
Mitochondrial Control in Inflammatory Gastrointestinal Diseases
Abstract
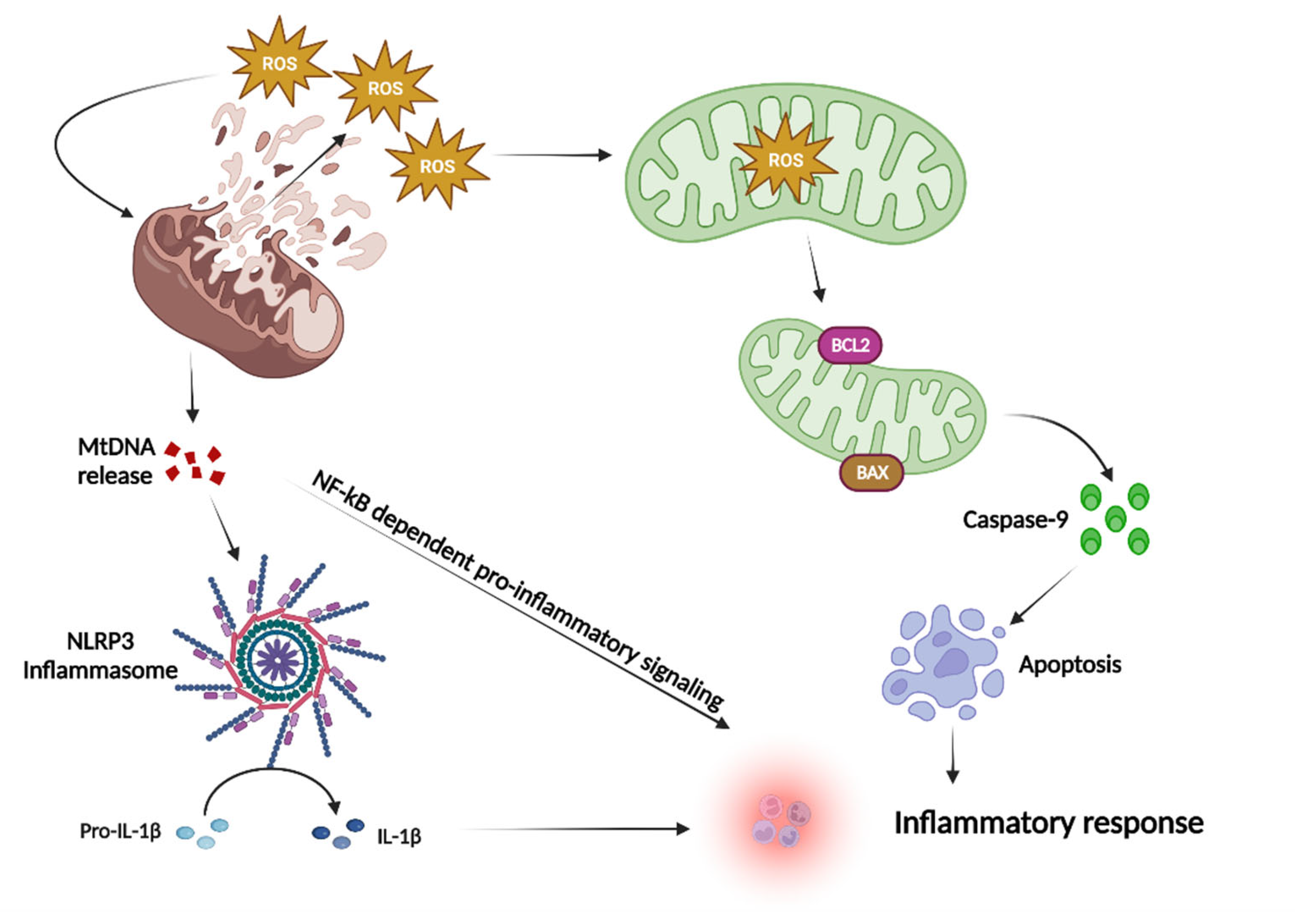
:1. Mitochondria-Mediated Inflammation
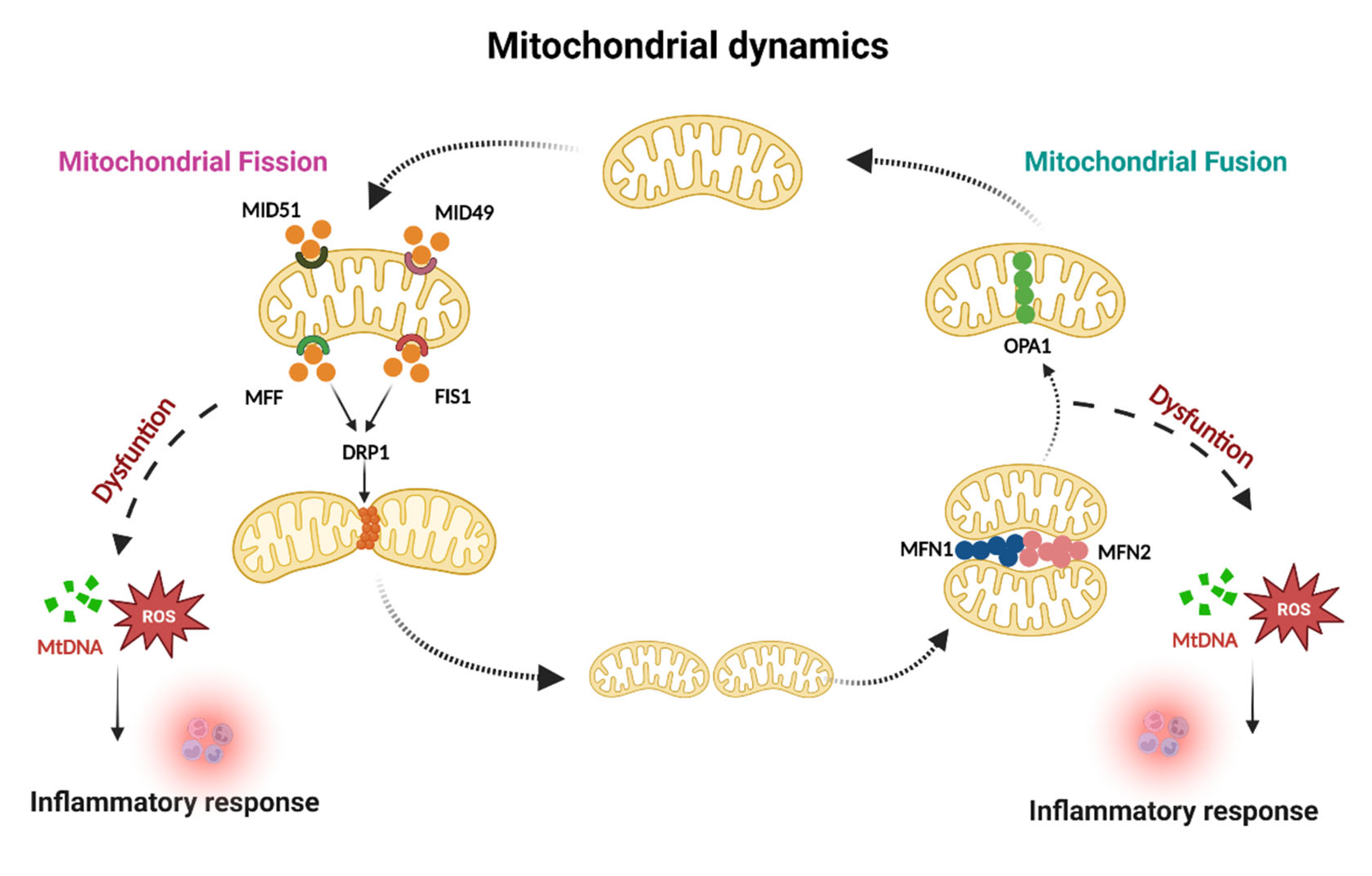
1.1. Mitochondrial Dynamics and Inflammation
1.1.1. Mitochondrial Fusion
1.1.2. Mitochondrial Fission
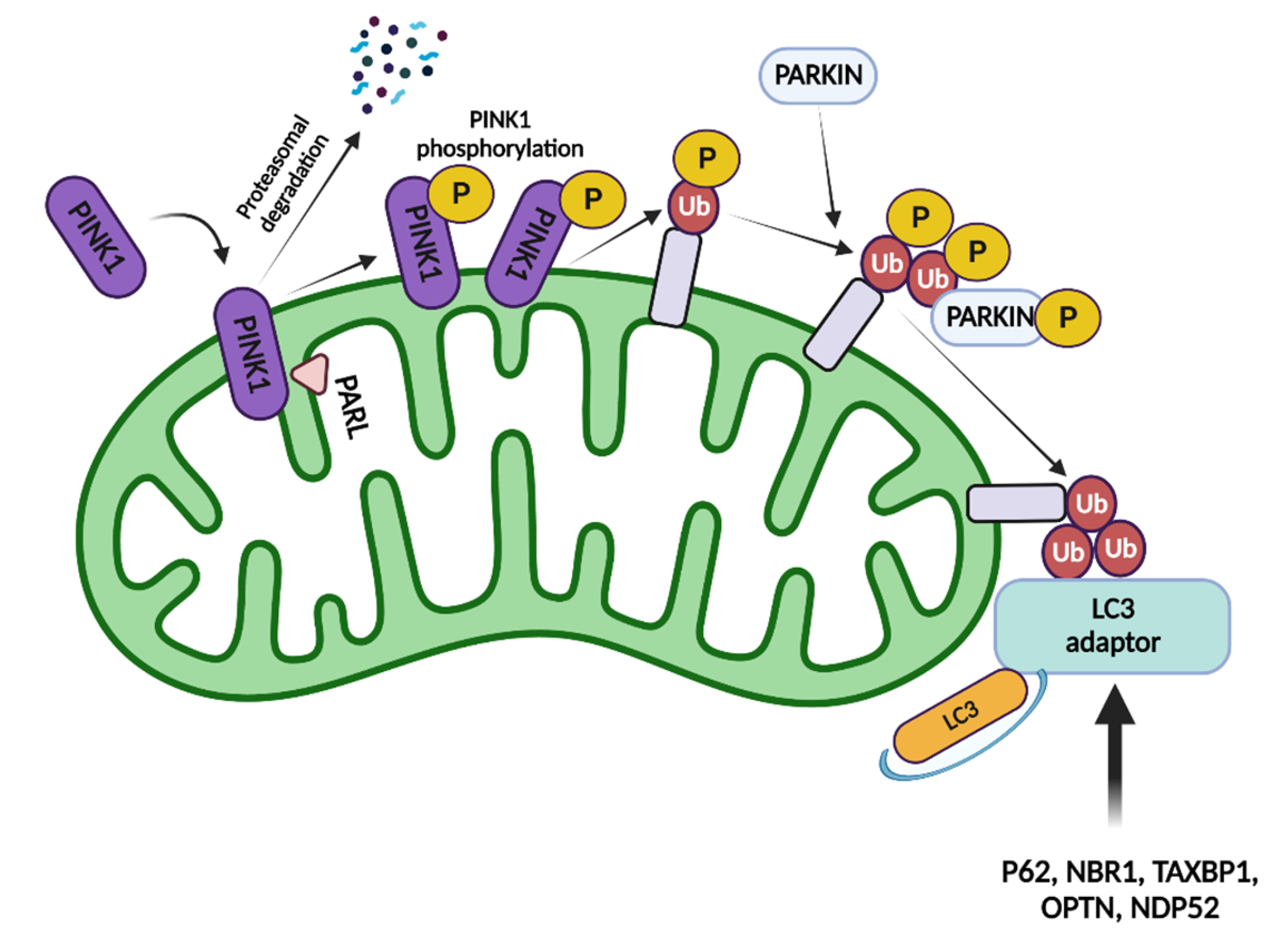
1.2. Mitophagy and Inflammation
1.2.1. Parkin/Pink-Mediated Mitophagy
1.2.2. Nix-Mediated Mitophagy
1.2.3. Mitophagy Dysfunction-Mediated Inflammation
1.3. Mitochondria and ROS
2. Gastrointestinal System Pathophysiology
2.1. Inflammatory Bowel Diseases
2.2. Colorectal Cancer
3. Mitochondrial Control of Inflammation in Gastrointestinal System Pathophysiology
3.1. Mitochondria and Inflammation in IBD
3.1.1. Mitophagy-Inflammation-IBD
3.1.2. ROS-Inflammation-IBD
3.1.3. Mitochondrial Dynamics-Inflammation-IBD
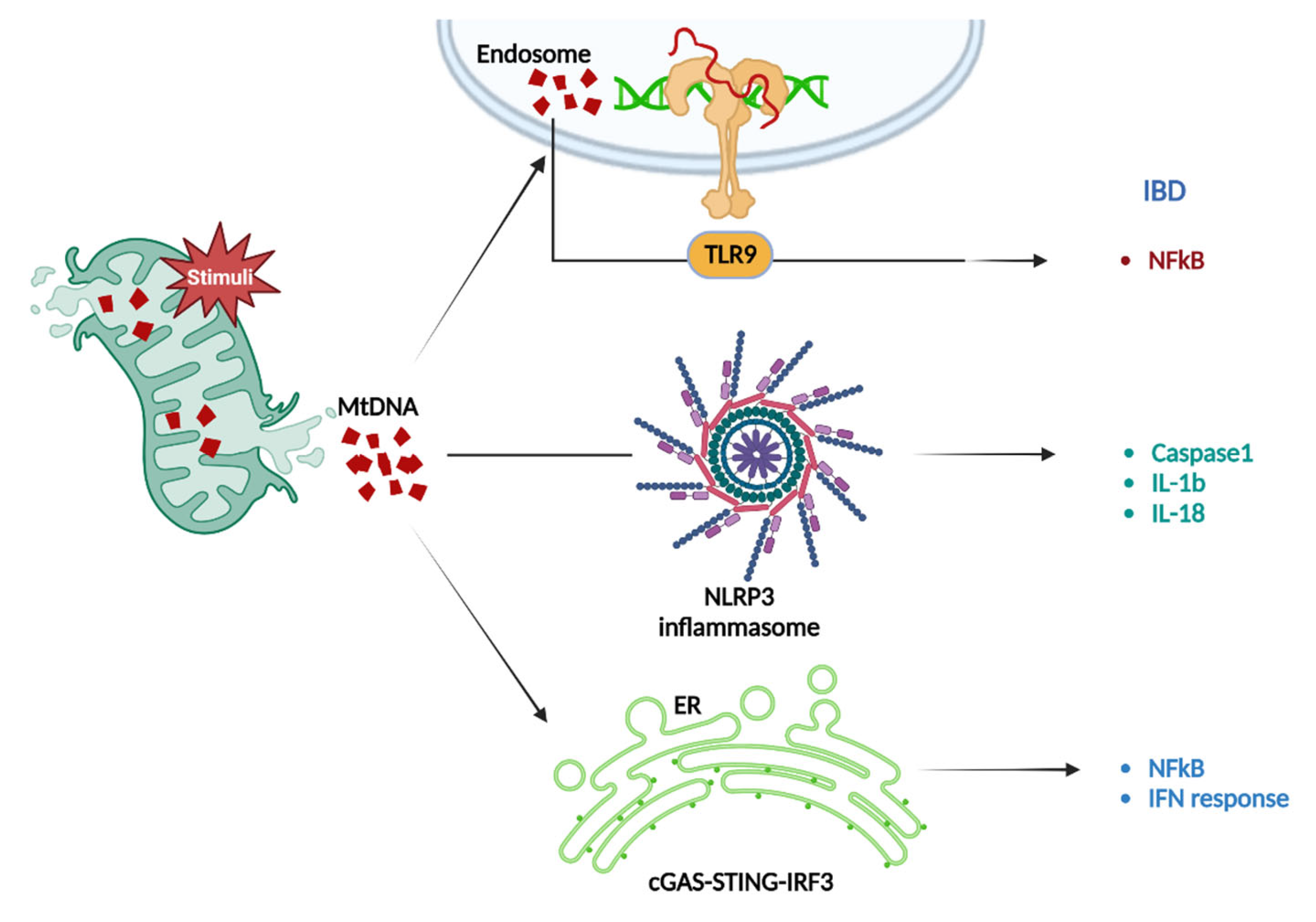
3.1.4. mtDNA-Induced Inflammation in IBD
3.2. Mitochondria and Inflammation in CRC
3.2.1. Mitophagy and Inflammation in CRC
3.2.2. ROS and Inflammation in CRC
3.2.3. Mitochondrial Dynamics and Inflammation in CRC
3.2.4. mtDNA-Mediated Inflammation in CRC
4. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IBD | inflammatory bowel disease |
| CRC | colorectal cancer disease |
| mtDNA | mitochondrial DNA |
| CD | Crohn’s disease |
| DSS | dextran sodium sulfate |
| HSP | heat shock protein |
| IMM | the inner mitochondrial membrane |
| Irgm1 | Immune-related GTPase family M protein |
| COX | cytochrome c oxidase |
| DAMP | damage-associated molecular pattern |
| Drp1 | dynamin-related protein 1 |
| DNBS | dinitrobenzene sulfonic acid |
| OMM | outer mitochondrial membrane |
| Opa1 | optic atrophy 1 |
| ROS | reactive oxygen species |
| TNF-a | tumour necrosis factor alpha |
| IFN-γ | interferon-γ |
| RNS | reactive nitrogen species |
| MID49/51 | mitochondrial dynamics proteins of 49 and 51 kDa |
| FIS1 | fission protein 1 |
| MFF | mitochondrial fission factor |
| DRP1 | dynamin-related protein 1 |
| GED | GTPase effector structural domain |
| Parkin | E3 Ubiquitin-Protein Ligase |
| Pink1 | PTEN Induced Kinase 1 |
| NDP52 | nuclear dot protein 52 |
| OPTN | optineurin |
| USP30 | ubiquitin-specific protease 30 |
| TAX1BP1 | TAX1 bindingprotein 1 |
| NBR1 | neighborof BRCA1 gene protein |
| LIR | LC3-interacting region |
| UBD | ubiquitin-binding domain |
| TBK1 | TANK-binding kinase 1 |
| UC | ulcerative colitis |
| ULK1 | Unc-51-like kinase 1 |
| OXPHOS | oxidative phosphorylation |
| ATP | adenosine triphosphate |
| MRC | mitochondrial respiratory chain |
| mtROS | Mitochondrial ROS |
| IRGM | immunity-related GTPase M |
| LRRK2 | leucine-rich repeat kinase 2 |
| Phb1 | prohibitin 1 |
| NF-κB | nuclear factor-kappa B |
| NRF2 | nuclear factor erythroid 2–related factor 2 |
| TLRs | Toll-like receptors |
| IRF3 | interferon regulatory factor 3 |
| TLR4 | TOLL-like receptor 4 |
| TRAP1 | Tumor necrosis factor type 1 |
References
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529 Pt 1, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Hevler, J.F.; Zenezeni Chiozzi, R.; Cabrera-Orefice, A.; Brandt, U.; Arnold, S.; Heck, A.J.R. Molecular characterization of a complex of apoptosis-inducing factor 1 with cytochrome c oxidase of the mitochondrial respiratory chain. Proc. Natl. Acad. Sci. USA 2021, 118, e2106950118. [Google Scholar] [CrossRef]
- Yoon, Y.G.; Koob, M.D.; Yoo, Y.H. Re-engineering the mitochondrial genomes in mammalian cells. Anat Cell Biol. 2010, 43, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C. Defects in mitochondrial DNA replication and human disease. Crit Rev. Biochem. Mol. Biol. 2012, 47, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Kuhlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 444. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.Y.W.; Wai, T.; Simonsen, A. Quality control of the mitochondrion. Dev. Cell 2021, 56, 881–905. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Luna-Sanchez, M.; Bianchi, P.; Quintana, A. Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: A Pathogen from Within. Int. J. Mol. Sci. 2021, 22, 8523. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division and fusion in metabolism. Curr. Opin. Cell Biol. 2015, 33, 111–118. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.H.; Li, Y.Y.; Jin, J. The essential functions of mitochondrial dynamics in immune cells. Cell Mol. Immunol. 2020, 17, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Madan, S.; Uttekar, B.; Chowdhary, S.; Rikhy, R. Mitochondria Lead the Way: Mitochondrial Dynamics and Function in Cellular Movements in Development and Disease. Front. Cell Dev. Biol. 2021, 9, 781933. [Google Scholar] [CrossRef] [PubMed]
- Serasinghe, M.N.; Chipuk, J.E. Mitochondrial Fission in Human Diseases. Handb. Exp. Pharmacol. 2017, 240, 159–188. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Sedlackova, L.; Korolchuk, V.I. Mitochondrial quality control as a key determinant of cell survival. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tan, J.; Miao, Y.; Zhang, Q. Mitochondrial Dynamics, Mitophagy, and Mitochondria-Endoplasmic Reticulum Contact Sites Crosstalk Under Hypoxia. Front. Cell Dev. Biol. 2022, 10, 848214. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.W.; Han, E.C.; Yoon, J.; Choi, C. The mitochondrial fusion-related proteins Mfn2 and OPA1 are transcriptionally induced during differentiation of bone marrow progenitors to immature dendritic cells. Mol. Cells 2015, 38, 89–94. [Google Scholar] [CrossRef]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Formosa, L.E.; Ryan, M.T. Mitochondrial fusion: Reaching the end of mitofusin’s tether. J. Cell Biol. 2016, 215, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Y.; Li, X.; Zhang, W.; Liu, Z.; Wu, M.; Pan, Q.; Liu, H. Emerging role of transcription factor EB in mitochondrial quality control. Biomed. Pharm. 2020, 128, 110272. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Shutt, T.E. The Role of Impaired Mitochondrial Dynamics in MFN2-Mediated Pathology. Front. Cell Dev. Biol. 2022, 10, 858286. [Google Scholar] [CrossRef] [PubMed]
