
Mitochondrial Role in Oncogenesis and Potential Chemotherapeutic Strategy of Mitochondrial Infusion in Breast Cancer

{kind=link}
Abstract
:1. Introduction
1.1. Epidemiology and Hallmarks of Cancer
1.2. Mitochondria: The Powerhouse
1.3. Mitochondria’s Role in Oncogenesis
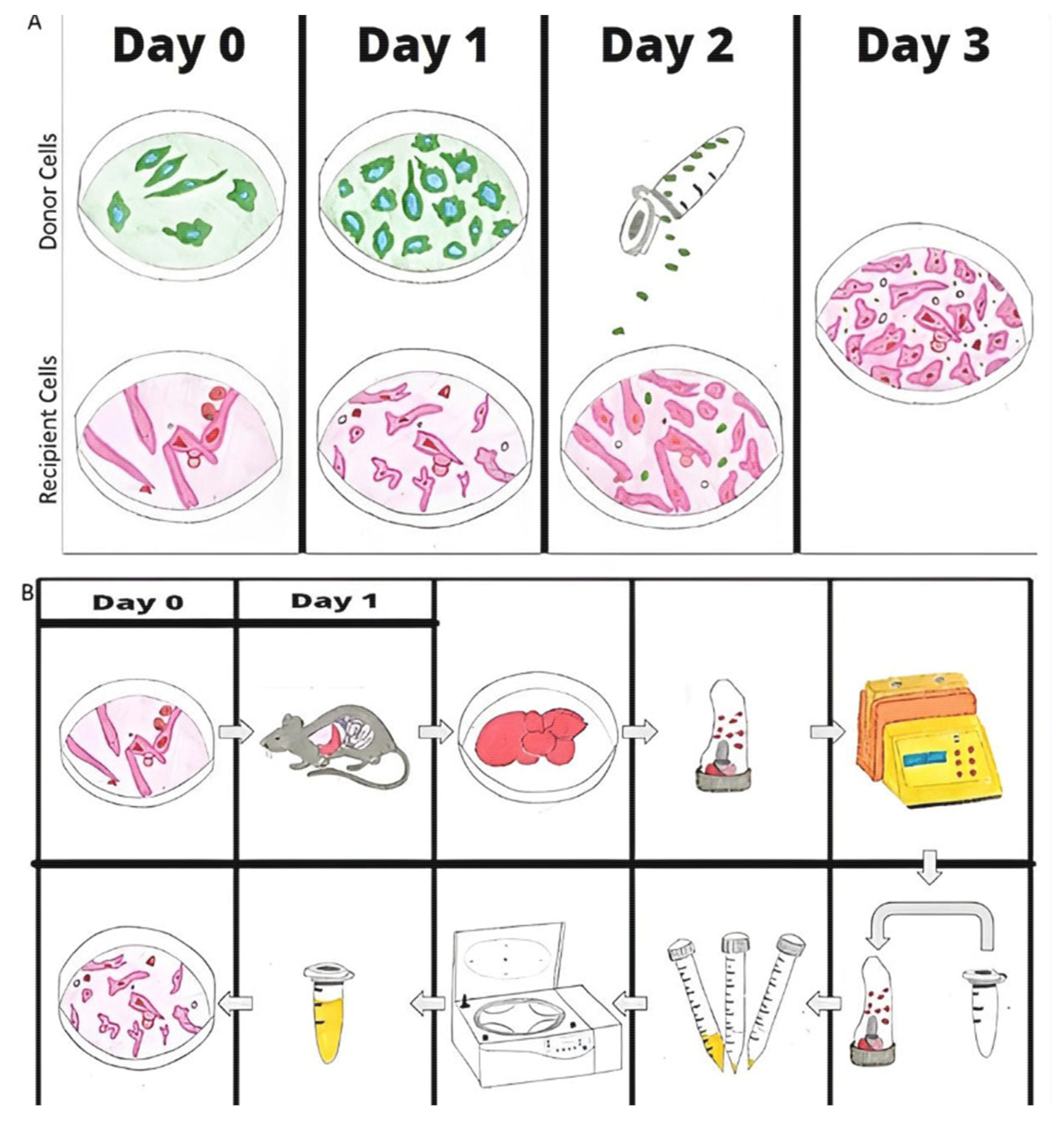
1.4. Mitochondrial Infusion
1.5. The “Good” of Mitochondrial Infusion
1.6. The “Bad” of Mitochondrial Infusion
2. Conclusions
The Past, Present, and Future
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chen, F.-M.; Huang, L.-J.; Ou-Yang, F.; Kan, J.-Y.; Kao, L.-C.; Hou, M.-F. Activation of mitochondrial unfolded protein response is associated with Her2-overexpression breast cancer. Breast Cancer Res. Treat. 2020, 183, 61–70. [Google Scholar] [CrossRef]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef]
- Howard, M.F.; Olopade, O.I. Epidemiology of Triple-Negative Breast Cancer: A Review. Cancer J. 2021, 27, 8–16. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.; Witkiewicz, A.K.; Howell, A.; Philp, N.J.; Pestell, R.G.; Lisanti, M.P. Mitochondrial metabolism in cancer metastasis. Cell Cycle 2012, 11, 1445–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reda, A.; Refaat, A.; Abd-Rabou, A.A.; Mahmoud, A.M.; Adel, M.; Sabet, S.; Ali, S.S. Role of mitochondria in rescuing glycolytically inhibited subpopulation of triple negative but not hor-mone-responsive breast cancer cells. Sci. Rep. 2019, 9, 13748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, L.; Soares, P.; Máximo, V.; Samuels, D.C. Somatic mitochondrial DNA mutations in cancer escape purifying selection and high pathogenicity mutations lead to the oncocytic phenotype: Pathogenicity analysis of reported somatic mtDNA mutations in tumors. BMC Cancer 2012, 12, 53. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Lunetti, P.; Di Giacomo, M.; Vergara, D.; De Domenico, S.; Maffia, M.; Zara, V.; Capobianco, L.; Ferramosca, A. Metabolic reprogramming in breast cancer results in distinct mitochondrial bioenergetics between luminal and basal subtypes. FEBS J. 2019, 286, 688–709. [Google Scholar] [CrossRef]
- Santidrian, A.F.; Matsuno-Yagi, A.; Ritland, M.; Seo, B.B.; LeBoeuf, S.E.; Gay, L.J.; Yagi, T.; Felding-Habermann, B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin. Investig. 2013, 123, 1068–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hy-poxia-inducible factor activation and tumorigenesis. Mol. Cell. Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.J.; Ma, W.; Starost, M.F.; Lago, C.U.; Lim, P.K.; Sack, M.N.; Kang, J.-G.; Wang, P.-Y.; Hwang, P.M. Ambient Oxygen Promotes Tumorigenesis. PLoS ONE 2011, 6, e19785. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef]
