

Identification of Molecular Determinants in iRhoms1 and 2 That Contribute to the Substrate Selectivity of Stimulated ADAM17
 , and
, and
Abstract
:1. Introduction
2. Results
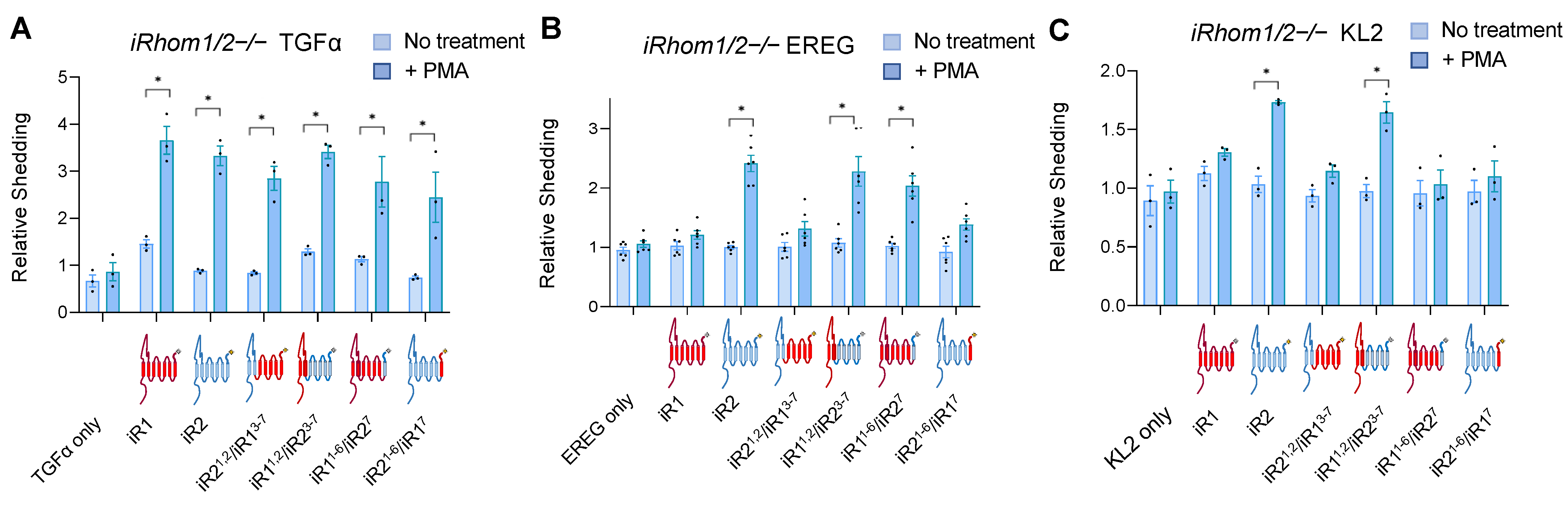
2.1. Chimeras between iRhom1 and iRhom2 Reveal a Key Contribution of the 7th Transmembrane Domain to the Substrate Selectivity of ADAM17
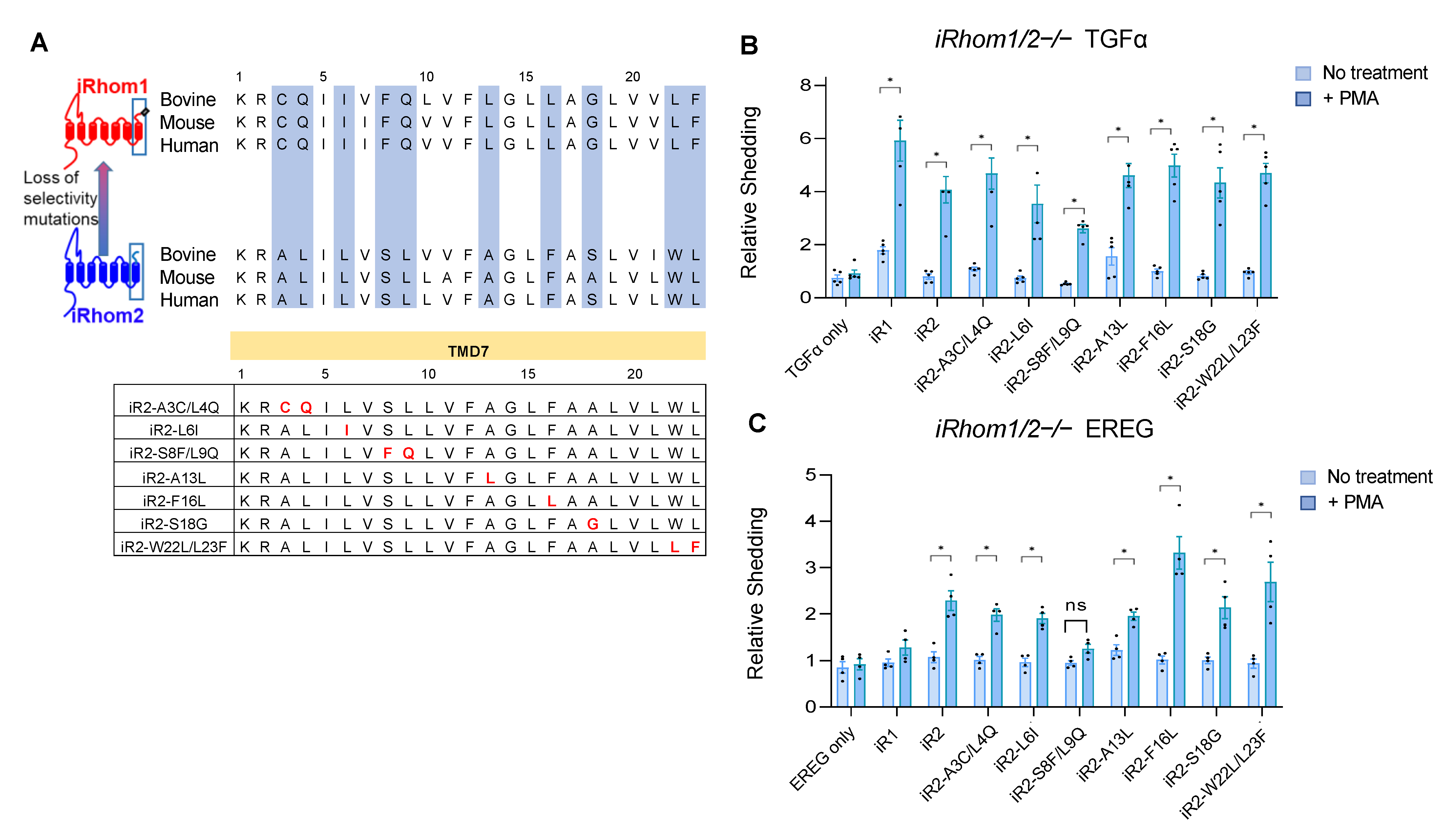
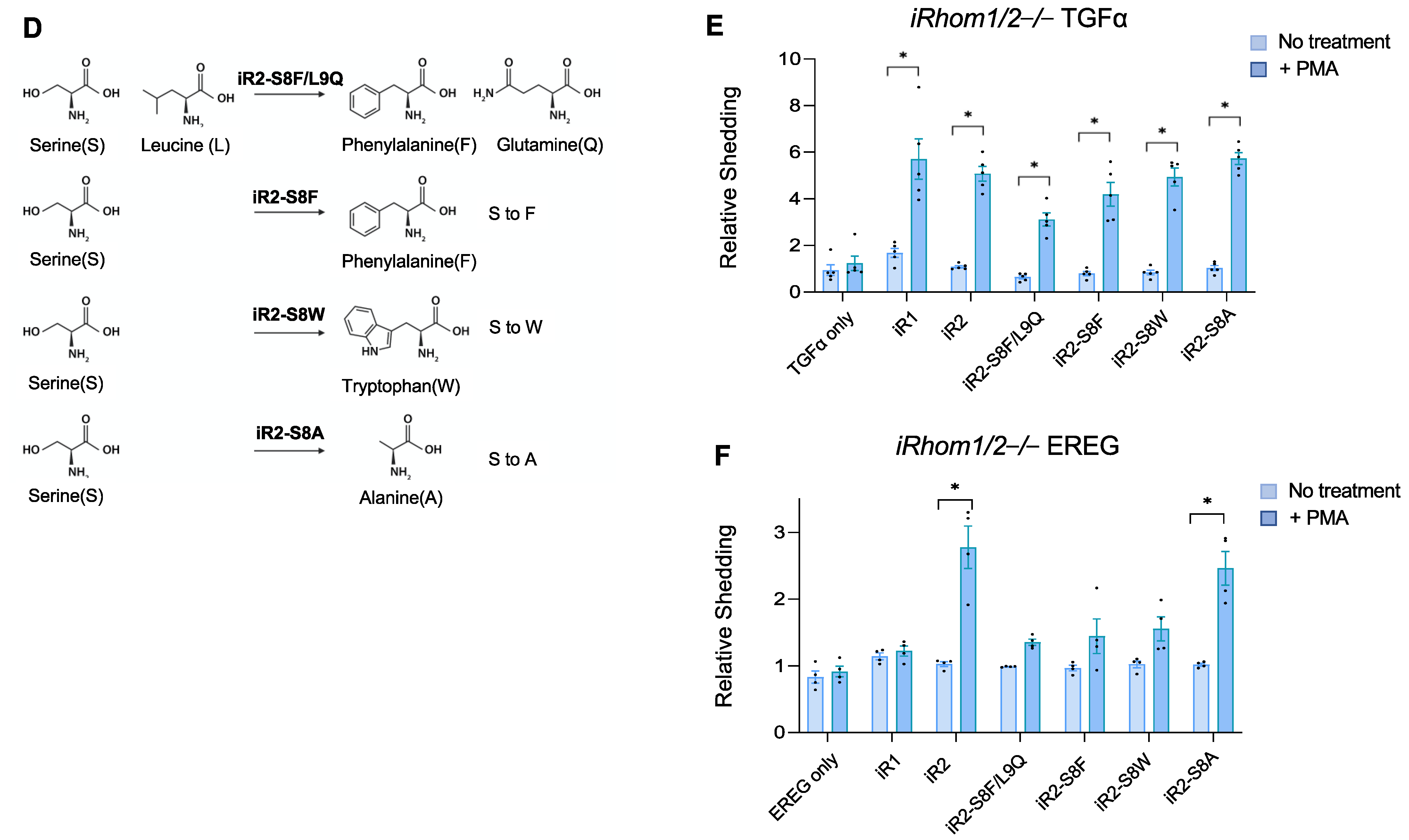
2.2. Specific Point Mutations Identify Amino Acid Residues in the TMD7 of iR2 That Are Required for Stimulated Shedding of EREG, but Not TGFα
2.3. A Gain-of-Function Point Mutation in the TMD7 of iR1 Allows It to Promote a Modest but Significant Stimulation of Shedding of the iR2-Selective EREG
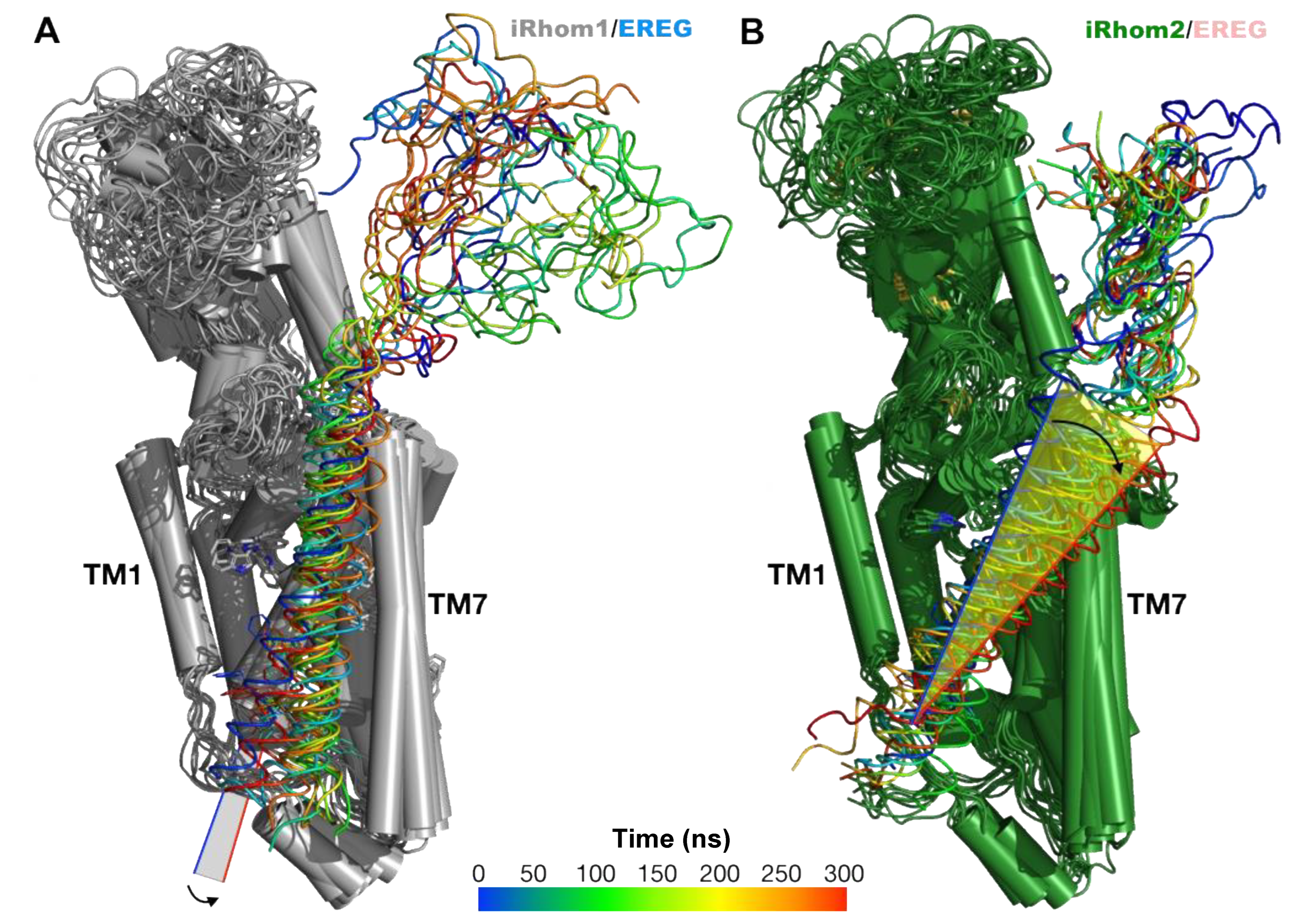
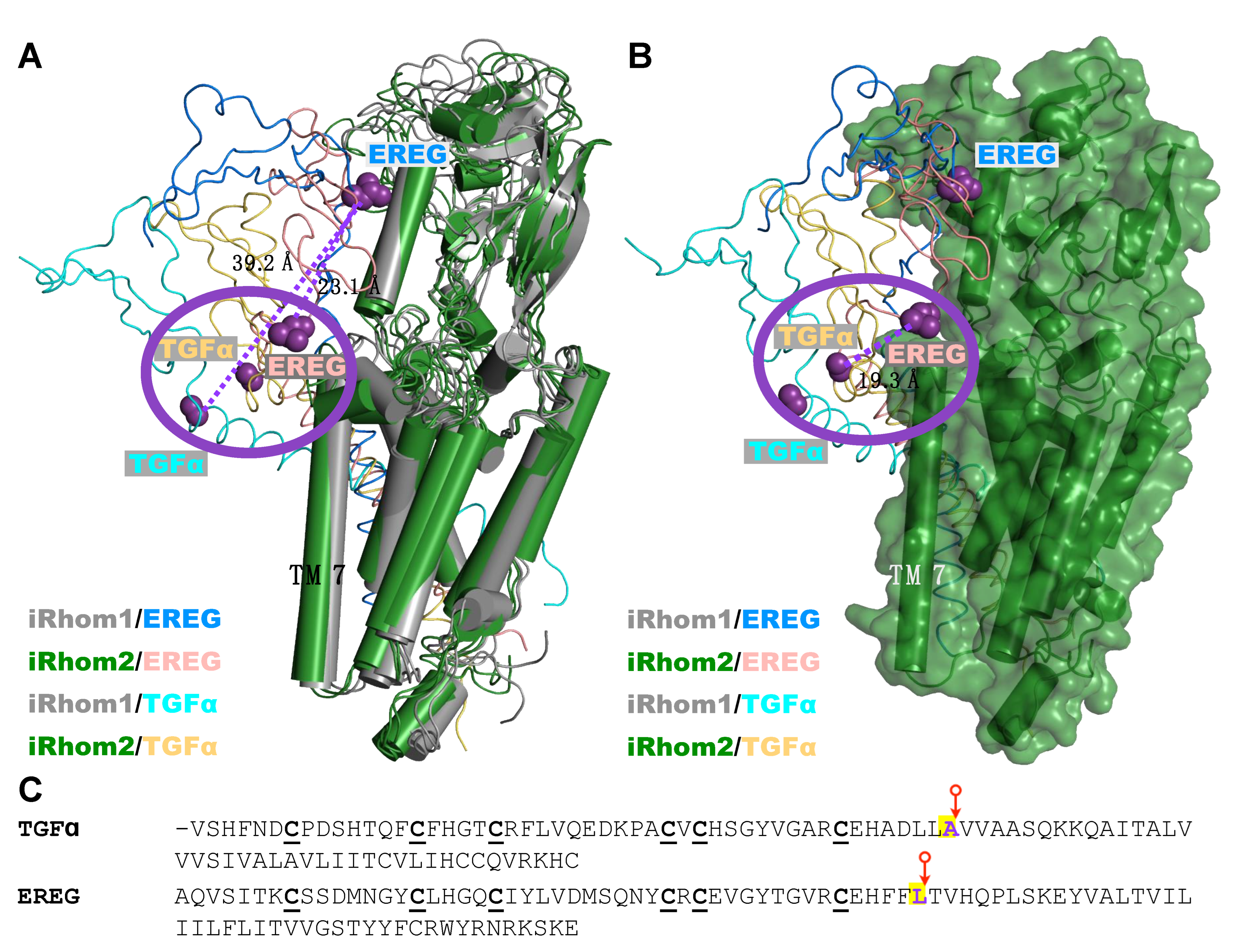
2.4. Computational Methods Suggest That Different Substrate Conformations Determine the Substrate Selectivity of ADAM17/iR2 versus ADAM17/iR1
2.5. Unbiased Molecular Dynamics Simulations of the Protein Complexes
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Cloning and Generation of iRhom1/2 and Substrate Mutants
4.3. Ectodomain Shedding Assays
4.4. Western Blot Analysis
4.5. Computational Modeling
5. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blobel, C.P. ADAMs: Key players in EGFR-signaling, development and disease. Nat. Rev. Mol. Cell. Bio. 2005, 6, 32–43. [Google Scholar] [CrossRef]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. ADAM17, shedding, TACE as therapeutic targets. Pharmacol. Res. 2013, 71, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Jin, S.-L.C.; Milla, M.E.; Burkhart, W.; Cartner, H.L.; Chen, W.-J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; Hoffman, C.R.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-a. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Black, R.