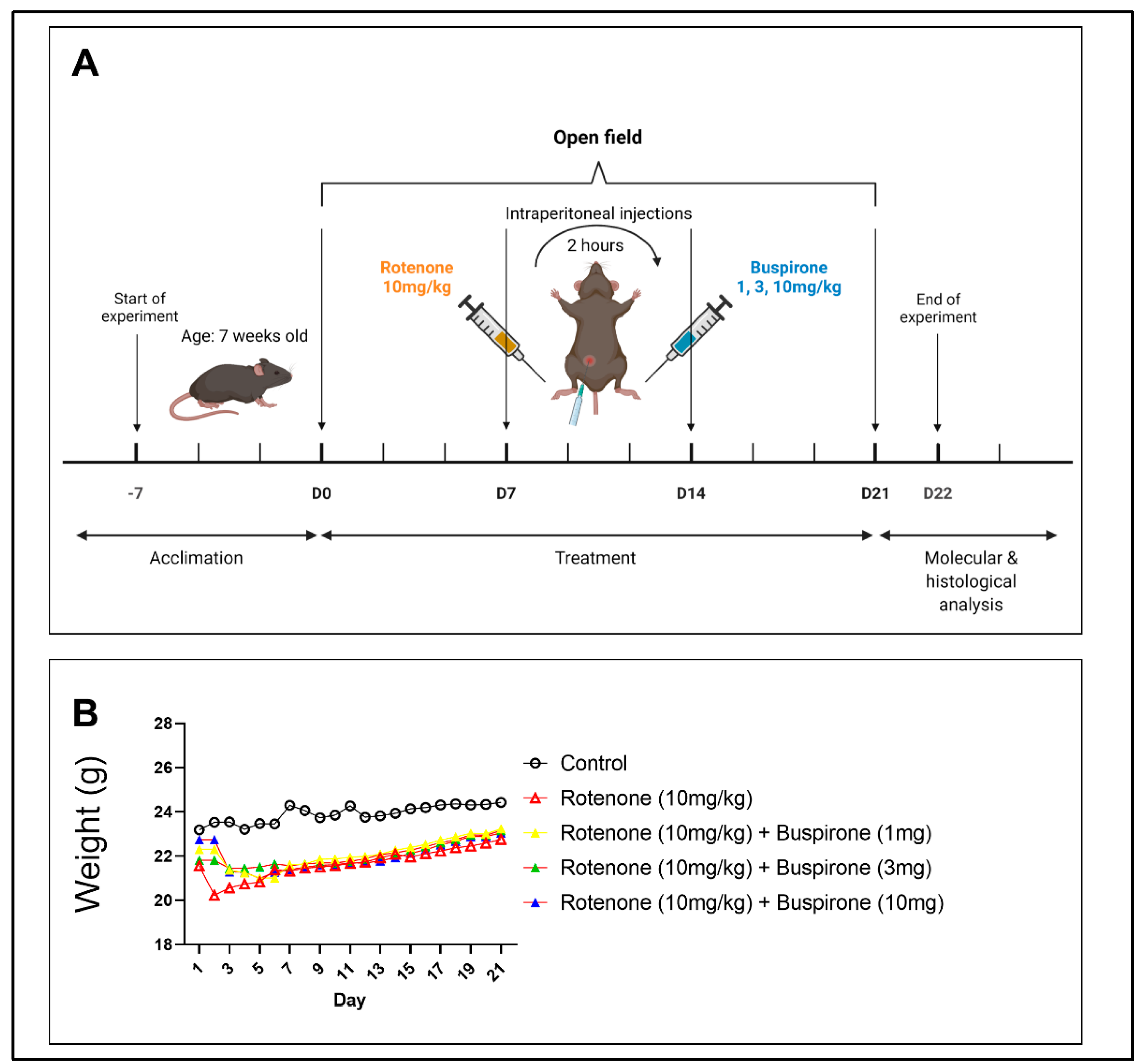
4.1. Animal Experiments
4.2. Experimental Protocol
4.3. Open Field
4.4. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
4.5. Western Blot
4.6. Immunohistochemistry
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.L.; Dragicevic, N.; Seifert, K.; Choi, D.Y.; Liu, M.; Kim, H.C.; Cass, W.A.; Sullivan, P.G.; Bing, G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J. Neurochem. 2007, 100, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Gamber, K.M. Animal Models of Parkinson’s Disease: New models provide greater translational and predictive value. BioTechniques 2016, 61, 210–211. [Google Scholar] [CrossRef] [Green Version]
- Radad, K.; Al-Shraim, M.; Al-Emam, A.; Wang, F.; Kranner, B.; Rausch, W.D.; Moldzio, R. Rotenone: From modelling to implication in Parkinson’s disease. Folia Neuropathol. 2019, 57, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef] [Green Version]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef] [Green Version]
- Castano, A.; Herrera, A.J.; Cano, J.; Machado, A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J. Neurochem. 1998, 70, 1584–1592. [Google Scholar] [CrossRef] [Green Version]
- Thomas Broome, S.; Louangaphay, K.; Keay, K.A.; Leggio, G.M.; Musumeci, G.; Castorina, A. Dopamine: An immune transmitter. Neural Regen. Res. 2020, 15, 2173–2185. [Google Scholar] [CrossRef]
- McKenna, F.; McLaughlin, P.J.; Lewis, B.J.; Sibbring, G.C.; Cummerson, J.A.; Bowen-Jones, D.; Moots, R.J. Dopamine receptor expression on human T- and B-lymphocytes, monocytes, neutrophils, eosinophils and NK cells: A flow cytometric study. J. Neuroimmunol. 2002, 132, 34–40. [Google Scholar] [CrossRef]
- Elgueta, D.; Aymerich, M.S.; Contreras, F.; Montoya, A.; Celorrio, M.; Rojo-Bustamante, E.; Riquelme, E.; Gonzalez, H.; Vasquez, M.; Franco, R.; et al. Pharmacologic antagonism of dopamine receptor D3 attenuates neurodegeneration and motor impairment in a mouse model of Parkinson’s disease. Neuropharmacology 2017, 113, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, D.; Contreras, F.; Prado, C.; Montoya, A.; Ugalde, V.; Chovar, O.; Villagra, R.; Henriquez, C.; Abellanas, M.A.; Aymerich, M.S.; et al. Dopamine Receptor D3 Expression Is Altered in CD4+ T-Cells From Parkinson’s Disease Patients and Its Pharmacologic Inhibition Attenuates the Motor Impairment in a Mouse Model. Front. Immunol. 2019, 10, 981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Perlmutter, J.S.; Benzinger, T.L.S.; Morris, J.C.; Xu, J. Dopamine D3 receptor: A neglected participant in Parkinson Disease pathogenesis and treatment? Ageing Res. Rev. 2020, 57, 100994. [Google Scholar] [CrossRef] [PubMed]