- Syu, J.-P.; Chi, J.-T.; Kung, H.-N. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget 2016, 7, 14659–14672. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Lei, J.H.; Bao, J.; Wang, H.; Hao, W.; Li, L.; Peng, C.; Masuda, T.; Miao, K.; Xu, J.; et al. BRCA1 Deficiency: BRCA1 Deficiency Impairs Mitophagy and Promotes Inflammasome Activation and Mammary Tumor Metastasis. Adv. Sci. 2020, 7, 1903616. [Google Scholar] [CrossRef]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2019, 21, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sami, E.; Paul, B.T.; Koziol, J.A.; ElShamy, W.M. The Immunosuppressive Microenvironment in BRCA1-IRIS–Overexpressing TNBC Tumors Is Induced by Bidirectional Interaction with Tumor-Associated Macrophages. Cancer Res. 2020, 80, 1102–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial Division Is Requisite to RAS-Induced Transformation and Targeted by Oncogenic MAPK Pathway Inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef] [Green Version]
- Amari, L.; Germain, M. Mitochondrial Extracellular Vesicles—Origins and Roles. Front. Mol. Neurosci. 2021, 14, 247. [Google Scholar] [CrossRef]
- Zampieri, L.; Silva-Almeida, C.; Rondeau, J.; Sonveaux, P. Mitochondrial Transfer in Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 3245. [Google Scholar] [CrossRef]
- Caicedo, A.; Fritz, V.; Brondello, J.-M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; de Fraipont, F.; Moisan, A.; Prouteau, C.A.; Boukhaddaoui, H.; et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 2015, 5, srep09073. [Google Scholar] [CrossRef] [Green Version]
- Rousselle, T.V.; Kuscu, C.; Kuscu, C.; Schlegel, K.; Huang, L.; Namwanje, M.; Eason, J.D.; Makowski, L.; Maluf, D.; Mas, V.; et al. FTY720 Regulates Mitochondria Biogenesis in Dendritic Cells to Prevent Kidney Ischemic Reperfusion Injury. Front. Immunol. 2020, 11, 1278. [Google Scholar] [CrossRef]
- Doulamis, I.P.; Guariento, A.; Duignan, T.; Kido, T.; Orfany, A.; Saeed, M.Y.; Weixler, V.H.; Blitzer, D.; Shin, B.; Snay, E.R.; et al. Mitochondrial transplantation by intra-arterial injection for acute kidney injury. Am. J. Physiol. Physiol. 2020, 319, F403–F413. [Google Scholar] [CrossRef]
- Fu, A.; Hou, Y.; Yu, Z.; Zhao, Z.; Liu, Z. Healthy mitochondria inhibit the metastatic melanoma in lungs. Int. J. Biol. Sci. 2019, 15, 2707–2718. [Google Scholar] [CrossRef]
- Kaipparettu, B.A.; Ma, Y.; Park, J.H.; Lee, T.L.; Zhang, Y.; Yotnda, P.; Creighton, C.J.; Chan, W.Y.; Wong, L.J.C. Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by sup-pressing oncogenic pathways. PLoS ONE 2013, 8, e61747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Liu, X.; Wang, B.; Wang, Z.; Liu, Y.; Di, C.; Si, J.; Li, H.; Wu, Q.; Xu, D.; et al. Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 2019, 9, 3595–3607. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-C.; Chang, H.-S.; Wu, Y.-C.; Cheng, W.-L.; Lin, T.-T.; Chang, H.-J.; Kuo, S.-J.; Chen, S.-T.; Liu, C.-S. Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.-P.; Elliott, R.L.; Head, J.F. Exogenous normal mammary epithelial mitochondria suppress glycolytic metabolism and glucose uptake of human breast cancer cells. Breast Cancer Res. Treat. 2015, 153, 519–529. [Google Scholar] [CrossRef]
