
Role of Cockayne Syndrome Group B Protein in Replication Stress: Implications for Cancer Therapy

Abstract
:1. Introduction
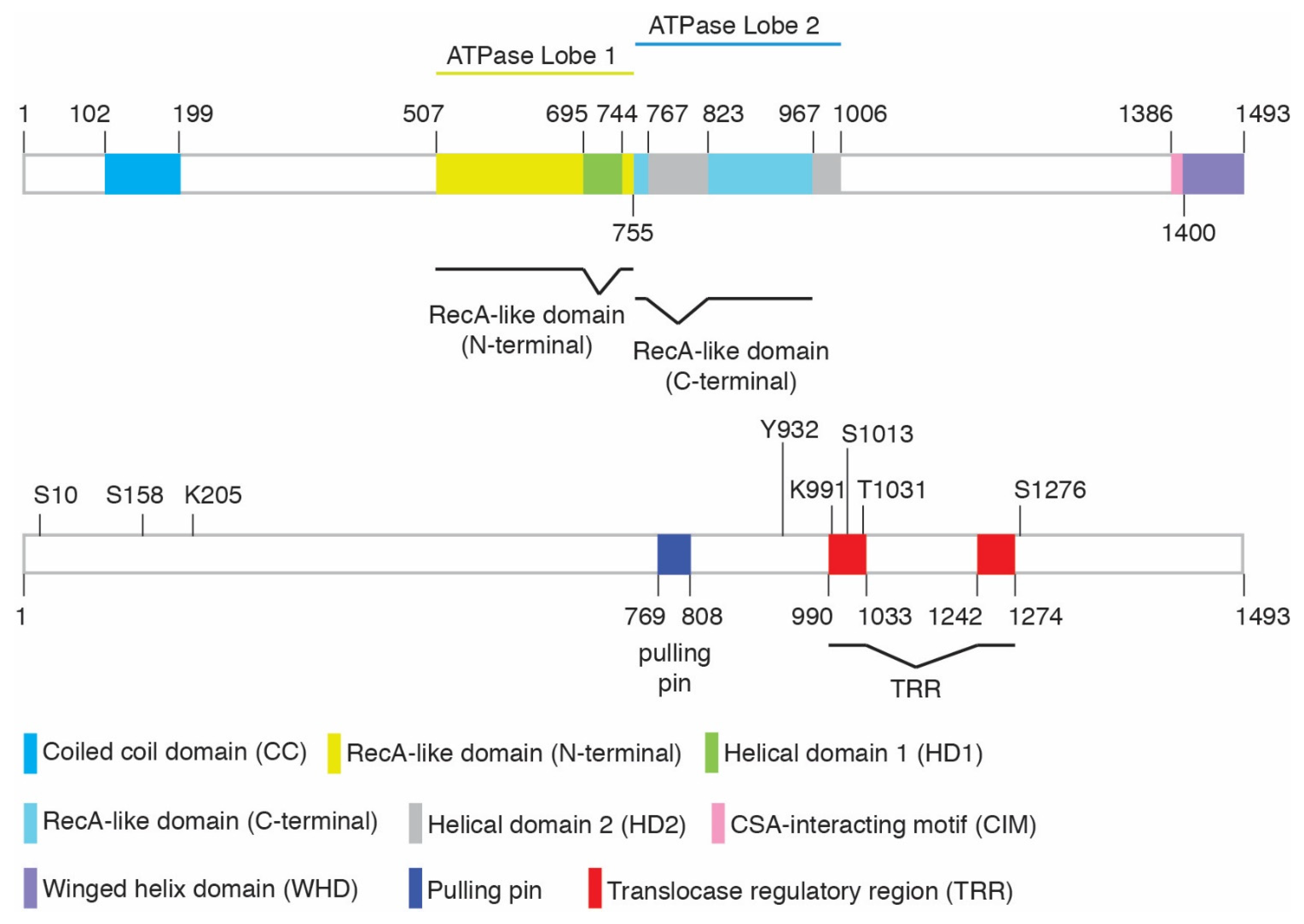
2. CSB and Chromatin Remodeling Activity
3. CSB-Deficient Cells and Their Sensitivity to Replication Stress-Inducing Agents
4. Association of CSB with Replication Forks in Unperturbed Conditions
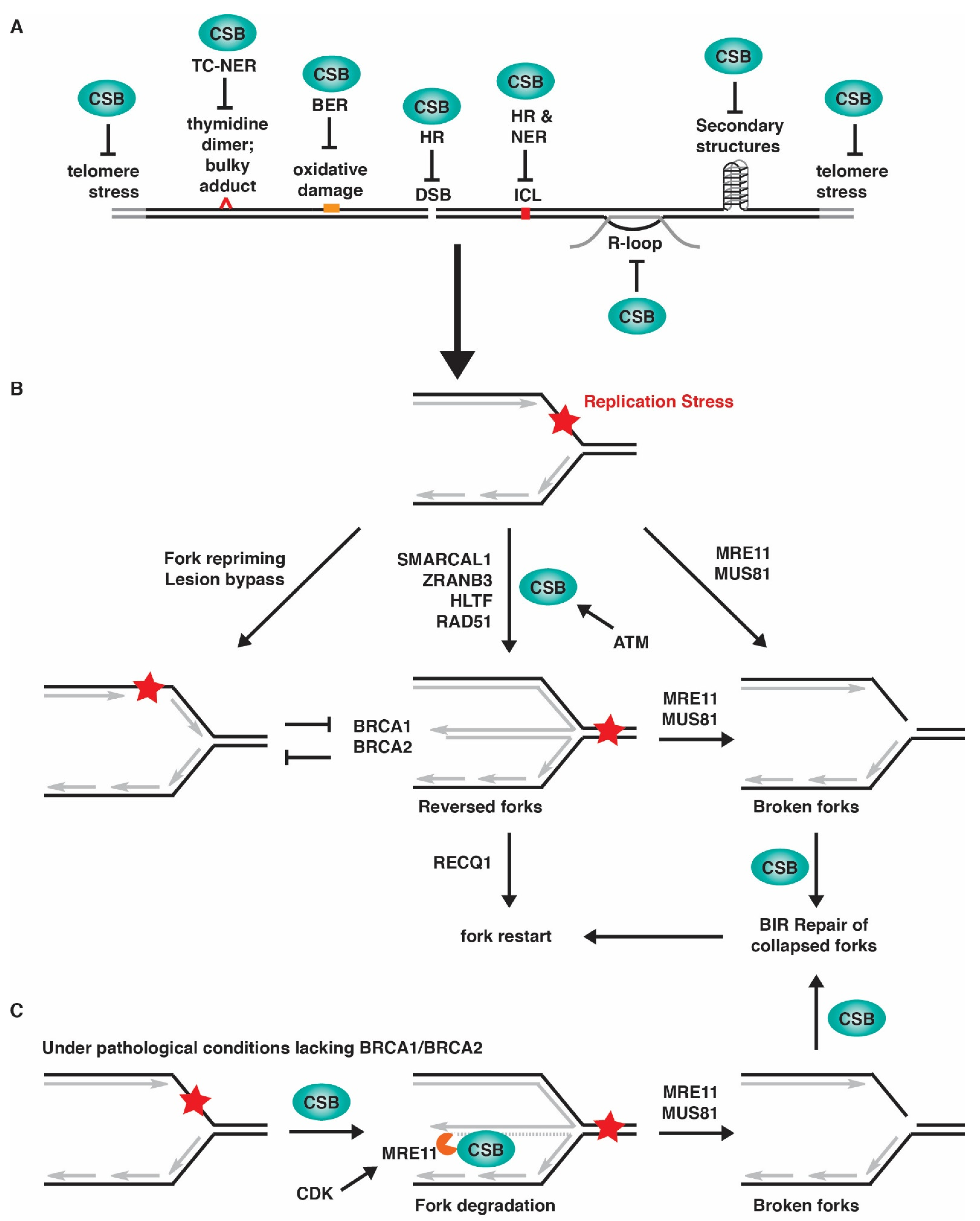
5. Association of CSB with Replication Forks in Response to Replication Stress
6. CSB and Its Role in Fork Reversal in Response to Replication Stress
7. CSB and Restart of Stalled Forks
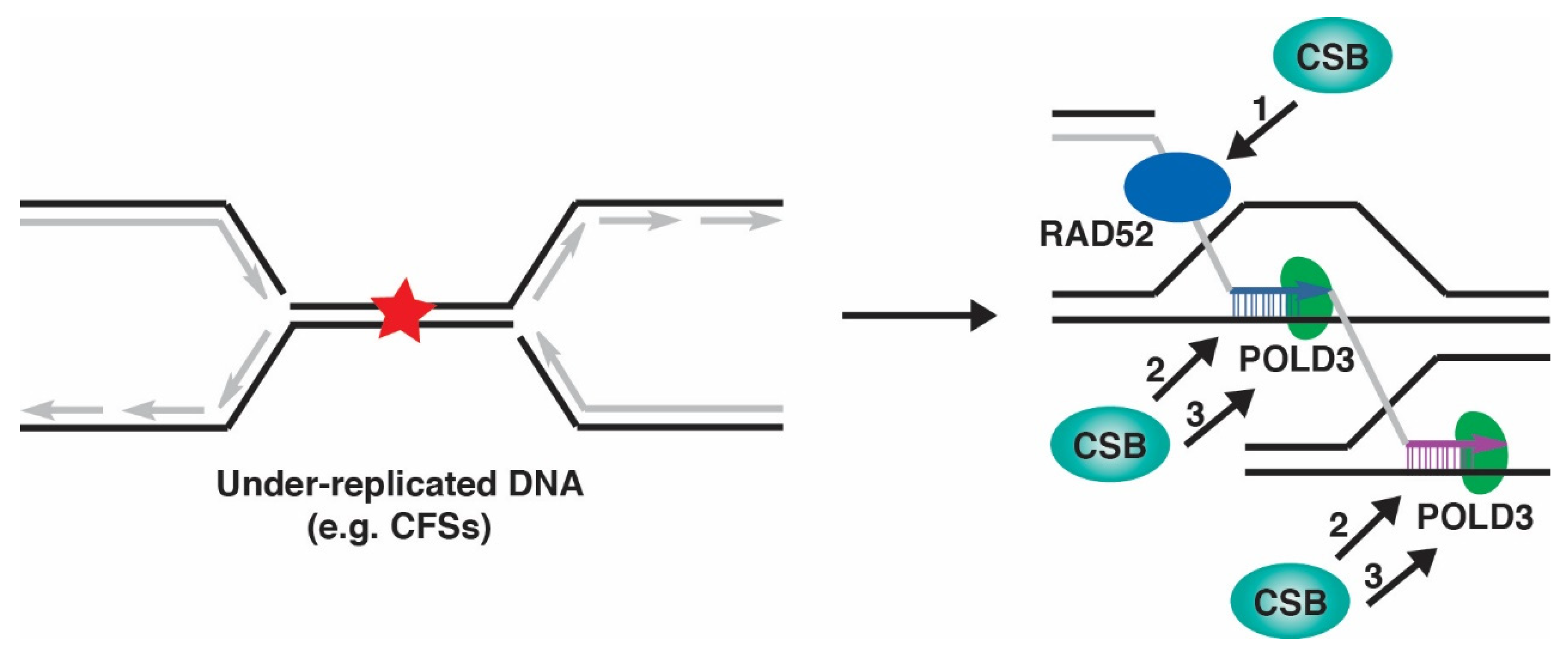
8. CSB and MiDAS
9. CSB and Telomeres
10. CSB and Its Implications in Targeted Therapy in Cancer
11. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baddeley, D.; Chagin, V.O.; Schermelleh, L.; Martin, S.; Pombo, A.; Carlton, P.M.; Gahl, A.; Domaing, P.; Birk, U.; Leonhardt, H.; et al. Meansurement of replication structures at the nanometer scale using super-resolution light microscopy. Nucleic Acids Res. 2010, 38, e8. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.; Langston, L.; Stillman, B. Principles and Concepts of DNA Replication in Bacteria, Archaea, and Eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5, a010108. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Joseph, S.A.; Taglialatela, A.; Leuzzi, G.; Huang, J.-W.; Cuella-Martin, R.; Ciccia, A. Time for remodeling: SNF2-family DNA translocases in replication fork metabolism and human disease. DNA Repair 2020, 95, 102943. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Cortez, D.; Lopes, M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 2020, 21, 633–651. [Google Scholar] [CrossRef]
- Conti, B.A.; Smogorzewska, A. Mechanisms of direct replication restart at stressed replisomes. DNA Repair 2020, 95, 102947. [Google Scholar] [CrossRef]
- Troelstra, C.; van Gool, A.; de Wit, J.; Vermeulen, W.; Bootsma, D.; Hoeijmakers, J.H. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell 1992, 71, 939–953. [Google Scholar] [CrossRef]
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013, 134, 161–170. [Google Scholar] [CrossRef]
- Laugel, V.; Dalloz, C.; Durand, M.; Sauvanaud, F.; Kristensen, U.; Vincent, M.C.; Pasquier, L.; Odent, S.; Cormier-Daire, V.; Gener, B.; et al. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum. Mutat. 2009, 31, 113–126. [Google Scholar] [CrossRef]
- Vélez-Cruz, R.; Egly, J.-M. Cockayne syndrome group B (CSB) protein: At the crossroads of transcriptional networks. Mech. Ageing Dev. 2013, 134, 234–242. [Google Scholar] [CrossRef]
- Batenburg, N.L.; Cui, S.; Walker, J.R.; Schellhorn, H.E.; Zhu, X.-D. The Winged Helix Domain of CSB Regulates RNAPII Occupancy at Promoter Proximal Pause Sites. Int. J. Mol. Sci. 2021, 22, 3379. [Google Scholar] [CrossRef] [PubMed]
- Selby, C.P.; Sancar, A. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl. Acad. Sci. USA 1997, 94, 11205–11209. [Google Scholar] [CrossRef] [PubMed]