- Raber, J.; Wienclaw, R.A.; Cataldo, L.J. Buspirone. In The Gale Encyclopedia of Mental Health; Gale: Detroit, MI, USA, 2012; pp. 264–267. [Google Scholar]
- Kim, S.W.; Fowler, J.S.; Skolnick, P.; Muench, L.; Kang, Y.; Shea, C.; Logan, J.; Kim, D.; Carter, P.; King, P.; et al. Therapeutic doses of buspirone block D3 receptors in the living primate brain–CORRIGENDUM. Int. J. Neuropsychopharmacol. 2014, 17, 1354. [Google Scholar] [CrossRef] [Green Version]
- Leggio, G.M.; Camillieri, G.; Platania, C.B.; Castorina, A.; Marrazzo, G.; Torrisi, S.A.; Nona, C.N.; D’Agata, V.; Nobrega, J.; Stark, H.; et al. Dopamine D3 receptor is necessary for ethanol consumption: An approach with buspirone. Neuropsychopharmacology 2014, 39, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Thomas Broome, S.; Fisher, T.; Faiz, A.; Keay, K.A.; Musumeci, G.; Al-Badri, G.; Castorina, A. Assessing the Anti-Inflammatory Activity of the Anxiolytic Drug Buspirone Using CRISPR-Cas9 Gene Editing in LPS-Stimulated BV-2 Microglial Cells. Cells 2021, 10, 1312. [Google Scholar] [CrossRef]
- Gao, H.-M.; Hong, J.-S.; Zhang, W.; Liu, B. Distinct Role for Microglia in Rotenone-Induced Degeneration of Dopaminergic Neurons. J. Neurosci. 2002, 22, 782–790. [Google Scholar] [CrossRef] [Green Version]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Liu, H.Y.; Yue, J.; Hu, L.N.; Cheng, L.F.; Wang, X.S.; Wang, X.J.; Feng, B. Chronic minocycline treatment reduces the anxiety-like behaviors induced by repeated restraint stress through modulating neuroinflammation. Brain Res. Bull. 2018, 143, 19–26. [Google Scholar] [CrossRef]
- Waschek, J.A. VIP and PACAP: Neuropeptide modulators of CNS inflammation, injury, and repair. Br. J. Pharmacol. 2013, 169, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Marzagalli, R.; Leggio, G.M.; Bucolo, C.; Pricoco, E.; Keay, K.A.; Cardile, V.; Castorina, S.; Salomone, S.; Drago, F.; Castorina, A. Genetic blockade of the dopamine D3 receptor enhances hippocampal expression of PACAP and receptors and alters their cortical distribution. Neuroscience 2016, 316, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Dhanalakshmi, C.; Janakiraman, U.; Manivasagam, T.; Justin Thenmozhi, A.; Essa, M.M.; Kalandar, A.; Khan, M.A.; Guillemin, G.J. Vanillin Attenuated Behavioural Impairments, Neurochemical Deficts, Oxidative Stress and Apoptosis Against Rotenone Induced Rat Model of Parkinson’s Disease. Neurochem. Res. 2016, 41, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, H.; Guo, X.; Ge, D.; Shi, Y.; Lu, X.; Lu, J.; Chen, J.; Ding, F.; Zhang, Q. Involvement of Akt/mTOR in the Neurotoxicity of Rotenone-Induced Parkinson’s Disease Models. Int. J. Environ. Res. Public Health 2019, 16, 3811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inden, M.; Kitamura, Y.; Takeuchi, H.; Yanagida, T.; Takata, K.; Kobayashi, Y.; Taniguchi, T.; Yoshimoto, K.; Kaneko, M.; Okuma, Y.; et al. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4-phenylbutyrate, a chemical chaperone. J. Neurochem. 2007, 101, 1491–1504. [Google Scholar] [CrossRef]