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloprotease disintegrin that releases tumour-necrosis factor-a from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Horiuchi, K.; Kimura, T.; Miyamoto, T.; Takaishi, H.; Okada, Y.; Toyama, Y.; Blobel, C.P. Cutting Edge: TNF-{alpha}-Converting Enzyme (TACE/ADAM17) Inactivation in Mouse Myeloid Cells Prevents Lethality from Endotoxin Shock. J. Immunol. 2007, 179, 2686–2689. [Google Scholar] [CrossRef] [Green Version]
- Issuree, P.D.; Maretzky, T.; McIlwain, D.R.; Monette, S.; Qing, X.; Lang, P.A.; Swendeman, S.L.; Park-Min, K.H.; Binder, N.; Kalliolias, G.D.; et al. iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J. Clin. Investig. 2013, 123, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Weskamp, G.; Zhou, H.M.; Higashiyama, S.; Peschon, J.J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR-ligands. J. Cell. Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Sunnarborg, S.W.; Hinkle, C.L.; Stevenson, M.; Russell, W.E.; Raska, C.S.; Peschon, J.J.; Castner, B.J.; Gerhart, M.J.; Paxton, R.J.; Black, R.A.; et al. Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J. Biol. Chem. 2002, 277, 12838–12845. [Google Scholar] [CrossRef] [Green Version]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russel, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Hall, K.C.; Hill, D.; Otero, M.; Plumb, D.A.; Froemel, D.; Dragomir, C.L.; Maretzky, T.; Boskey, A.; Crawford, H.C.; Selleri, L.; et al. ADAM17 Controls Endochondral Ossification by Regulating Terminal Differentiation of Chondrocytes. Mol. Cell. Biol. 2013, 33, 3077–3090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Horiuchi, K.; Kimura, T.; Mizuno, S.; Yoda, M.; Morioka, H.; Akiyama, H.; Threadgill, D.; Okada, Y.; Toyama, Y.; et al. Conditional Inactivation of TNFalpha-Converting Enzyme in Chondrocytes Results in an Elongated Growth Plate and Shorter Long Bones. PLoS ONE 2013, 8, e54853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaydon, D.C.; Biancheri, P.; Di, W.L.; Plagnol, V.; Cabral, R.M.; Brooke, M.A.; van Heel, D.A.; Ruschendorf, F.; Toynbee, M.; Walne, A.; et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl. J. Med. 2011, 365, 1502–1508. [Google Scholar] [CrossRef] [Green Version]