- Giamogante, F.; Barazzuol, L.; Brini, M.; Cali, T. ER-Mitochondria Contact Sites Reporters: Strengths and Weaknesses of the Available Approaches. Int. J. Mol. Sci. 2020, 21, 8157. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd; Kitsis, R.N. The mitochondrial dynamism-mitophagy-cell death interactome: Multiple roles performed by members of a mitochondrial molecular ensemble. Circ. Res. 2015, 116, 167–182. [Google Scholar] [CrossRef]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Cao, H.; Wang, G.; Lyu, Y.; Sun, X.; An, J.; Wu, Z.; Huang, Q.; Liu, B.; Xing, J. Drp1-mediated mitochondrial fission promotes cell proliferation through crosstalk of p53 and NF-kappaB pathways in hepatocellular carcinoma. Oncotarget 2016, 7, 65001–65011. [Google Scholar] [CrossRef] [PubMed]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Lendahl, U.; Nister, M.; Zhao, J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front. Endocrinol. 2020, 11, 374. [Google Scholar] [CrossRef]
- Otera, H.; Mihara, K. Discovery of the membrane receptor for mitochondrial fission GTPase Drp1. Small GTPases 2011, 2, 167–172. [Google Scholar] [CrossRef]
- Otera, H.; Miyata, N.; Kuge, O.; Mihara, K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J. Cell Biol. 2016, 212, 531–544. [Google Scholar] [CrossRef]
- Ihenacho, U.K.; Meacham, K.A.; Harwig, M.C.; Widlansky, M.E.; Hill, R.B. Mitochondrial Fission Protein 1: Emerging Roles in Organellar Form and Function in Health and Disease. Front. Endocrinol. 2021, 12, 660095. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Kage, F.; Higgs, H.N. Mff oligomerization is required for Drp1 activation and synergy with actin filaments during mitochondrial division. Mol. Biol. Cell 2021, 32, ar5. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef]
- Carinci, M.; Vezzani, B.; Patergnani, S.; Ludewig, P.; Lessmann, K.; Magnus, T.; Casetta, I.; Pugliatti, M.; Pinton, P.; Giorgi, C. Different Roles of Mitochondria in Cell Death and Inflammation: Focusing on Mitochondrial Quality Control in Ischemic Stroke and Reperfusion. Biomedicines 2021, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Liu, Y.; Li, Z.; Zhu, P.; Zhao, M. Mitochondria Related Cell Death Modalities and Disease. Front. Cell Dev. Biol. 2022, 10, 832356. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.L.; Gustafsson, A.B. Mitochondrial autophagy—An essential quality control mechanism for myocardial homeostasis. Circ. J. 2013, 77, 2449–2454. [Google Scholar] [CrossRef]
- Lahiri, V.; Hawkins, W.D.; Klionsky, D.J. Watch What You (Self-) Eat: Autophagic Mechanisms that Modulate Metabolism. Cell Metab. 2019, 29, 803–826. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Jacoupy, M.; Corvol, J.C.; Corti, O. PINK1/Parkin-Dependent Mitochondrial Surveillance: From Pleiotropy to Parkinson’s Disease. Front. Mol. Neurosci. 2017, 10, 120. [Google Scholar] [CrossRef]
- Marongiu, R.; Spencer, B.; Crews, L.; Adame, A.; Patrick, C.; Trejo, M.; Dallapiccola, B.; Valente, E.M.; Masliah, E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J. Neurochem. 2009, 108, 1561–1574. [Google Scholar] [CrossRef]
- Yamada, T.; Dawson, T.M.; Yanagawa, T.; Iijima, M.; Sesaki, H. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 2019, 15, 2012–2018. [Google Scholar] [CrossRef]
- Wauer, T.; Swatek, K.N.; Wagstaff, J.L.; Gladkova, C.; Pruneda, J.N.; Michel, M.A.; Gersch, M.; Johnson, C.M.; Freund, S.M.; Komander, D. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 2015, 34, 307–325. [Google Scholar] [CrossRef]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.R.; Martinez, A.; Lane, J.D.; Mayor, U.; Clague, M.J.; Urbe, S. USP30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. EMBO Rep. 2015, 16, 618–627. [Google Scholar] [CrossRef]
- Bogorodskiy, A.; Okhrimenko, I.; Burkatovskii, D.; Jakobs, P.; Maslov, I.; Gordeliy, V.; Dencher, N.A.; Gensch, T.; Voos, W.; Altschmied, J.; et al. Role of Mitochondrial Protein Import in Age-Related Neurodegenerative and Cardiovascular Diseases. Cells 2021, 10, 3528. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef]
- Ramirez, A.; Old, W.; Selwood, D.L.; Liu, X. Cannabidiol activates PINK1-Parkin-dependent mitophagy and mitochondrial-derived vesicles. Eur. J. Cell Biol. 2022, 101, 151185. [Google Scholar] [CrossRef] [PubMed]
- Vizziello, M.; Borellini, L.; Franco, G.; Ardolino, G. Disruption of Mitochondrial Homeostasis: The Role of PINK1 in Parkinson’s Disease. Cells 2021, 10, 3022. [Google Scholar] [CrossRef] [PubMed]
- Rasool, S.; Soya, N.; Truong, L.; Croteau, N.; Lukacs, G.L.; Trempe, J.F. PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep. 2018, 19, e44981. [Google Scholar] [CrossRef] [PubMed]
- Sauve, V.; Sung, G.; MacDougall, E.J.; Kozlov, G.; Saran, A.; Fakih, R.; Fon, E.A.; Gehring, K. Structural basis for feedforward control in the PINK1/Parkin pathway. EMBO J. 2022, 41, e109460. [Google Scholar] [CrossRef]
- Madruga, E.; Maestro, I.; Martinez, A. Mitophagy Modulation, a New Player in the Race against ALS. Int. J. Mol. Sci. 2021, 22, 740. [Google Scholar] [CrossRef] [PubMed]
- Wesch, N.; Kirkin, V.; Rogov, V.V. Atg8-Family Proteins-Structural Features and Molecular Interactions in Autophagy and Beyond. Cells 2020, 9, 2008. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herhaus, L. TBK1 (TANK-binding kinase 1)-mediated regulation of autophagy in health and disease. Matrix Biol. 2021, 100–101, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ma, Q.; Siraj, S.; Ney, P.A.; Liu, J.; Liao, X.; Yuan, Y.; Li, W.; Liu, L.; Chen, Q. Nix-mediated mitophagy regulates platelet activation and life span. Blood Adv. 2019, 3, 2342–2354. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Ray, R.; Chen, G.; Vande Velde, C.; Cizeau, J.; Park, J.H.; Reed, J.C.; Gietz, R.D.; Greenberg, A.H. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J. Biol. Chem. 2000, 275, 1439–1448. [Google Scholar] [CrossRef]
- Kollek, M.; Muller, A.; Egle, A.; Erlacher, M. Bcl-2 proteins in development, health, and disease of the hematopoietic system. FEBS J. 2016, 283, 2779–2810. [Google Scholar] [CrossRef]