- D’Amico, A.G.; Scuderi, S.; Leggio, G.M.; Castorina, A.; Drago, F.; D’Agata, V. Increased hippocampal CREB phosphorylation in dopamine D3 receptor knockout mice following passive avoidance conditioning. Neurochem. Res. 2013, 38, 2516–2523. [Google Scholar] [CrossRef]
- Prut, L.; Belzung, C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. Eur. J. Pharmacol. 2003, 463, 3–33. [Google Scholar] [CrossRef]
- Leggio, G.M.; Torrisi, S.A.; Castorina, A.; Platania, C.B.; Impellizzeri, A.A.; Fidilio, A.; Caraci, F.; Bucolo, C.; Drago, F.; Salomone, S. Dopamine D3 receptor-dependent changes in alpha6 GABAA subunit expression in striatum modulate anxiety-like behaviour: Responsiveness and tolerance to diazepam. Eur. Neuropsychopharmacol. 2015, 25, 1427–1436. [Google Scholar] [CrossRef]
- Schneider, R.B.; Auinger, P.; Tarolli, C.G.; Iourinets, J.; Gil-Diaz, M.C.; Richard, I.H. A trial of buspirone for anxiety in Parkinson’s disease: Safety and tolerability. Park. Relat. Disord. 2020, 81, 69–74. [Google Scholar] [CrossRef]
- Maramai, S.; Gemma, S.; Brogi, S.; Campiani, G.; Butini, S.; Stark, H.; Brindisi, M. Dopamine D3 Receptor Antagonists as Potential Therapeutics for the Treatment of Neurological Diseases. Front. Neurosci. 2016, 10, 451. [Google Scholar] [CrossRef] [Green Version]
- Le Foll, B.; Payer, D.; Di Ciano, P.; Guranda, M.; Nakajima, S.; Tong, J.; Mansouri, E.; Wilson, A.A.; Houle, S.; Meyer, J.H.; et al. Occupancy of Dopamine D3 and D2 Receptors by Buspirone: A [11C]-(+)-PHNO PET Study in Humans. Neuropsychopharmacology 2016, 41, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, P.; Sheng, W.S.; Hu, S.; Prasad, S.; Lokensgard, J.R. Differential Cytokine-Induced Responses of Polarized Microglia. Brain Sci. 2021, 11, 1482. [Google Scholar] [CrossRef] [PubMed]
- Castorina, A.; Vogiatzis, M.; Kang, J.W.M.; Keay, K.A. PACAP and VIP expression in the periaqueductal grey of the rat following sciatic nerve constriction injury. Neuropeptides 2019, 74, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, I.P.; de Paiva, I.H.R.; Duarte-Silva, E.P.; de Melo, M.G.; da Silva, R.S.; Oliveira, W.H.; da Silveira Andrade da Costa, B.L.; Peixoto, C.A. Metformin and fluoxetine improve depressive-like behavior in a murine model of Parkinsons disease through the modulation of neuroinflammation, neurogenesis and neuroplasticity. Int. Immunopharmacol. 2021, 102, 108415. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kong, D.; Zhou, Q.; Wei, G.; Song, J.; Liang, Y.; Du, G. Baicalein alleviates depression-like behavior in rotenone- induced Parkinson’s disease model in mice through activating the BDNF/TrkB/CREB pathway. Biomed. Pharmacother. 2021, 140, 111556. [Google Scholar] [CrossRef]