- Bandsma, R.H.; van Goor, H.; Yourshaw, M.; Horlings, R.K.; Jonkman, M.F.; Scholvinck, E.H.; Karrenbeld, A.; Scheenstra, R.; Komhoff, M.; Rump, P.; et al. Loss of ADAM17 is associated with severe multiorgan dysfunction. Hum. Pathol. 2015, 46, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Imoto, I.; Saito, M.; Suga, K.; Kohmoto, T.; Otsu, M.; Horiuchi, K.; Nakayama, H.; Higashiyama, S.; Sugimoto, M.; Sasaki, A.; et al. Functionally confirmed compound heterozygous ADAM17 missense loss-of-function variants cause neonatal inflammatory skin and bowel disease 1. Sci. Rep. 2021, 11, 9552. [Google Scholar] [CrossRef]
- Franzke, C.W.; Cobzaru, C.; Triantafyllopoulou, A.; Loffek, S.; Horiuchi, K.; Threadgill, D.W.; Kurz, T.; van Rooijen, N.; Bruckner-Tuderman, L.; Blobel, C.P. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J. Exp. Med. 2012, 209, 1105–1119. [Google Scholar] [CrossRef] [Green Version]
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Cichy, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Lichtenberger, B.M.; Gerber, P.A.; Holcmann, M.; Buhren, B.A.; Amberg, N.; Smolle, V.; Schrumpf, H.; Boelke, E.; Ansari, P.; Mackenzie, C.; et al. Epidermal EGFR controls cutaneous host defense and prevents inflammation. Sci. Transl. Med. 2013, 5, 199ra111. [Google Scholar] [CrossRef]
- Sibilia, M.; Wagner, B.; Hoebertz, A.; Elliott, C.; Marino, S.; Jochum, W.; Wagner, E.F. Mice humanised for the EGF receptor display hypomorphic phenotypes in skin, bone and heart. Development 2003, 130, 4515–4525. [Google Scholar] [CrossRef] [Green Version]