- Stevnsner, T.; Muftuoglu, M.; Aamann, M.D.; Bohr, V.A. The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mech. Ageing Dev. 2008, 129, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Mulderrig, L.; Garaycoechea, J.I.; Tuong, Z.K.; Millington, C.L.; Dingler, F.A.; Ferdinand, J.R.; Gaul, L.; Tadross, J.A.; Arends, M.J.; O’Rahilly, S.; et al. Aldehyde-driven transcriptional stress triggers an anorexic DNA damage response. Nature 2021, 600, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, N.L.; Thompson, E.L.; Hendrickson, E.A.; Zhu, X.-D. Cockayne syndrome group B protein regulates DNA double-strand break repair and checkpoint activation. EMBO J. 2015, 34, 1399–1416. [Google Scholar] [CrossRef]
- Batenburg, N.L.; Walker, J.R.; Noordermeer, S.M.; Moatti, N.; Durocher, D.; Zhu, X.-D. ATM and CDK2 control chromatin remodeler CSB to inhibit RIF1 in DSB repair pathway choice. Nat. Commun. 2017, 8, 1921. [Google Scholar] [CrossRef]
- Teng, Y.; Yadav, T.; Duan, M.; Tan, J.; Xiang, Y.; Gao, B.; Xu, J.; Liang, Z.; Liu, Y.; Nakajima, S.; et al. ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat. Commun. 2018, 9, 4115. [Google Scholar] [CrossRef]
- Feng, E.; Batenburg, N.L.; Walker, J.R.; Ho, A.; Mitchell, T.R.H.; Qin, J.; Zhu, X.-D. CSB cooperates with SMARCAL1 to maintain telomere stability in ALT cells. J. Cell Sci. 2020, 133, jcs234914. [Google Scholar] [CrossRef]
- Batenburg, N.L.; Mitchell, T.R.H.; Leach, D.M.; Rainbow, A.J.; Zhu, X.-D. Cockayne Syndrome group B protein interacts with TRF2 and regulates telomere length and stability. Nucleic Acids Res. 2012, 40, 9661–9674. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. Mitochondrial deficiency in Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Paccosi, E.; Costanzo, F.; Costantino, M.; Balzerano, A.; Monteonofrio, L.; Soddu, S.; Prantera, G.; Brancorsini, S.; Egly, J.-M.; Proietti-De-Santis, L. The Cockayne syndrome group A and B proteins are part of a ubiquitin–proteasome degradation complex regulating cell division. Proc. Natl. Acad. Sci. USA 2020, 117, 30498–30508. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, N.L.; Mersaoui, S.Y.; Walker, J.R.; Coulombe, Y.; Hammond-Martel, I.; Wurtele, H.; Masson, J.-Y.; Zhu, X.-D. Cockayne syndrome group B protein regulates fork restart, fork progression, and MRE11-dependent fork degradation in BRCA1/2-deficient cells. Nucleic Acid Res. 2021, 49, 12836–12854. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Baptiste, B.A.; Okur, M.N.; Bohr, V.A. Current and emerging roles of Cockayne syndrome group B (CSB) protein. Nucleic Acids Res. 2021, 49, 2418–2434. [Google Scholar] [CrossRef]
- Paccosi, E.; Proietti-De-Santis, L. The emerging role of Cockayne group A and B proteins in ubiquitin/proteasome-directed protein degradation. Mech. Ageing Dev. 2021, 195, 111466. [Google Scholar] [CrossRef] [PubMed]
- Vessoni, A.T.; Guerra, C.C.C.; Kajitani, G.S.; Nascimento, L.L.S.; Garcia, C.C.M. Cockayne Syndrome: The many challenges and approaches to understand a multifaceted disease. Genet. Mol. Biol. 2020, 43, e20190085. [Google Scholar] [CrossRef]