- Kundu, M.; Lindsten, T.; Yang, C.Y.; Wu, J.; Zhao, F.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.A.; Ameenudeen, S.; Kumar, A.; Hemalatha, S.; Ahmed, N.; Ali, N.; AlAsmari, A.F.; Aashique, M.; Waseem, M. Emerging Role of Mitophagy in Inflammatory Diseases: Cellular and Molecular Episodes. Curr. Pharm. Des. 2020, 26, 485–491. [Google Scholar] [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Gkikas, I.; Palikaras, K.; Tavernarakis, N. The Role of Mitophagy in Innate Immunity. Front. Immunol. 2018, 9, 1283. [Google Scholar] [CrossRef]
- Esteban-Martinez, L.; Sierra-Filardi, E.; McGreal, R.S.; Salazar-Roa, M.; Marino, G.; Seco, E.; Durand, S.; Enot, D.; Grana, O.; Malumbres, M.; et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017, 36, 1688–1706. [Google Scholar] [CrossRef]
- Baldanta, S.; Fernandez-Escobar, M.; Acin-Perez, R.; Albert, M.; Camafeita, E.; Jorge, I.; Vazquez, J.; Enriquez, J.A.; Guerra, S. ISG15 governs mitochondrial function in macrophages following vaccinia virus infection. PLoS Pathog. 2017, 13, e1006651. [Google Scholar] [CrossRef] [PubMed]
- Almansa-Ordonez, A.; Bellido, R.; Vassena, R.; Barragan, M.; Zambelli, F. Oxidative Stress in Reproduction: A Mitochondrial Perspective. Biology 2020, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg Species Apex 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Shields, H.J.; Traa, A.; Van Raamsdonk, J.M. Beneficial and Detrimental Effects of Reactive Oxygen Species on Lifespan: A Comprehensive Review of Comparative and Experimental Studies. Front. Cell Dev. Biol. 2021, 9, 628157. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.A.; Perez de la Lastra, J.M.; Plou, F.J.; Perez-Lebena, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta 2016, 1862, 576–591. [Google Scholar] [CrossRef]
- Li, H.; Luo, Y.F.; Wang, Y.S.; Yang, Q.; Xiao, Y.L.; Cai, H.R.; Xie, C.M. Using ROS as a Second Messenger, NADPH Oxidase 2 Mediates Macrophage Senescence via Interaction with NF-kappaB during Pseudomonas aeruginosa Infection. Oxid. Med. Cell Longev. 2018, 2018, 9741838. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Wang, X.; Du, J.; Gu, Z.; Zhao, Y. Reactive Oxygen Species-Regulating Strategies Based on Nanomaterials for Disease Treatment. Adv. Sci. 2021, 8, 2002797. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect Biol. 2013, 5, a008714. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Letai, A. Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci. 2009, 122, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, Y.; Hu, Q.; Bai, X.C.; Huang, W.; Yan, C.; Scheres, S.H.; Shi, Y. Atomic structure of the apoptosome: Mechanism of cytochrome c- and dATP-mediated activation of Apaf-1. Genes Dev. 2015, 29, 2349–2361. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Yang, R.; Hao, J.; Wang, J.; Sun, C.; Fesik, S.W.; Wu, J.C.; Tomaselli, K.J.; Armstrong, R.C. Regulation of the Apaf-1/caspase-9 apoptosome by caspase-3 and XIAP. J. Biol. Chem. 2003, 278, 8091–8098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cande, C.; Cecconi, F.; Dessen, P.; Kroemer, G. Apoptosis-inducing factor (AIF): Key to the conserved caspase-independent pathways of cell death? J. Cell Sci. 2002, 115, 4727–4734. [Google Scholar] [CrossRef]
- Kieffer, D.A.; Martin, R.J.; Adams, S.H. Impact of Dietary Fibers on Nutrient Management and Detoxification Organs: Gut, Liver, and Kidneys. Adv. Nutr. 2016, 7, 1111–1121. [Google Scholar] [CrossRef]
- Avramidou, M.; Angst, F.; Angst, J.; Aeschlimann, A.; Rossler, W.; Schnyder, U. Epidemiology of gastrointestinal symptoms in young and middle-aged Swiss adults: Prevalences and comorbidities in a longitudinal population cohort over 28 years. BMC Gastroenterol. 2018, 18, 21. [Google Scholar] [CrossRef]
- Lee, S.H.; Kwon, J.E.; Cho, M.L. Immunological pathogenesis of inflammatory bowel disease. Intestig. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Barbara, G.; Barbaro, M.R.; Fuschi, D.; Palombo, M.; Falangone, F.; Cremon, C.; Marasco, G.; Stanghellini, V. Inflammatory and Microbiota-Related Regulation of the Intestinal Epithelial Barrier. Front. Nutr. 2021, 8, 718356. [Google Scholar] [CrossRef]
- Hnatyszyn, A.; Hryhorowicz, S.; Kaczmarek-Rys, M.; Lis, E.; Slomski, R.; Scott, R.J.; Plawski, A. Colorectal carcinoma in the course of inflammatory bowel diseases. Hered Cancer Clin. Pract 2019, 17, 18. [Google Scholar] [CrossRef]
- Fournier, B.M.; Parkos, C.A. The role of neutrophils during intestinal inflammation. Mucosal. Immunol. 2012, 5, 354–366. [Google Scholar] [CrossRef]
- Yalchin, M.; Baker, A.M.; Graham, T.A.; Hart, A. Predicting Colorectal Cancer Occurrence in IBD. Cancers 2021, 13, 2908. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Pang, X.; Li, L.; Zeng, Z.; Chen, M.; Zhang, S. Ubiquitin-specific proteases in inflammatory bowel disease-related signalling pathway regulation. Cell Death Dis. 2022, 13, 139. [Google Scholar] [CrossRef]
- Lin, Y.Y.; Zhao, J.M.; Ji, Y.J.; Ma, Z.; Zheng, H.D.; Huang, Y.; Cui, Y.H.; Lu, Y.; Wu, H.G. Typical ulcerative colitis treated by herbs-partitioned moxibustion: A case report. World J. Clin. Cases 2020, 8, 1515–1524. [Google Scholar] [CrossRef]
- Choi, J.K.; Kim, D.W.; Shin, S.Y.; Park, E.C.; Kang, J.G. Effect of Ulcerative Colitis on Incidence of Colorectal Cancer: Results from the Nationwide Population-Based Cohort Study (2003–2013). J. Cancer 2016, 7, 681–686. [Google Scholar] [CrossRef]
- Queliza, K.; Ihekweazu, F.D.; Schady, D.; Jensen, C.; Kellermayer, R. Granulomatous Upper Gastrointestinal Inflammation in Pediatric Ulcerative Colitis. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Fousekis, F.S.; Katsanos, K.H.; Theopistos, V.I.; Baltayiannis, G.; Kosmidou, M.; Glantzounis, G.; Christou, L.; Tsianos, E.V.; Christodoulou, D.K. Hepatobiliary and pancreatic manifestations in inflammatory bowel diseases: A referral center study. BMC Gastroenterol. 2019, 19, 48. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Watts, D.A.; Satsangi, J. The genetic jigsaw of inflammatory bowel disease. Gut 2002, 50 (Suppl. 3), III31–III36. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Environmental risk factors for inflammatory bowel diseases: A review. Dig. Dis. Sci. 2015, 60, 290–298. [Google Scholar] [CrossRef]