- Lora, J.; Weskamp, G.; Li, T.M.; Maretzky, T.; Shola, D.T.N.; Monette, S.; Lichtenthaler, S.F.; Lu, T.T.; Yang, C.; Blobel, C.P. Targeted truncation of the ADAM17 cytoplasmic domain in mice results in protein destabilization and a hypomorphic phenotype. J. Biol. Chem. 2021, 296, 100733. [Google Scholar] [CrossRef]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Maretzky, T.; Weskamp, G.; Monette, S.; Qing, X.; Issuree, P.D.; Crawford, H.C.; McIlwain, D.R.; Mak, T.W.; Salmon, J.E.; et al. iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 6080–6085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maretzky, T.; McIlwain, D.R.; Issuree, P.D.; Li, X.; Malapeira, J.; Amin, S.; Lang, P.A.; Mak, T.W.; Blobel, C.P. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. USA 2013, 110, 11433–11438. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Lang, P.A.; Maretzky, T.; Hamada, K.; Ohishi, K.; Maney, S.K.; Berger, T.; Murthy, A.; Duncan, G.; Xu, H.C.; et al. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 2012, 335, 229–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 2012, 335, 225–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christova, Y.; Adrain, C.; Bambrough, P.; Ibrahim, A.; Freeman, M. Mammalian iRhoms have distinct physiological functions including an essential role in TACE regulation. EMBO Rep. 2013, 14, 884–890. [Google Scholar] [CrossRef]
- Fang, R.; Haxaire, C.; Otero, M.; Lessard, S.; Weskamp, G.; McIlwain, D.R.; Mak, T.W.; Lichtenthaler, S.F.; Blobel, C.P. Role of iRhoms 1 and 2 in Endochondral Ossification. Int. J. Mol. Sci. 2020, 21, 8732. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Maretzky, T.; Perez-Aguilar, J.M.; Monette, S.; Weskamp, G.; Le Gall, S.; Beutler, B.; Weinstein, H.; Blobel, C.P. Structural modeling defines transmembrane residues in ADAM17 that are crucial for Rhbdf2/ADAM17-dependent proteolysis. J. Cell. Sci. 2017, 130, 868–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.; Li, X.; Maretzky, T.; Perez-Aguilar, J.M.; McIlwain, D.; Xie, Y.; Zheng, Y.; Mak, T.W.; Weinstein, H.; Blobel, C.P. Substrate-selective protein ectodomain shedding by ADAM17 and iRhom2 depends on their juxtamembrane and transmembrane domains. FASEB J. 2020, 34, 4956–4969. [Google Scholar] [CrossRef] [Green Version]