- Spyropoulou, Z.; Papaspyropoulos, A.; Lagopati, N.; Myrianthopoulos, V.; Georgakilas, A.G.; Fousteri, M.; Kotsinas, A.; Gorgoulis, V.G. Cockayne Syndrome Group B (CSB): The Regulatory Framework Governing the Multifunctional Protein and Its Plausible Role in Cancer. Cells 2021, 10, 866. [Google Scholar] [CrossRef]
- Flaus, A.; Martin, D.M.A.; Barton, G.J.; Owen-Hughes, T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acid Res. 2006, 34, 2887–2905. [Google Scholar] [CrossRef]
- Seeber, A.; Hauer, M.; Gasser, S.M. Nucleosome remodelers in double-strand break repair. Curr. Opin. Genet. Dev. 2013, 23, 174–184. [Google Scholar] [CrossRef]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef]
- Batenburg, N.L.; Qin, J.; Walker, J.R.; Zhu, X.-D. Efficient UV repair requires disengagement of the CSB winged helix domain from the CSB ATPase domain. DNA Repair 2018, 68, 58–67. [Google Scholar] [CrossRef]
- Imam, S.Z.; Indig, F.E.; Cheng, W.-H.; Saxena, S.P.; Stevnsner, T.; Kufe, D.; Bohr, V.A. Cockayne syndrome protein B interacts with and is phosphorylated by c-Abl tyrosine kinase. Nucleic Acids Res. 2007, 35, 4941–4951. [Google Scholar] [CrossRef]
- Cui, S.; Walker, J.R.; Batenburg, N.L.; Zhu, X.-D. Cockayne syndrome group B protein uses its DNA translocase activity to promote mitotic DNA synthesis. DNA Repair 2022, 116, 103354. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, N.L.; Walker, J.R.; Coulombe, Y.; Sherker, A.; Masson, J.-Y.; Zhu, X.-D. CSB interacts with BRCA1 in late S/G2 to promote MRN- and CtIP-mediated DNA end resection. Nucleic Acids Res. 2019, 47, 10678–10692. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; D’Souza, R.C.J.; Tyanova, S.; Schaab, C.; Wiśniewski, J.R.; Cox, J.; Mann, M. Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative Phosphoproteomics Identifies Substrates and Functional Modules of Aurora and Polo-Like Kinase Activities in Mitotic Cells. Sci. Signal. 2011, 4, rs5. [Google Scholar] [CrossRef]
- Sin, Y.; Tanaka, K.; Saijo, M. The C-terminal Region and SUMOylation of Cockayne Syndrome Group B Protein Play Critical Roles in Transcription-coupled Nucleotide Excision Repair. J. Biol. Chem. 2016, 291, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Ranes, M.; Boeing, S.; Wang, Y.; Wienholz, F.; Menoni, H.; Walker, J.; Encheva, V.; Chakravarty, P.; Mari, P.-O.; Stewart, A.; et al. A ubiquitylation site in Cockayne syndrome B required for repair of oxidative DNA damage, but not for transcription-coupled nucleotide excision repair. Nucleic Acids Res. 2016, 44, 5246–5255. [Google Scholar] [CrossRef] [PubMed]
- Scheibye-Knudsen, M.; Ramamoorthy, M.; Sykora, P.; Maynard, S.; Lin, P.-C.; Minor, R.K.; Wilson, D.M., 3rd; Cooper, M.; Spencer, R.; de Cabo, R.; et al. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 2012, 209, 855–869. [Google Scholar] [CrossRef]
- Nicolai, S.; Filippi, S.; Caputo, M.; Cipak, L.; Gregan, J.; Ammerer, G.; Frontini, M.; Willems, D.; Prantera, G.; Balajee, A.S.; et al. Identification of Novel Proteins Co-Purifying with Cockayne Syndrome Group B (CSB) Reveals Potential Roles for CSB in RNA Metabolism and Chromatin Dynamics. PLoS ONE 2015, 10, e0128558. [Google Scholar]