- Cui, G.; Yuan, A. A Systematic Review of Epidemiology and Risk Factors Associated With Chinese Inflammatory Bowel Disease. Front. Med. 2018, 5, 183. [Google Scholar] [CrossRef]
- Wei, Y.; Lu, C.; Chen, J.; Cui, G.; Wang, L.; Yu, T.; Yang, Y.; Wu, W.; Ding, Y.; Li, L.; et al. High salt diet stimulates gut Th17 response and exacerbates TNBS-induced colitis in mice. Oncotarget 2017, 8, 70–82. [Google Scholar] [CrossRef]
- Lagishetty, V.; Misharin, A.V.; Liu, N.Q.; Lisse, T.S.; Chun, R.F.; Ouyang, Y.; McLachlan, S.M.; Adams, J.S.; Hewison, M. Vitamin D deficiency in mice impairs colonic antibacterial activity and predisposes to colitis. Endocrinology 2010, 151, 2423–2432. [Google Scholar] [CrossRef]
- Zhou, J.; Zheng, R.; Zhang, S.; Zeng, H.; Wang, S.; Chen, R.; Sun, K.; Li, M.; Gu, J.; Zhuang, G.; et al. Colorectal cancer burden and trends: Comparison between China and major burden countries in the world. Chin. J. Cancer Res. 2021, 33, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Emilsson, L.; Roelstraete, B.; Ludvigsson, J.F. Risk of colorectal cancer in first degree relatives of patients with colorectal polyps: Nationwide case-control study in Sweden. BMJ 2021, 373, n877. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Holtedahl, K.; Borgquist, L.; Donker, G.A.; Buntinx, F.; Weller, D.; Campbell, C.; Mansson, J.; Hammersley, V.; Braaten, T.; Parajuli, R. Symptoms and signs of colorectal cancer, with differences between proximal and distal colon cancer: A prospective cohort study of diagnostic accuracy in primary care. BMC Fam. Pract 2021, 22, 148. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Castelli, G. Colorectal cancer: Genetic abnormalities, tumor progression, tumor heterogeneity, clonal evolution and tumor-initiating cells. Med. Sci. 2018, 6, 31. [Google Scholar] [CrossRef]
- Park, Y.; Lee, J.; Oh, J.H.; Shin, A.; Kim, J. Dietary patterns and colorectal cancer risk in a Korean population: A case-control study. Medicine 2016, 95, e3759. [Google Scholar] [CrossRef]
- Schepens, M.A.; Schonewille, A.J.; Vink, C.; van Schothorst, E.M.; Kramer, E.; Hendriks, T.; Brummer, R.J.; Keijer, J.; van der Meer, R.; Bovee-Oudenhoven, I.M. Supplemental calcium attenuates the colitis-related increase in diarrhea, intestinal permeability, and extracellular matrix breakdown in HLA-B27 transgenic rats. J. Nutr. 2009, 139, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Shaw, E.; Farris, M.S.; Stone, C.R.; Derksen, J.W.G.; Johnson, R.; Hilsden, R.J.; Friedenreich, C.M.; Brenner, D.R. Effects of physical activity on colorectal cancer risk among family history and body mass index subgroups: A systematic review and meta-analysis. BMC Cancer 2018, 18, 71. [Google Scholar] [CrossRef]
- Ho, G.T.; Theiss, A.L. Mitochondria and Inflammatory Bowel Diseases: Toward a Stratified Therapeutic Intervention. Annu Rev. Physiol. 2022, 84, 435–459. [Google Scholar] [CrossRef]
- Xia, B.; Crusius, J.; Meuwissen, S.; Pe?a, A. Inflammatory bowel disease: Definition, epidemiology, etiologic aspects, and immunogenetic studies. World J. Gastroenterol. 1998, 4, 446–458. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020, 16, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Stappenbeck, T.S.; Rioux, J.D.; Mizoguchi, A.; Saitoh, T.; Huett, A.; Darfeuille-Michaud, A.; Wileman, T.; Mizushima, N.; Carding, S.; Akira, S.; et al. Crohn disease: A current perspective on genetics, autophagy and immunity. Autophagy 2011, 7, 355–374. [Google Scholar] [CrossRef]
- Novak, E.A.; Mollen, K.P. Mitochondrial dysfunction in inflammatory bowel disease. Front. Cell Dev. Biol. 2015, 3, 62. [Google Scholar] [CrossRef]
- Okamoto, K.; Kondo-Okamoto, N.; Ohsumi, Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell 2009, 17, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Fracchiolla, D. Activation and targeting of ATG8 protein lipidation. Cell Discov. 2020, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, R.; Loebbermann, J.; Nakaya, H.I.; Khan, N.; Ma, H.; Gama, L.; Machiah, D.K.; Lawson, B.; Hakimpour, P.; Wang, Y.C.; et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature 2016, 531, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Kabi, A.; Nickerson, K.P.; Homer, C.R.; McDonald, C. Digesting the genetics of inflammatory bowel disease: Insights from studies of autophagy risk genes. Inflamm. Bowel. Dis. 2012, 18, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gulati, A.S.; Cantillana, V.; Henry, S.C.; Schmidt, E.A.; Daniell, X.; Grossniklaus, E.; Schoenborn, A.A.; Sartor, R.B.; Taylor, G.A. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiology. Gastrointest. Liver Physiol. 2013, 305, G573–G584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zheng, L.; McGovern, D.P.; Hamill, A.M.; Ichikawa, R.; Kanazawa, Y.; Luu, J.; Kumagai, K.; Cilluffo, M.; Fukata, M.; et al. Myeloid ATG16L1 Facilitates Host-Bacteria Interactions in Maintaining Intestinal Homeostasis. J. Immunol. 2017, 198, 2133–2146. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, C.; Yu, M.; Cui, J.; Liu, L.; Xu, Z. ULK1 and JNK are involved in mitophagy incurred by LRRK2 G2019S expression. Protein Cell 2013, 4, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Zhang, F.; Wang, W.; Zhao, W.; Zhou, J.; Feng, Y.; Wu, J.; Li, M.; Bai, X.; Zeng, Z.; et al. Heat Shock Transcription Factor 2 Promotes Mitophagy of Intestinal Epithelial Cells Through PARL/PINK1/Parkin Pathway in Ulcerative Colitis. Front. Pharmacol. 2022, 13, 893426. [Google Scholar] [CrossRef] [PubMed]
- Vincent, G.; Novak, E.A.; Siow, V.S.; Cunningham, K.E.; Griffith, B.D.; Comerford, T.E.; Mentrup, H.L.; Stolz, D.B.; Loughran, P.; Ranganathan, S.; et al. Nix-Mediated Mitophagy Modulates Mitochondrial Damage During Intestinal Inflammation. Antioxid. Redox Signal. 2020, 33, 1–19. [Google Scholar] [CrossRef]