- Grieve, A.G.; Xu, H.; Kunzel, U.; Bambrough, P.; Sieber, B.; Freeman, M. Phosphorylation of iRhom2 at the plasma membrane controls mammalian TACE-dependent inflammatory and growth factor signalling. Elife 2017, 6, e23968. [Google Scholar] [CrossRef]
- Cavadas, M.; Oikonomidi, I.; Gaspar, C.J.; Burbridge, E.; Badenes, M.; Felix, I.; Bolado, A.; Hu, T.; Bileck, A.; Gerner, C.; et al. Phosphorylation of iRhom2 Controls Stimulated Proteolytic Shedding by the Metalloprotease ADAM17/TACE. Cell Rep. 2017, 21, 745–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikonomidi, I.; Burbridge, E.; Cavadas, M.; Sullivan, G.; Collis, B.; Naegele, H.; Clancy, D.; Brezinova, J.; Hu, T.; Bileck, A.; et al. iTAP, a novel iRhom interactor, controls TNF secretion by policing the stability of iRhom/TACE. Elife 2018, 7, e35032. [Google Scholar] [CrossRef]
- Kunzel, U.; Grieve, A.G.; Meng, Y.; Sieber, B.; Cowley, S.A.; Freeman, M. FRMD8 promotes inflammatory and growth factor signalling by stabilising the iRhom/ADAM17 sheddase complex. Elife 2018, 7, e35012. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, S.M.; Maretzky, T.; Issuree, P.D.A.; Niu, X.-D.; Reiss, K.; Saftig, P.; Khokha, R.; Lundell, D.; Blobel, C.P. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell. Sci. 2010, 123, 3913–3922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef]
- Black, R.A.; Doedens, J.R.; Mahimkar, R.; Johnson, R.; Guo, L.; Wallace, A.; Virca, D.; Eisenman, J.; Slack, J.; Castner, B.; et al. Substrate specificity and inducibility of TACE (tumour necrosis factor alpha-converting enzyme) revisited: The Ala-Val preference, and induced intrinsic activity. Biochem. Soc. Symp. 2003, 70, 39–52. [Google Scholar]