- Aamann, M.D.; Muftuoglu, M.; Bohr, V.A.; Stevnsner, T. Multiple interaction partners for Cockayne syndrome proteins: Implications for genome and transcriptome maintenance. Mech. Ageing Dev. 2013, 134, 212–224. [Google Scholar] [CrossRef]
- Anindya, R.; Mari, P.-O.; Kristensen, U.; Kool, H.; Giglia-Mari, G.; Mullenders, L.H.; Fousteri, M.; Vermeulen, W.; Egly, J.-M.; Svejstrup, J.Q. A Ubiquitin-Binding Domain in Cockayne Syndrome B Required for Transcription-Coupled Nucleotide Excision Repair. Mol. Cell 2010, 38, 637–648. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.S.; Sato, Y.; Yamagata, A.; Goto-Ito, S.; Saijo, M.; Fukai, S. Structural basis of ubiquitin recognition by the winged-helix domain of Cockayne syndrome group B protein. Nucleic Acids Res. 2019, 47, 3784–3794. [Google Scholar] [CrossRef] [PubMed]
- Kokic, G.; Wagner, F.R.; Chernev, A.; Urlaub, H.; Cramer, P. Structural basis of human transcription–DNA repair coupling. Nature 2021, 598, 368–372. [Google Scholar] [CrossRef] [PubMed]
- van de Weegen, Y.; Golan-Berman, H.; Mevissen, T.E.T.; Apelt, K.; González-Prieto, R.; Goedhart, J.; Heilbrun, E.E.; Vertegaal, A.C.O.; van den Heuvel, D.; Walter, J.C.; et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 2020, 11, 2104. [Google Scholar] [CrossRef]
- Citterio, E.; Rademakers, S.; van der Horst, G.T.; van Gool, A.J.; Hoeijmakers, J.H.; Vermeulen, W. Biochemical and Biological Characterization of Wild-type and ATPase-deficient Cockayne Syndrome B Repair Protein. J. Biol. Chem. 1998, 273, 11844–11851. [Google Scholar] [CrossRef] [PubMed]
- Selby, C.P.; Sancar, A. Human Transcription-Repair Coupling Factor CSB/ERCC6 Is a DNA-stimulated ATPase but Is Not a Helicase and Does Not Disrupt the Ternary Transcription Complex of Stalled RNA Polymerase II. J. Biol. Chem. 1997, 272, 1885–1890. [Google Scholar] [CrossRef]
- Citterio, E.; Van Den Boom, V.; Schnitzler, G.; Kanaar, R.; Bonte, E.; Kingston, R.E.; Hoeijmakers, J.H.; Vermeulen, W. ATP-Dependent Chromatin Remodeling by the Cockayne Syndrome B DNA Repair-Transcription-Coupling Factor. Mol. Cell. Biol. 2000, 20, 7643–7653. [Google Scholar] [CrossRef]
- Lake, R.J.; Geyko, A.; Hemashettar, G.; Zhao, Y.; Fan, H.-Y. UV-Induced Association of the CSB Remodeling Protein with Chromatin Requires ATP-Dependent Relief of N-Terminal Autorepression. Mol. Cell 2010, 37, 235–246. [Google Scholar] [CrossRef]
- Kharbanda, S.; Yuan, Z.-M.; Weichselbaum, R.; Kufe, D. Determination of cell fate by c-Abl activation in the response to DNA damage. Oncogene 1998, 17, 3309–3318. [Google Scholar] [CrossRef]
- Liebelt, F.; Schimmel, J.; Verlaan-de Vries, M.; Klemann, E.; van Royen, M.E.; van der Weegen, Y.; Luijsterburg, M.S.; Mullenders, L.H.; Pines, A.; Vermeulen, W.; et al. Transcription-coupled nucleotide excision repair is coordinated by ubiquitin and SUMO in response to ultraviolet irradiation. Nucleic Acids Res. 2020, 48, 231–248. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Chaudhuri, A.R.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Flohr, C.; Bürkle, A.; Radicella, J.P.; Epe, B. Poly(ADP-ribosyl)ation accelerates DNA repair in a pathway dependent on Cockayne syndrome B protein. Nucleic Acids Res. 2003, 31, 5332–5337. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; von Kobbe, C.; Harrigan, J.A.; Indig, F.E.; Christiansen, M.; Stevnsner, T.; Bohr, V.A. Cooperation of the Cockayne Syndrome Group B Protein and Poly(ADP-Ribose) Polymerase 1 in the Response to Oxidative Stress. Mol. Cell. Biol. 2005, 25, 7625–7636. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A High Fat Diet and NAD+ Rescue Premature Aging in Cockayne Syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef]
- Lake, R.J.; Bilkis, R.; Fan, H.-Y. Dynamic Interplay between Cockayne Syndrome Protein B and Poly(ADP-Ribose) Polymerase 1 during Oxidative DNA Damage Repair. Biomedicines 2022, 10, 361. [Google Scholar] [CrossRef]
- Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; Tutt, A.N.J.; Lord, C.J. Opinion: PARP inhibitors in cancer-what do we still need to know? Open Biol. 2022, 12, 220118. [Google Scholar] [CrossRef]
- Brickner, J.R.; Garzon, J.L.; Cimprich, K.A. Walking a tightrope: The complex balancing act of R-loops in genome stability. Mol. Cell 2022, 82, 2267–2297. [Google Scholar] [CrossRef]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Vesela, E.; Chroma, K.; Turi, Z.; Mistrik, M. Common Chemical Inductors of Replication Stress: Focus on Cell-Based Studies. Biomolecules 2017, 7, 19. [Google Scholar] [CrossRef]
- de Waard, H.; de Wit, J.; Gorgels, T.G.M.F.; van den Aardweg, G.; Andressoo, J.-O.; Vermeij, M.; van Steeg, H.; Hoeijmakers, J.H.J.; van der Horst, G.T. Cell type-specific hypersensitivity to oxidative damage in CSB and XPA mice. DNA Repair 2003, 2, 13–25. [Google Scholar] [CrossRef]
- Squires, S.; Ryan, A.J.; Strutt, H.L.; Johnson, R.T. Hypersensitivity of Cockayne’s syndrome cells to camptothecin is associated with the generation of abnormally high levels of double strand breaks in nascent DNA. Cancer Res. 1993, 53, 2012–2019. [Google Scholar]
- Newman, J.C.; Bailey, A.D.; Weiner, A.M. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc. Natl. Acad. Sci. USA 2006, 103, 9613–9618. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Morón, E.; Calderón-Montaño, J.M.; Pastor, N.; Höglund, A.; Ruiz-Castizon, Á.; Domínguez, I.; López-Lázaro, M.; Hajji, N.; Helleday, T.; Mateos, S.; et al. The Cockayne syndrome protein B is involved in the repair of 5-AZA-2′-deoxycytidine-induced DNA lesions. Oncotarget 2018, 9, 35069–35084. [Google Scholar] [CrossRef] [PubMed]
- Caputo, M.; Frontini, M.; Velez-Cruz, R.; Nicolai, S.; Prantera, G.; Proietti-De-Santis, L. The CSB repair factor is overexpressed in cancer cells, increases apoptotic resistance, and promotes tumor growth. DNA Repair 2013, 12, 293–299. [Google Scholar] [CrossRef]
- Furuta, T.; Ueda, T.; Aune, G.; Sarasin, A.; Kraemer, K.H.; Pommier, Y. Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res. 2002, 62, 4899–4902. [Google Scholar]
- McKay, B.C.; Becerril, C.; Ljungman, M. P53 plays a protective role against UV- and cisplatin-induced apoptosis in transcription-coupled repair proficient fibroblasts. Oncogene 2001, 20, 6805–6808. [Google Scholar] [CrossRef]