- Signorile, A.; Sgaramella, G.; Bellomo, F.; De Rasmo, D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Theiss, A.L.; Jenkins, A.K.; Okoro, N.I.; Klapproth, J.M.; Merlin, D.; Sitaraman, S.V. Prohibitin inhibits tumor necrosis factor alpha-induced nuclear factor-kappa B nuclear translocation via the novel mechanism of decreasing importin alpha3 expression. Mol. Biol. Cell 2009, 20, 4412–4423. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.N.; Panopoulos, M.; Neumann, W.L.; Turner, K.; Cantarel, B.L.; Thompson-Snipes, L.; Dassopoulos, T.; Feagins, L.A.; Souza, R.F.; Mills, J.C.; et al. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 2020, 69, 1928–1938. [Google Scholar] [CrossRef]
- Theiss, A.L.; Vijay-Kumar, M.; Obertone, T.S.; Jones, D.P.; Hansen, J.M.; Gewirtz, A.T.; Merlin, D.; Sitaraman, S.V. Prohibitin is a novel regulator of antioxidant response that attenuates colonic inflammation in mice. Gastroenterology 2009, 137, 199–208.e6. [Google Scholar] [CrossRef]
- Gierynska, M.; Szulc-Dabrowska, L.; Struzik, J.; Mielcarska, M.B.; Gregorczyk-Zboroch, K.P. Integrity of the Intestinal Barrier: The Involvement of Epithelial Cells and Microbiota-A Mutual Relationship. Animals 2022, 12, 145. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.C.; Bevins, C.L. Paneth cells: Maestros of the small intestinal crypts. Annu. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef]
- Nedelsky, N.; Kuballa, P.; Castoreno, A.B.; Xavier, R.J. Inflammatory Bowel Disease at the Intersection of Autophagy and Immunity: Insights from Human Genetics. In Molecular Genetics of Inflammatory Bowel Disease; Hedin, C., Rioux, J.D., D’Amato, M., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 305–328. [Google Scholar] [CrossRef]
- Matsuzawa-Ishimoto, Y.; Shono, Y.; Gomez, L.E.; Hubbard-Lucey, V.M.; Cammer, M.; Neil, J.; Dewan, M.Z.; Lieberman, S.R.; Lazrak, A.; Marinis, J.M.; et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 2017, 214, 3687–3705. [Google Scholar] [CrossRef]
- Vona, R.; Pallotta, L.; Cappelletti, M.; Severi, C.; Matarrese, P. The Impact of Oxidative Stress in Human Pathology: Focus on Gastrointestinal Disorders. Antioxidants 2021, 10, 201. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef]
- Krzystek-Korpacka, M.; Kempinski, R.; Bromke, M.A.; Neubauer, K. Oxidative Stress Markers in Inflammatory Bowel Diseases: Systematic Review. Diagnostics 2020, 10, 601. [Google Scholar] [CrossRef] [PubMed]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Moghadam, Z.M.; Henneke, P.; Kolter, J. From Flies to Men: ROS and the NADPH Oxidase in Phagocytes. Front. Cell Dev. Biol. 2021, 9, 628991. [Google Scholar] [CrossRef]
- Swanson, P.A., 2nd; Kumar, A.; Samarin, S.; Vijay-Kumar, M.; Kundu, K.; Murthy, N.; Hansen, J.; Nusrat, A.; Neish, A.S. Enteric commensal bacteria potentiate epithelial restitution via reactive oxygen species-mediated inactivation of focal adhesion kinase phosphatases. Proc. Natl. Acad. Sci. USA 2011, 108, 8803–8808. [Google Scholar] [CrossRef] [PubMed]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid Med. Cell Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Bannaga, A.S.; Selinger, C.P. Inflammatory bowel disease and anxiety: Links, risks, and challenges faced. Clin. Exp. Gastroenterol. 2015, 8, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, P.K.; Singh, K.; Singh, N.; Jaggi, A.S. A review on chemical-induced inflammatory bowel disease models in rodents. Korean J. Physiol. Pharmacol. 2014, 18, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.; Kim, H.S.; Kwon, T.H.; Palikhe, A.; Zaw, T.S.; Jeong, J.H.; Sohn, U.D. Anti-inflammatory Effects of Flavonoids on TNBS-induced Colitis of Rats. Korean J. Physiol. Pharmacol. 2015, 19, 43–50. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed]
- Zielinska, A.K.; Salaga, M.; Siwinski, P.; Wlodarczyk, M.; Dziki, A.; Fichna, J. Oxidative Stress Does Not Influence Subjective Pain Sensation in Inflammatory Bowel Disease Patients. Antioxidants 2021, 10, 1237. [Google Scholar] [CrossRef] [PubMed]
- Piechota-Polanczyk, A.; Fichna, J. Review article: The role of oxidative stress in pathogenesis and treatment of inflammatory bowel diseases. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 605–620. [Google Scholar] [CrossRef]
- Wu, X.; Huang, L.; Liu, J. Relationship between oxidative stress and nuclear factor-erythroid-2-related factor 2 signaling in diabetic cardiomyopathy (Review). Exp. Ther Med. 2021, 22, 678. [Google Scholar] [CrossRef]
- Kikuchi, N.; Ishii, Y.; Morishima, Y.; Yageta, Y.; Haraguchi, N.; Itoh, K.; Yamamoto, M.; Hizawa, N. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir Res. 2010, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Liu, X.; Luo, Y.; Jiang, S.; Liu, H.; You, F.; Zheng, C.; Wu, J. Apoptosis Exerts a Vital Role in the Treatment of Colitis-Associated Cancer by Herbal Medicine. Front. Pharmacol. 2020, 11, 438. [Google Scholar] [CrossRef]
- Yang, N.; Xia, Z.; Shao, N.; Li, B.; Xue, L.; Peng, Y.; Zhi, F.; Yang, Y. Carnosic acid prevents dextran sulfate sodium-induced acute colitis associated with the regulation of the Keap1/Nrf2 pathway. Sci. Rep. 2017, 7, 11036. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; de la Mata, M.; Cotan, D.; Oropesa-Avila, M.; de Lavera, I.; Alvarez-Cordoba, M.; Luzon-Hidalgo, R.; Sanchez-Alcazar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Thai, P.N.; Lu, S.; Pu, J.; Bers, D.M. Intrafibrillar and perinuclear mitochondrial heterogeneity in adult cardiac myocytes. J. Mol. Cell Cardiol 2019, 136, 72–84. [Google Scholar] [CrossRef]
- Li, D.; Yang, S.; Xing, Y.; Pan, L.; Zhao, R.; Zhao, Y.; Liu, L.; Wu, M. Novel Insights and Current Evidence for Mechanisms of Atherosclerosis: Mitochondrial Dynamics as a Potential Therapeutic Target. Front. Cell Dev. Biol. 2021, 9, 673839. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Siracusa, R.; Cordaro, M.; Peritore, A.F.; Gugliandolo, E.; Mancuso, G.; Midiri, A.; Di Paola, R.; Cuzzocrea, S. Therapeutic potential of dinitrobenzene sulfonic acid (DNBS)-induced colitis in mice by targeting IL-1beta and IL-18. Biochem. Pharmacol. 2018, 155, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Klos, P.; Dabravolski, S.A. The Role of Mitochondria Dysfunction in Inflammatory Bowel Diseases and Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 11673. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Qvit, N.; Su, Y.C.; Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef]