- Doedens, J.R.; Mahimkar, R.M.; Black, R.A. TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem. Biophys. Res. Commun. 2003, 308, 331–338. [Google Scholar] [CrossRef]
- Seegar, T.C.M.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the alpha-Secretase ADAM10. Cell 2017, 171, 1638–1648.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; Saunders, C.; Senftleben, A.; Heidbuechel, J.P.W.; Halwachs, B.; Bolik, J.; Hedemann, N.; Röder, C.; Bauerschlag, D.; Rose-John, S.; et al. Tetraspanin 8 Subfamily Members Regulate Substrate-Specificity of a Disintegrin and Metalloprotease 17. Cells 2022, 11, 2683. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol 2018, 12, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Kuo, D.; Ding, J.; Cohn, I.S.; Zhang, F.; Wei, K.; Rao, D.A.; Rozo, C.; Sokhi, U.K.; Shanaj, S.; Oliver, D.J.; et al. HBEGF(+) macrophages in rheumatoid arthritis induce fibroblast invasiveness. Sci. Transl. Med. 2019, 11, eaau8587. [Google Scholar] [CrossRef]
- Jing, C.; Jin, Y.H.; You, Z.; Qiong, Q.; Jun, Z. Prognostic value of amphiregulin and epiregulin mRNA expression in metastatic colorectal cancer patients. Oncotarget 2016, 7, 55890–55899. [Google Scholar] [CrossRef] [Green Version]
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015, 42, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Busser, B.; Sancey, L.; Brambilla, E.; Coll, J.L.; Hurbin, A. The multiple roles of amphiregulin in human cancer. Biochim. Biophys. Acta 2011, 1816, 119–131. [Google Scholar] [CrossRef]
- Cheng, W.L.; Feng, P.H.; Lee, K.Y.; Chen, K.Y.; Sun, W.L.; Van Hiep, N.; Luo, C.S.; Wu, S.M. The Role of EREG/EGFR Pathway in Tumor Progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef] [PubMed]
- Gnosa, S.; Puig-Blasco, L.; Piotrowski, K.B.; Freiberg, M.L.; Savickas, S.; Madsen, D.H.; Auf dem Keller, U.; Kronqvist, P.; Kveiborg, M. ADAM17-mediated EGFR ligand shedding directs macrophage promoted cancer cell invasion. JCI Insight 2022, 7, e155296. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, N.; Horiuchi, K.; Becherer, J.D.; Toyama, Y.; Besmer, P.; Blobel, C.P. Different ADAMs have distinct influences on Kit ligand processing: Phorbol-ester-stimulated ectodomain shedding of Kitl1 by ADAM17 is reduced by ADAM19. J. Cell Sci 2007, 120, 943–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Weskamp, G.; Zheng, Y.; Chesneau, V.; Horiuchi, K.; Blobel, C.P. A sensitive method to monitor ectodomain shedding of ligands of the epidermal growth factor receptor. In Epidermal Growth Factor: Methods and Protocols; Patel, T.B., Bertics, P.J., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2006; Volume 327, pp. 99–113. [Google Scholar]
- Perez-Aguilar, J.M.; Shan, J.; LeVine, M.V.; Khelashvili, G.; Weinstein, H. A functional selectivity mechanism at the serotonin-2A GPCR involves ligand-dependent conformations of intracellular loop 2. J. Am. Chem. Soc. 2014, 136, 16044–16054. [Google Scholar] [CrossRef] [Green Version]
- Perez-Aguilar, J.M.; Kang, S.G.; Zhang, L.; Zhou, R. Modeling and Structural Characterization of the Sweet Taste Receptor Heterodimer. ACS Chem. Neurosci. 2019, 10, 4579–4592. [Google Scholar] [CrossRef]
- Rangel-Galvan, M.; Rangel, A.; Romero-Mendez, C.; Davila, E.M.; Castro, M.E.; Caballero, N.A.; Melendez Bustamante, F.J.; Sanchez-Gaytan, B.L.; Meza, U.; Perez-Aguilar, J.M. Inhibitory Mechanism of the Isoflavone Derivative Genistein in the Human CaV3.3 Channel. ACS Chem Neurosci 2021, 12, 651–659. [Google Scholar] [CrossRef]
- Davila, E.M.; Patricio, F.; Rebolledo-Bustillo, M.; Garcia-Gomez, D.; Hernandez, J.C.G.; Sanchez-Gaytan, B.L.; Limon, I.D.; Perez-Aguilar, J.M. Interacting binding insights and conformational consequences of the differential activity of cannabidiol with two endocannabinoid-activated G-protein-coupled receptors. Front. Pharmacol. 2022, 13, 945935. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Direction |
|---|---|---|
| iR2-A3C/L4Q | 5′-GTACCGCAAGCGATGCCAGATCCTCGTGTCG-3′ | Forward |