- Song, I.-S.; Jeong, Y.J.; Jeong, S.H.; Kim, J.E.; Han, J.; Kim, T.-H.; Jang, S.-W. Modulation of Mitochondrial ERβ Expression Inhibits Triple-Negative Breast Cancer Tumor Progression by Activating Mitochondrial Function. Cell. Physiol. Biochem. 2019, 52, 468–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.C.; Chang, H.S.; Wu, Y.C.; Cheng, W.L.; Lin, T.T.; Chang, H.J.; Chen, S.T.; Liu, C.S. Antitumor Actions of Intratumoral Delivery of Membrane-Fused Mitochondria in a Mouse Model of Tri-ple-Negative Breast Cancers. Onco-Targets Ther. 2020, 13, 5241–5255. [Google Scholar] [CrossRef]
- Ma, M.; Lin, X.; Liu, H.; Zhang, R.; Chen, R. Suppression of DRP1-mediated mitophagy increases the apoptosis of hepatocellular carcinoma cells in the setting of chemotherapy. Oncol. Rep. 2020, 43, 1010–1018. [Google Scholar] [CrossRef]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [Green Version]
- Guariento, A.; Piekarski, B.L.; Doulamis, I.P.; Blitzer, D.; Ferraro, A.M.; Harrild, D.M.; Zurakowski, D.; Pedro, J.; McCully, J.D.; Emani, S.M. Autologous mitochondrial transplantation for cardiogenic shock in pediatric patients following ische-mia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2020, 162, 992–1001. [Google Scholar] [CrossRef]
- Boukelmoune, N.; Chiu, G.S.; Kavelaars, A.; Heijnen, C.J. Mitochondrial transfer from mesenchymal stem cells to neural stem cells protects against the neurotoxic effects of cisplatin. Acta Neuropathol. Commun. 2018, 6, 139. [Google Scholar] [CrossRef]
- Wang, X.; Gerdes, H.-H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.-F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 2017, 6, e22187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.S.; Baty, J.W.; Dong, L.-F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells without Mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 2018, 11, 11. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, P.S.; Castelow, C.; Patel, D.S.; Bhattacharya, S.K.; Kuscu, C.; Kuscu, C.; Makowski, L.; Eason, J.D.; Bajwa, A. Mitochondrial Role in Oncogenesis and Potential Chemotherapeutic Strategy of Mitochondrial Infusion in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 12993. https://doi.org/10.3390/ijms232112993
Patel PS, Castelow C, Patel DS, Bhattacharya SK, Kuscu C, Kuscu C, Makowski L, Eason JD, Bajwa A. Mitochondrial Role in Oncogenesis and Potential Chemotherapeutic Strategy of Mitochondrial Infusion in Breast Cancer. International Journal of Molecular Sciences. 2022; 23(21):12993. https://doi.org/10.3390/ijms232112993
Chicago/Turabian StylePatel, Prisha S., Christopher Castelow, Disha S. Patel, Syamal K. Bhattacharya, Cem Kuscu, Canan Kuscu, Liza Makowski, James D. Eason, and Amandeep Bajwa. 2022. "Mitochondrial Role in Oncogenesis and Potential Chemotherapeutic Strategy of Mitochondrial Infusion in Breast Cancer" International Journal of Molecular Sciences 23, no. 21: 12993. https://doi.org/10.3390/ijms232112993