- Ivashko-Pachima, Y.; Gozes, I. Activity-dependent neuroprotective protein (ADNP)-end-binding protein (EB) interactions regulate microtubule dynamics toward protection against tauopathy. Prog. Mol. Biol. Transl. Sci. 2021, 177, 65–90. [Google Scholar] [CrossRef]
- Burke, N.N.; Kerr, D.M.; Moriarty, O.; Finn, D.P.; Roche, M. Minocycline modulates neuropathic pain behaviour and cortical M1-M2 microglial gene expression in a rat model of depression. Brain Behav. Immun. 2014, 42, 147–156. [Google Scholar] [CrossRef]
- Wang, J.; Lai, S.; Li, G.; Zhou, T.; Wang, B.; Cao, F.; Chen, T.; Zhang, X.; Chen, Y. Microglial activation contributes to depressive-like behavior in dopamine D3 receptor knockout mice. Brain Behav. Immun. 2020, 83, 226–238. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M.; Murakami, S.; Takeshima, M.; Torigoe, N.; Kitamura, Y.; Miyoshi, K. Targeting 5-HT1A receptors in astrocytes to protect dopaminergic neurons in Parkinsonian models. Neurobiol. Dis. 2013, 59, 244–256. [Google Scholar] [CrossRef]
- Cheng, J.P.; Leary, J.B.; Sembhi, A.; Edwards, C.M.; Bondi, C.O.; Kline, A.E. 5-hydroxytryptamine1A (5-HT1A) receptor agonists: A decade of empirical evidence supports their use as an efficacious therapeutic strategy for brain trauma. Brain Res. 2016, 1640, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Liang, P.; Wang, K.; Jia, J.; Wang, X. Serotonin 1A receptor agonist modulation of motor deficits and cortical oscillations by NMDA receptor interaction in parkinsonian rats. Neuropharmacology 2022, 203, 108881. [Google Scholar] [CrossRef]
- Krabbe, G.; Matyash, V.; Pannasch, U.; Mamer, L.; Boddeke, H.W.; Kettenmann, H. Activation of serotonin receptors promotes microglial injury-induced motility but attenuates phagocytic activity. Brain Behav. Immun. 2012, 26, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Isooka, N.; Imafuku, F.; Sun, J.; Kikuoka, R.; Furukawa, C.; Asanuma, M. Chronic Systemic Exposure to Low-Dose Rotenone Induced Central and Peripheral Neuropathology and Motor Deficits in Mice: Reproducible Animal Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3254. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Hwang, J.J.; Poston, K.L. Episodic recognition memory and the hippocampus in Parkinson’s disease: A review. Cortex 2019, 113, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Carey, G.; Gormezoglu, M.; de Jong, J.J.A.; Hofman, P.A.M.; Backes, W.H.; Dujardin, K.; Leentjens, A.F.G. Neuroimaging of Anxiety in Parkinson’s Disease: A Systematic Review. Mov. Disord. 2021, 36, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef]