| 5′-CGACACGAGGATCTGGCATCGCTTGCGGTAC-3′ | Reverse | |
| iR2-L6I | 5′-CGAGCCCTCATCATCGTGTCGCTGC-3′ | Forward |
| 5′-GCAGCGACACGATGATGAGGGCTCG-3′ | Reverse | |
| iR2-S8F/L9Q | 5′-CCTCATCCTCGTGTTCCAGCTGGTCTTTG-3′ | Forward |
| 5′-CAAAGACCAGCTGGAACACGAGGATGAGG-3′ | Reverse | |
| iR2-A13L | 5′-CTGCTGGTCTTTCTTGGGCTCTTTGC-3′ | Forward |
| 5′-GCAAAGAGCCCAAGAAAGACCAGCAG-3′ | Reverse | |
| Ir2-F16L | 5′-GCTGGGCTCTTAGCTTCCCTGGTG-3′ | Forward |
| 5′-CACCAGGGAAGCTAAGAGCCCAGC-3′ | Reverse | |
| iR2-S18G | 5′-GCTCTTTGCTGGCCTGGTGCTGTGG-3′ | Forward |
| 5′-CCACAGCACCAGGCCAGCAAAGAGC-3′ | Reverse | |
| iR2-W22L/L23F | 5′-CCTGGTGCTGCTGTTCTACATCTACC-3′ | Forward |
| 5′-GGTAGATGTAGAACAGCAGCACCAGG-3′ | Reverse | |
| iR1-F8S | 5′-CAGATCATCATCTCCCAGGTCGTCTTCC-3′ | Forward |
| 5′-GGAAGACGACCTGGGAGATGATGATCTG-3′ | Reverse | |
| iR1-L16F | 5′-CTGGGCCTGTTTGCCGGCCTGGTG-3′ | Forward |
| 5′-CACCAGGCCGGCAAACAGGCCCAG-3′ | Reverse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Dávila, E.M.; Li, X.; Tang, B.; Rabinowitsch, A.I.; Perez-Aguilar, J.M.; Blobel, C.P. Identification of Molecular Determinants in iRhoms1 and 2 That Contribute to the Substrate Selectivity of Stimulated ADAM17. Int. J. Mol. Sci. 2022, 23, 12796. https://doi.org/10.3390/ijms232112796
Zhao Y, Dávila EM, Li X, Tang B, Rabinowitsch AI, Perez-Aguilar JM, Blobel CP. Identification of Molecular Determinants in iRhoms1 and 2 That Contribute to the Substrate Selectivity of Stimulated ADAM17. International Journal of Molecular Sciences. 2022; 23(21):12796. https://doi.org/10.3390/ijms232112796
Chicago/Turabian StyleZhao, Yi, Eliud Morales Dávila, Xue Li, Beiyu Tang, Ariana I. Rabinowitsch, Jose Manuel Perez-Aguilar, and Carl P. Blobel. 2022. "Identification of Molecular Determinants in iRhoms1 and 2 That Contribute to the Substrate Selectivity of Stimulated ADAM17" International Journal of Molecular Sciences 23, no. 21: 12796. https://doi.org/10.3390/ijms232112796