- Mancini, N.L.; Goudie, L.; Xu, W.; Sabouny, R.; Rajeev, S.; Wang, A.; Esquerre, N.; Al Rajabi, A.; Jayme, T.S.; van Tilburg Bernandes, E.; et al. Perturbed Mitochondrial Dynamics Is a Novel Feature of Colitis That Can Be Targeted to Lessen Disease. Cell Mol. Gastroenterol. Hepatol. 2020, 10, 287–307. [Google Scholar] [CrossRef]
- Cunningham, K.E.; Vincent, G.; Sodhi, C.P.; Novak, E.A.; Ranganathan, S.; Egan, C.E.; Stolz, D.B.; Rogers, M.B.; Firek, B.; Morowitz, M.J.; et al. Peroxisome Proliferator-activated Receptor-gamma Coactivator 1-alpha (PGC1alpha) Protects against Experimental Murine Colitis. J. Biol. Chem. 2016, 291, 10184–10200. [Google Scholar] [CrossRef]
- Castejon, M.L.; Alarcon-de-la-Lastra, C.; Rosillo, M.A.; Montoya, T.; Fernandez-Bolanos, J.G.; Gonzalez-Benjumea, A.; Sanchez-Hidalgo, M. A New Peracetylated Oleuropein Derivative Ameliorates JoInt. Inflammation and Destruction in a Murine Collagen-Induced Arthritis Model via Activation of the Nrf-2/Ho-1 Antioxidant Pathway and Suppression of MAPKs and NF-kappaB Activation. Nutrients 2021, 13, 311. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Tsukamoto, H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology 2016, 150, 1704–1709. [Google Scholar] [CrossRef]
- Boyapati, R.K.; Dorward, D.A.; Tamborska, A.; Kalla, R.; Ventham, N.T.; Doherty, M.K.; Whitfield, P.D.; Gray, M.; Loane, J.; Rossi, A.G.; et al. Mitochondrial DNA Is a Pro-Inflammatory Damage-Associated Molecular Pattern Released During Active IBD. Inflamm. Bowel. Dis. 2018, 24, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef] [PubMed]
- Vrablicova, Z.; Tomova, K.; Tothova, L.; Babickova, J.; Gromova, B.; Konecna, B.; Liptak, R.; Hlavaty, T.; Gardlik, R. Nuclear and Mitochondrial Circulating Cell-Free DNA Is Increased in Patients With Inflammatory Bowel Disease in Clinical Remission. Front. Med. 2020, 7, 593316. [Google Scholar] [CrossRef]
- Boyapati, R.K.; Rossi, A.G.; Satsangi, J.; Ho, G.T. Gut mucosal DAMPs in IBD: From mechanisms to therapeutic implications. Mucosal. Immunol. 2016, 9, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Carlos, D.; Costa, F.R.; Pereira, C.A.; Rocha, F.A.; Yaochite, J.N.; Oliveira, G.G.; Carneiro, F.S.; Tostes, R.C.; Ramos, S.G.; Zamboni, D.S.; et al. Mitochondrial DNA Activates the NLRP3 Inflammasome and Predisposes to Type 1 Diabetes in Murine Model. Front. Immunol. 2017, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef]
- Onody, A.; Veres-Szekely, A.; Pap, D.; Rokonay, R.; Szebeni, B.; Sziksz, E.; Oswald, F.; Veres, G.; Cseh, A.; Szabo, A.J.; et al. Interleukin-24 regulates mucosal remodeling in inflammatory bowel diseases. J. Transl. Med. 2021, 19, 237. [Google Scholar] [CrossRef]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2022. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Zhong, M.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020, 52, 475–486.e475. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Dagda, R.K.; Chu, C.T. Monitoring mitophagy in neuronal cell cultures. Methods Mol. Biol. 2011, 793, 325–339. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J.; Liu, C.; Wang, Q.; Yan, J.; Hui, L.; Jia, Q.; Shan, H.; Tao, L.; Zhang, M. The Role of Mitophagy in Regulating Cell Death. Oxid Med. Cell Longev. 2021, 2021, 6617256. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Kitagishi, Y.; Kobayashi, M. Function and characteristics of PINK1 in mitochondria. Oxid Med. Cell Longev. 2013, 2013, 601587. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Yin, K.; Lee, J.; Liu, Z.; Kim, H.; Martin, D.R.; Wu, D.; Liu, M.; Xue, X. Mitophagy protein PINK1 suppresses colon tumor growth by metabolic reprogramming via p53 activation and reducing acetyl-CoA production. Cell Death Differ. 2021, 28, 2421–2435. [Google Scholar] [CrossRef]
- Lucafo, M.; Curci, D.; Franzin, M.; Decorti, G.; Stocco, G. Inflammatory Bowel Disease and Risk of Colorectal Cancer: An Overview From Pathophysiology to Pharmacological Prevention. Front. Pharmacol. 2021, 12, 772101. [Google Scholar] [CrossRef] [PubMed]
- da Silva-Camargo, C.C.V.; Svoboda Baldin, R.K.; Costacurta Polli, N.L.; Agostinho, A.P.; Olandosk, M.; de Noronha, L.; Sotomaior, V.S. Parkin protein expression and its impact on survival of patients with advanced colorectal cancer. Cancer Biol. Med. 2018, 15, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Magaji, B.A.; Moy, F.M.; Roslani, A.C.; Law, C.W. Survival rates and predictors of survival among colorectal cancer patients in a Malaysian tertiary hospital. BMC Cancer 2017, 17, 339. [Google Scholar] [CrossRef]
- He, L.; Gu, K. Tanshinone IIA regulates colorectal cancer apoptosis via attenuation of Parkinmediated mitophagy by suppressing AMPK/Skp2 pathways. Mol. Med. Rep. 2018, 18, 1692–1703. [Google Scholar] [CrossRef]
- Liyanagamage, D.; Martinus, R.D. Role of Mitochondrial Stress Protein HSP60 in Diabetes-Induced Neuroinflammation. Mediators Inflamm. 2020, 2020, 8073516. [Google Scholar] [CrossRef]
- Inigo, J.R.; Chandra, D. The mitochondrial unfolded protein response (UPR(mt)): Shielding against toxicity to mitochondria in cancer. J. Hematol. Oncol. 2022, 15, 98. [Google Scholar] [CrossRef]
- Guo, J.; Zhu, S.; Deng, H.; Xu, R. HSP60-knockdown suppresses proliferation in colorectal cancer cells via activating the adenine/AMPK/mTOR signaling pathway. Oncol. Lett. 2021, 22, 630. [Google Scholar] [CrossRef]
- Vabulas, R.M.; Ahmad-Nejad, P.; da Costa, C.; Miethke, T.; Kirschning, C.J.; Hacker, H.; Wagner, H. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J. Biol. Chem. 2001, 276, 31332–31339. [Google Scholar] [CrossRef]