- Enoiu, M.; Jiricny, J.; Schärer, O.D. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 2012, 40, 8953–8964. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, C.; Wu, H.; Xie, Y.; Gao, H.; Zhang, X. CSB affected on the sensitivity of lung cancer cells to platinum-based drugs through the global decrease of let-7 and miR-29. BMC Cancer 2019, 19, 948. [Google Scholar] [CrossRef]
- Iyama, T.; Lee, S.Y.; Berquist, B.R.; Gileadi, O.; Bohr, V.A.; Seidman, M.M.; McHugh, P.J.; Wilson, D.M., 3rd. CSB interacts with SNM1A and promotes DNA interstrand crosslink processing. Nucleic Acids Res. 2015, 43, 247–258. [Google Scholar] [CrossRef]
- Wong, H.-K.; Muftuoglu, M.; Beck, G.; Imam, S.Z.; Bohr, V.A.; Wilson, D.M., 3rd. Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007, 35, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- van Oosterwijk, M.F.; Filon, R.; de Groot, A.J.; van Zeeland, A.A.; Mullenders, L.H. Lack of transcription-coupled repair of acetylaminofluorene DNA adducts in human fibroblasts contrasts their efficient inhibition of transcription. J. Biol. Chem. 1998, 273, 13599–13604. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.H.; Chu, E.H.Y. Effects of DNA damaging agents on cultured fibroblasts derived from patients with Cockayne syndrome. Mutat. Res. 1979, 59, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Sunesen, M.; Selzer, R.R.; Brosh, R.M., Jr.; Balajee, A.S.; Stevnsner, T.; Bohr, V.A. Molecular characterization of an acidic region deletion mutant of Cockayne syndrome group B protein. Nucleic Acid Res. 2000, 28, 3151–3159. [Google Scholar] [CrossRef]
- Tuo, J.; Müftüoglu, M.; Chen, C.; Jaruga, P.; Selzer, R.R.; Brosh, R.M., Jr.; Rodriguez, H.; Dizdaroglu, M.; Bohr, V.A. The Cockayne Syndrome group B gene product is involved in general genome base excision repair of 8-hyroxyguanine in DNA. J. Biol. Chem. 2001, 276, 45772–45779. [Google Scholar] [CrossRef]
- Muftuoglu, M.; Selzer, R.; Tuo, J.; Brosh, R.M., Jr.; Bohr, V.A. Phenotypic consequences of mutations in the conserved motifs of the putative helicase domain of the human Cockayne Syndrome Group B gene. Gene 2002, 283, 27–40. [Google Scholar] [CrossRef]
- Alabert, C.; Bukowski-Wills, J.-C.; Lee, S.-B.; Kustatscher, G.; Nakamura, K.; de Lima Alves, F.; Menard, P.; Mejlvang, J.; Rappsilber, J.; Groth, A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 2014, 16, 281–291. [Google Scholar] [CrossRef]
- Wessel, S.R.; Mohni, K.N.; Luzwick, J.W.; Dungrawala, H.; Cortez, D. Functional Analysis of the Replication Fork Proteome Identifies BET Proteins as PCNA Regulators. Cell Rep. 2019, 28, 3497–3509.e4. [Google Scholar] [CrossRef]
- Roy, S.; Luzwick, J.W.; Schlacher, K. SIRF: Quantitative in situ analysis of protein interactions at DNA replication forks. J. Cell Biol. 2018, 217, 1521–1536. [Google Scholar] [CrossRef]