- Yu, R.; Li, J.; Lin, Z.; Ouyang, Z.; Huang, X.; Reglodi, D.; Vaudry, D. TAT-tagging of VIP exerts positive allosteric modulation of the PAC1 receptor and enhances VIP neuroprotective effect in the MPTP mouse model of Parkinson’s disease. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129626. [Google Scholar] [CrossRef]
- Yang, S.; Yang, J.; Yang, Z.; Chen, P.; Fraser, A.; Zhang, W.; Pang, H.; Gao, X.; Wilson, B.; Hong, J.S.; et al. Pituitary adenylate cyclase-activating polypeptide (PACAP) 38 and PACAP4–6 are neuroprotective through inhibition of NADPH oxidase: Potent regulators of microglia-mediated oxidative stress. J. Pharmacol Exp. Ther. 2006, 319, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Karunia, J.; Niaz, A.; Mandwie, M.; Thomas Broome, S.; Keay, K.A.; Waschek, J.A.; Al-Badri, G.; Castorina, A. PACAP and VIP Modulate LPS-Induced Microglial Activation and Trigger Distinct Phenotypic Changes in Murine BV2 Microglial Cells. Int. J. Mol. Sci. 2021, 22, 10947. [Google Scholar] [CrossRef]
- Bello-Arroyo, E.; Roque, H.; Marcos, A.; Orihuel, J.; Higuera-Matas, A.; Desco, M.; Caiolfa, V.R.; Ambrosio, E.; Lara-Pezzi, E.; Gomez-Gaviro, M.V. MouBeAT: A New and Open Toolbox for Guided Analysis of Behavioral Tests in Mice. Front. Behav. Neurosci. 2018, 12, 201. [Google Scholar] [CrossRef] [Green Version]
- Mandwie, M.; Karunia, J.; Niaz, A.; Keay, K.A.; Musumeci, G.; Rennie, C.; McGrath, K.; Al-Badri, G.; Castorina, A. Metformin Treatment Attenuates Brain Inflammation and Rescues PACAP/VIP Neuropeptide Alterations in Mice Fed a High-Fat Diet. Int. J. Mol. Sci. 2021, 22, 13660. [Google Scholar] [CrossRef] [PubMed]
- Crowe, A.R.; Yue, W. Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio. Protoc. 2019, 9, e3465. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Parkinson’s Disease | Buspirone |
|---|---|---|
| 1 | Saline (vehicle) | Saline (vehicle) |
| 2 | Rotenone 10 mg/kg | Saline (vehicle) |
| 3 1 | Saline (vehicle) | Buspirone 1 mg/kg |
| 41 | Saline (vehicle) | Buspirone 3 mg/kg |
| 5 1 | Saline (vehicle) | Buspirone 10 mg/kg |
| 6 | Rotenone 10 mg/kg | Buspirone 1 mg/kg |
| 7 | Rotenone 10 mg/kg | Buspirone 3 mg/kg |
| 8 | Rotenone 10 mg/kg | Buspirone 10 mg/kg |
| Accession # | Gene | Primer Sequence (5′-3′) | Length (bp) |
|---|---|---|---|
| NM_009377.3 | Tyrosine hydroxylase (TH) | Fwd GCCCTACCAAGATCAAACCTAC Rev ATACGAGAGGCATAGTTCCTGA | 93 |
| NM_010020.3 | Dopamine transporter (DAT) | Fwd ATGACATCAAGCAGATGACTGG Rev CACGACCACATACAGAAGGAAG | 95 |
| NM_001131020.1 | Glial fibrillary acidic protein (GFAP) | Fwd GAGATTCGCACTCAATACGAGG Rev CTGCAAACTTAGACCGATACCA | 79 |