- Kim, H.; Yang, J.; Kim, M.J.; Choi, S.; Chung, J.R.; Kim, J.M.; Yoo, Y.H.; Chung, J.; Koh, H. Tumor Necrosis Factor Receptor-associated Protein 1 (TRAP1) Mutation and TRAP1 Inhibitor Gamitrinib-triphenylphosphonium (G-TPP) Induce a Forkhead Box O (FOXO)-dependent Cell Protective Signal from Mitochondria. J. Biol. Chem. 2016, 291, 1841–1853. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Lai, L.A.; Brentnall, T.A.; Pan, S. Biomarkers for colitis-associated colorectal cancer. World J. Gastroenterol. 2016, 22, 7882–7891. [Google Scholar] [CrossRef] [PubMed]
- Purushottam Dharaskar, S.; Paithankar, K.; Kanugovi Vijayavittal, A.; Shabbir Kara, H.; Amere Subbarao, S. Mitochondrial chaperone, TRAP1 modulates mitochondrial dynamics and promotes tumor metastasis. Mitochondrion 2020, 54, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Wang, X.; Gan, S.; Tang, X.; Kang, X.; Zhu, S. The Mitochondrial Chaperone TRAP1 as a Candidate Target of Oncotherapy. Front. Oncol. 2020, 10, 585047. [Google Scholar] [CrossRef]
- Matassa, D.S.; Agliarulo, I.; Avolio, R.; Landriscina, M.; Esposito, F. TRAP1 Regulation of Cancer Metabolism: Dual Role as Oncogene or Tumor Suppressor. Genes 2018, 9, 195. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- De Almeida, C.V.; Taddei, A.; Amedei, A. The controversial role of Enterococcus faecalis in colorectal cancer. Therap. Adv. Gastroenterol. 2018, 11, 1756284818783606. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef]
- Sorolla, M.A.; Hidalgo, I.; Sorolla, A.; Montal, R.; Pallise, O.; Salud, A.; Parisi, E. Microenvironmental Reactive Oxygen Species in Colorectal Cancer: Involved Processes and Therapeutic Opportunities. Cancers 2021, 13, 5037. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.R.; Chang, D.K. Colorectal cancer in inflammatory bowel disease: The risk, pathogenesis, prevention and diagnosis. World J. Gastroenterol. 2014, 20, 9872–9881. [Google Scholar] [CrossRef] [PubMed]
- Rogler, G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014, 345, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, S.; Cao, Y.; Tian, X.; Zeng, R.; Liao, D.F.; Cao, D. Oxidative Stress and Carbonyl Lesions in Ulcerative Colitis and Associated Colorectal Cancer. Oxid. Med. Cell Longev. 2016, 2016, 9875298. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Uddin, M.N.; Hancock, J. The Role of Oxidative Stress and Its Counteractive Utility in Colorectal Cancer (CRC). Cancers 2020, 12, 3336. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Li, M.; Xu, J.Y.; Xiao, H.; Yang, X.; Fan, J.X.; Wu, K.; Chen, S. Aberrant ROS Mediate Cell Cycle and Motility in Colorectal Cancer Cells Through an Oncogenic CXCL14 Signaling Pathway. Front. Pharmacol. 2021, 12, 764015. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Zhang, R.; Kim, G.Y.; Bae, S.C.; Hyun, J.W. Epigenetic changes induced by oxidative stress in colorectal cancer cells: Methylation of tumor suppressor RUNX3. Tumour Biol. 2012, 33, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef]
- Khor, T.O.; Huang, M.T.; Prawan, A.; Liu, Y.; Hao, X.; Yu, S.; Cheung, W.K.; Chan, J.Y.; Reddy, B.S.; Yang, C.S.; et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev. Res. 2008, 1, 187–191. [Google Scholar] [CrossRef]
- He, F.; Antonucci, L.; Karin, M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis 2020, 41, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Hyun, Y.J.; Zhen, A.X.; Cho, S.J.; Ahn, M.J.; Yi, J.M.; Hyun, J.W. Luteolin promotes apoptotic cell death via upregulation of Nrf2 expression by DNA demethylase and the interaction of Nrf2 with p53 in human colon cancer cells. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Li, S.; Yang, G.; Zhu, X.; Cheng, L.; Sun, Y.; Zhao, Z. Combination of rapamycin and garlic-derived S-allylmercaptocysteine induces colon cancer cell apoptosis and suppresses tumor growth in xenograft nude mice through autophagy/p62/Nrf2 pathway. Oncol. Rep. 2017, 38, 1637–1644. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Kim, W.I.; Bae, J.H.; Cho, M.K.; Lee, S.H.; Nam, H.S.; Choi, I.H.; Cho, S.W. Overexpression of Nrf2 promotes colon cancer progression via ERK and AKT signaling pathways. Ann. Surg. Treat. Res. 2020, 98, 159–167. [Google Scholar] [CrossRef]
- Haque, E.; Karim, M.R.; Salam Teeli, A.; Smiech, M.; Leszczynski, P.; Winiarczyk, D.; Parvanov, E.D.; Atanasov, A.G.; Taniguchi, H. Molecular Mechanisms Underlying Hepatocellular Carcinoma Induction by Aberrant NRF2 Activation-Mediated Transcription Networks: Interaction of NRF2-KEAP1 Controls the Fate of Hepatocarcinogenesis. Int. J. Mol. Sci. 2020, 21, 5378. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Donquiles, C.; Alonso-Molero, J.; Fernandez-Villa, T.; Vilorio-Marques, L.; Molina, A.J.; Martin, V. The NRF2 transcription factor plays a dual role in colorectal cancer: A systematic review. PLoS ONE 2017, 12, e0177549. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Gundamaraju, R.; Lu, W.; Manikam, R. Revisiting Mitochondria Scored Cancer Progression and Metastasis. Cancers 2021, 13, 432. [Google Scholar] [CrossRef]
- Slattery, M.L.; Mullany, L.E.; Sakoda, L.; Samowitz, W.S.; Wolff, R.K.; Stevens, J.R.; Herrick, J.S. The NF-kappaB signalling pathway in colorectal cancer: Associations between dysregulated gene and miRNA expression. J. Cancer Res. Clin. Oncol. 2018, 144, 269–283. [Google Scholar] [CrossRef]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria-ros crosstalk in the control of cell death and aging. J. Signal. Transduct 2012, 2012, 329635. [Google Scholar] [CrossRef] [PubMed]
- Eberle, J.; Hossini, A.M. Expression and function of bcl-2 proteins in melanoma. Curr. Genomics 2008, 9, 409–419. [Google Scholar] [CrossRef]
- Wu, Z.; Huang, X.; Feng, Y.; Handschin, C.; Feng, Y.; Gullicksen, P.S.; Bare, O.; Labow, M.; Spiegelman, B.; Stevenson, S.C. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc. Natl. Acad. Sci. USA 2006, 103, 14379–14384. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003, Correction in Nat. Cell Biol. 2014, 16, 1125. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Jung, J.W.; Jung, J.; Han, Y.; Suh, D.H.; Kim, H.S.; Dhanasekaran, D.N.; Song, Y.S. PGC1alpha induced by reactive oxygen species contributes to chemoresistance of ovarian cancer cells. Oncotarget 2017, 8, 60299–60311. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Han, Y.S.; Lee, S.H. PGC-1alpha Controls Mitochondrial Biogenesis in Drug-Resistant Colorectal Cancer Cells by Regulating Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2019, 20, 1707. [Google Scholar] [CrossRef] [PubMed]