- Yu, A.; Fan, H.-Y.; Liao, D.; Bailey, A.D.; Weiner, A.M. Activation of p53 or Loss of the Cockayne Syndrome Group B Repair Protein Causes Metaphase Fragility of Human U1, U2, and 5S Genes. Mol. Cell 2000, 5, 801–810. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Scheibye-Knudsen, M.; Tseng, A.; Jensen, M.B.; Scheibye-Alsing, K.; Fang, E.F.; Iyama, T.; Bharti, S.K.; Marosi, K.; Froetscher, L.; Kassahun, H.; et al. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl. Acad. Sci. USA 2016, 113, 12502–12507. [Google Scholar] [CrossRef] [PubMed]
- Adelman, K.; Lis, J.T. Promoter-proximal pausing of RNA polymerase II: Emerging roles in metazoans. Nat. Rev. Genet. 2012, 13, 720–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, J.; McGlynn, P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009, 37, 3475–3492. [Google Scholar] [CrossRef]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Quinet, A.; Tirman, S.; Cybulla, E.; Meroni, A.; Vindigni, A. To skip or not to skip: Choosing repriming to tolerate DNA damage. Mol. Cell 2021, 81, 649–658. [Google Scholar] [CrossRef]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.-W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e8. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Chaudhuri, A.R.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef]
- Lemaçon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, 860. [Google Scholar] [CrossRef]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Técher, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881.e7. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Bétous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blastyák, A.; Hajdú, I.; Unk, I.; Haracska, L. Role of Double-Stranded DNA Translocase Activity of Human HLTF in Replication of Damaged DNA. Mol. Cell. Biol. 2010, 30, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Bugreev, D.V.; Rossi, M.J.; Mazin, A.V. Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res. 2011, 39, 2153–2164. [Google Scholar] [CrossRef] [PubMed]
- Gari, K.; Décaillet, C.; Delannoy, M.; Wu, L.; Constantinou, A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl. Acad. Sci. USA 2008, 105, 16107–16112. [Google Scholar] [CrossRef]
- Chavez, D.A.; Greer, B.H.; Eichman, B.F. The HIRAN domain of helicase-like transcription factor positions the DNA translocase motor to drive efficient DNA fork progression. J. Biol. Chem. 2018, 293, 8484–8494. [Google Scholar] [CrossRef]
- Muftuoglu, M.; Sharma, S.; Thorslund, T.; Stevnsner, T.; Soerensen, M.M.; Brosh, R.M., Jr.; Bohr, V.A. Cockayne syndrome group B protein has novel strand annealing and exchange activities. Nucleic Acids Res. 2006, 34, 295–304. [Google Scholar] [CrossRef]
- Vindigni, A.; Lopes, M. Combining electron microscopy with single molecular DNA fiber approaches to study DNA replication dynamics. Biophys. Chem. 2017, 225, 3–9. [Google Scholar] [CrossRef]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef]
- Vujanovic, M.; Krietsch, J.; Raso, M.C.; Terraneo, N.; Zellweger, R.; Schmid, J.A.; Taglialatela, A.; Huang, J.-W.; Holland, C.L.; Zwicky, K.; et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell 2017, 67, 882–890.e5. [Google Scholar] [CrossRef]