| NM_001082960.1 | CD11b | Fwd GAGCAGGGGTCATTCGCTAC Rev GCTGGCTTAGATGCGATGGT | 94 |
| NM_008361.4 | Interleukin-1β (IL-1β) | Fwd GCTACCTGTGTCTTTCCCGT Rev CATCTCGGAGCCTGTAGTGC | 164 |
| NM_007482.3 | Arginase-1 (Arg1) | Fwd ACAAGACAGGGCTCCTTTCAG Rev TTAAAGCCACTGCCGTGTTC | 105 |
| NM_011434.2 | Superoxidase dismutase (SOD1) | Fwd CAATGGTGGTCCATGAGAAACA Rev CCCAGCATTTCCAGTCTTTGTA | 77 |
| NM_001199177.1 | Mitochondrial dynamin like GTPase (OPA1) | Fwd GCCCTTCTCTTGTTAGGTTCAC Rev ACACCTTCCTGTAATGCTTGTC | 88 |
| NM_007540.4 | Brain derived neurotrophic factor (BDNF) | Fwd CGAGTGGGTCACAGCGGCAG Rev GCCCCTGCAGCCTTCCTTGG | 160 |
| NM_001310086.1 | Activity-dependent neuroprotective protein (ADNP) | Fwd GTGACATTGGGTTGGAATACTGT Rev AGGTTTTGTCCGATAGTCCTGA | 149 |
| NM_016989.2 | Pituitary adenylate cyclase-activating polypeptide (PACAP) | Fwd AGGCTTACGATCAGGACGGA Rev CTCCTGTCGGCTGGGTAGTA | 121 |
| NM_053991.1 | Vasoactive intestinal peptide (VIP) | Fwd CCTGGCGATCCTGACACTCT Rev CTGCAGCCTGTCATCCAACC | 100 |
| NM_213557.1 | 18S ribosomal subunit (s18) | Fwd GGCGGAAAATAGCCTTCGCT Rev AGCCCTCTTGGTGAGGTCAA | 101 |
| Antibody | Dilution | Source (Cat. #) |
|---|---|---|
| Tyrosine hydroxylase (TH) | 1:200 (WB) 1:500 (IHC-P) | Abcam (ab112) |
| Dopamine transporter (DAT) | 1:1000 | Abcam (ab128848) |
| Glial fibrillary acidic protein (GFAP) | 1:1000 | Abcam (ab68428) |
| Ionized calcium binding protein (Iba1) | 1:1000 | Sigma Aldrich (SAB2702364) |
| Mitochondrial dynamin like GTPase (OPA1) | 1:1000 | GeneTex (GTX129917) |
| Superoxidase dismutase (SOD1) | 1:1000 | GeneTex (GTX100554) |
| Pituitary adenylate cyclase-activating polypeptide (PACAP) | 1:1000 | GeneTex (GTX37576) |
| Vasoactive intestinal peptide (VIP) | 1:1000 | GeneTex (GTX129461) |
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | 1:1000 | BioRad (VPA00187) |
| Goat anti-Rabbit IgG HRP | 1:10,000 | BioRad (STAR208P) |
| Goat anti-Mouse IgG (H + L)-HRP | 1:10,000 | BioRad (1706516) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas Broome, S.; Castorina, A. The Anxiolytic Drug Buspirone Prevents Rotenone-Induced Toxicity in a Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 1845. https://doi.org/10.3390/ijms23031845
Thomas Broome S, Castorina A. The Anxiolytic Drug Buspirone Prevents Rotenone-Induced Toxicity in a Mouse Model of Parkinson’s Disease. International Journal of Molecular Sciences. 2022; 23(3):1845. https://doi.org/10.3390/ijms23031845
Chicago/Turabian StyleThomas Broome, Sarah, and Alessandro Castorina. 2022. "The Anxiolytic Drug Buspirone Prevents Rotenone-Induced Toxicity in a Mouse Model of Parkinson’s Disease" International Journal of Molecular Sciences 23, no. 3: 1845. https://doi.org/10.3390/ijms23031845