- Emery, J.M.; Ortiz, R.M. Mitofusin 2: A link between mitochondrial function and substrate metabolism? Mitochondrion 2021, 61, 125–137. [Google Scholar] [CrossRef]
- Cheng, X.; Zhou, D.; Wei, J.; Lin, J. Cell-cycle arrest at G2/M and proliferation inhibition by adenovirus-expressed mitofusin-2 gene in human colorectal cancer cell lines. Neoplasma 2013, 60, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.T.; Zhang, X.Y.; Cao, R.; Sun, L.; Jing, W.; Zhao, J.Z.; Zhang, S.L.; Huang, L.T.; Han, C.B. Effects of Dynamin-related Protein 1 Regulated Mitochondrial Dynamic Changes on Invasion and Metastasis of Lung Cancer Cells. J. Cancer 2019, 10, 4045–4053. [Google Scholar] [CrossRef] [PubMed]
- Inoue-Yamauchi, A.; Oda, H. Depletion of mitochondrial fission factor DRP1 causes increased apoptosis in human colon cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 81–85. [Google Scholar] [CrossRef]
- Tailor, D.; Hahm, E.R.; Kale, R.K.; Singh, S.V.; Singh, R.P. Sodium butyrate induces DRP1-mediated mitochondrial fusion and apoptosis in human colorectal cancer cells. Mitochondrion 2014, 16, 55–64. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Cui, H.; Huang, P.; Wang, Z.; Zhang, Y.; Zhang, Z.; Xu, W.; Wang, X.; Han, Y.; Guo, X. Association of decreased mitochondrial DNA content with the progression of colorectal cancer. BMC Cancer 2013, 13, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, H.B.; Kaiser, J.T.; Chan, D.C. The mitochondrial transcription and packaging factor Tfam imposes a U-turn on mitochondrial DNA. Nat. Struct Mol. Biol. 2011, 18, 1290–1296. [Google Scholar] [CrossRef]
- Yang, S.; He, X.; Zhao, J.; Wang, D.; Guo, S.; Gao, T.; Wang, G.; Jin, C.; Yan, Z.; Wang, N.; et al. Mitochondrial transcription factor A plays opposite roles in the initiation and progression of colitis-associated cancer. Cancer Commun 2021, 41, 695–714. [Google Scholar] [CrossRef] [PubMed]
- Sun, J. Impact of bacterial infection and intestinal microbiome on colorectal cancer development. Chin. Med. J. 2022, 135, 400–408. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Chon, H.J.; Kim, H.; Noh, J.H.; Yang, H.; Lee, W.S.; Kong, S.J.; Lee, S.J.; Lee, Y.S.; Kim, W.R.; Kim, J.H.; et al. STING signaling is a potential immunotherapeutic target in colorectal cancer. J. Cancer 2019, 10, 4932–4938. [Google Scholar] [CrossRef] [PubMed]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Effect | Reference |
|---|---|---|
| ATG7 | ATG7 deletion in intestinal anti-gen-presenting cells causes mitochondrial dysfunction and oxidative stress, and subsequently exacerbate inflammatory Th17 response | [126] |
| ATG16L1 | ATG16L1 deficiency induces abundant ROS production and mitophagy defect, which enhanced colitis in mouse model via increasing Il-1β and TNFα mRNA expression | [129] |
| HSF2 | HSF2 promotes mitophagy by reducing ROS and alleviates mucosal inflammation through inhibiting NLRP3 inflammasome via PARL/PINK1/PARKIN pathway | [133] |
| NIX | NIX deficient induces mitophagy defect and subsequently inflammatory features and loss of mucosal integrity in mouse experiments | [134] |
| PHB | PHB deletion in intestinal epithelial cells causes mitochondrial dysfunction and mitophagy defect, resulting in enhanced TNF-α, IL-1β, and reduced IL-10 mRNA expression, exacerbating the inflammatory response | [137] |
| IRGM1 | Knockdown IRGM1 could affect inflammatory response by regulating mitophagy in intestinal epithelial cells and Paneth cells | [128] |
| NRF2 | Oleuropein treatment alleviates intestinal inflammation by activating NRF2/HO-1 pathway and reducing iNOS, TNF-α and MCP-1 mRNA expression in DSS-induced IBD modle | [172] |
| PGC1α | PGC1α deficiency in intestinal epithelium shows more severe inflammation, reduces mitochondrial mass, and increases ROS production | [173] |
| Gene | Effect | Reference |
|---|---|---|
| PINK1 | PINK1 overexpression enhances mitophagy, reduces glycolysis, and increases mitochondrial respiration in mouse colon tumor cells. In contract, PINK1 deletion increases pro-inflammatory mRNA expression and colon tumorigenesis. | [191] |
| HSP60 | HSP60 knockdown inhibits cell proliferation through disrupting mitochondrial homeostasis in CRC. Also, HSP60 can combine with TRAP1 and activates inflammatory pathways, subsequently release the TNFα | [198] |
| NRF2 | ROS-induced NRF2 overexpression exacerbate inflammatory response and tumor formation in colonic tissues. In addition, high NRF2 level alleviated apoptosis and uncontrolled proliferation in tissue exposed to inflammatory macrophages | [217] |
| PGC-1α | Hypoxia induces PGC-1α expression, which is associated with enhanced motility, proliferation, and spheroid formation in CRC cells. | [231] |
| MFN2 | MFN2 inhibits CRC cells proliferation through promoting cell-cycle arrest at G2/M phase and increasing active caspase-3 levels | [234] |
| DROP1 | Slicing DROP1 inhibits human colon cancer cells and promotes its apoptosis through cytochrome C release and decreasing mitochondrial membrane potential | [237] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sui, G.-Y.; Wang, F.; Lee, J.; Roh, Y.S. Mitochondrial Control in Inflammatory Gastrointestinal Diseases. Int. J. Mol. Sci. 2022, 23, 14890. https://doi.org/10.3390/ijms232314890
Sui G-Y, Wang F, Lee J, Roh YS. Mitochondrial Control in Inflammatory Gastrointestinal Diseases. International Journal of Molecular Sciences. 2022; 23(23):14890. https://doi.org/10.3390/ijms232314890
Chicago/Turabian StyleSui, Guo-Yan, Feng Wang, Jin Lee, and Yoon Seok Roh. 2022. "Mitochondrial Control in Inflammatory Gastrointestinal Diseases" International Journal of Molecular Sciences 23, no. 23: 14890. https://doi.org/10.3390/ijms232314890