- Fugger, K.; Mistrik, M.; Neelsen, K.J.; Yao, Q.; Zellweger, R.; Kousholt, A.N.; Haahr, P.; Chu, W.K.; Bartek, J.; Lopes, M.; et al. FBH1 Catalyzes Regression of Stalled Replication Forks. Cell Rep. 2015, 10, 1749–1757. [Google Scholar] [CrossRef]
- Bai, G.; Kermi, C.; Stoy, H.; Schiltz, C.J.; Bacal, J.; Zaino, A.M.; Hadden, M.K.; Eichman, B.F.; Lopes, M.; Cimprich, K.A. HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol. Cell 2020, 78, 1237–1251.e7. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Reynolds, J.J.; Winczura, A.; Blackford, A.N.; Borel, V.; Miller, E.S.; Zlatanou, A.; Nieminuszczy, J.; Ryan, E.L.; Davies, N.J.; et al. BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks. Mol. Cell 2015, 59, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Przetocka, S.; Porro, A.; Bolck, H.A.; Walker, C.; Lezaja, A.; Trenner, A.; von Aesch, C.; Himmels, S.F.; D’Andrea, A.D.; Ceccaldi, R.; et al. CtIP-Mediated Fork Protection Synergizes with BRCA1 to Suppress Genomic Instability upon DNA Replication Stress. Mol. Cell 2018, 72, 568–582.e6. [Google Scholar] [CrossRef]
- Bainbridge, L.J.; Teague, R.; Doherty, A.J. Repriming DNA synthesis: An intrinsic restart pathway that maintains efficient genome replication. Nucleic Acids Res. 2021, 49, 4831–4847. [Google Scholar] [CrossRef]
- Tirman, S.; Cybulla, E.; Quinet, A.; Meroni, A.; Vindigni, A. PRIMPOL ready, set, reprime! Crit. Rev. Biochem. Mol. Biol. 2021, 56, 17–30. [Google Scholar] [CrossRef]
- Quinet, A.; Tirman, S.; Jackson, J.; Šviković, S.; Lemaçon, D.; Carvajal-Maldonado, D.; González-Acosta, D.; Vessoni, A.T.; Cybulla, E.; Wood, M.; et al. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol. Cell 2020, 77, 461–474.e9. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Cong, K.; Panzarino, N.J.; Nayak, S.; Calvo, J.; Deng, B.; Zhu, L.J.; Morocz, M.; Hegedus, L.; Haracska, L.; et al. Opposing Roles of FANCJ and HLTF Protect Forks and Restrain Replication during Stress. Cell Rep. 2018, 24, 3251–3261. [Google Scholar] [CrossRef]
- Pasero, P.; Vindigni, A. Nucleases Acting at Stalled Forks: How to Reboot the Replication Program with a Few Shortcuts. Annu. Rev. Genet. 2017, 51, 477–499. [Google Scholar] [CrossRef]
- Dehé, P.-M.; Gaillard, P.-H.L. Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell Biol. 2017, 18, 315–330. [Google Scholar] [CrossRef]
- Kramara, J.; Osia, B.; Malkova, A. Break-Induced Replication: The Where, The Why, and The How. Trends Genet. 2018, 34, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef]
- Mocanu, C.; Karanika, E.; Fernández-Casañas, M.; Herbert, A.; Olukoga, T.; Özgürses, M.E.; Chan, K.-L. DNA replication is highly resilient and persistent under the challenge of mild replication stress. Cell Rep. 2022, 39, 110701. [Google Scholar] [CrossRef] [PubMed]
- Özer, Ö.; Hickson, I.D. Pathways for maintenance of telomeres and common fragile sites during DNA replication stress. Open Biol. 2018, 8, 180018. [Google Scholar] [CrossRef]
- Mocanu, C.; Chan, K.-L. Mind the replication gap. R. Soc. Open Sci. 2021, 8, 201932. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef]
- Di Marco, S.; Hasanova, Z.; Kanagaraj, R.; Chappidi, N.; Altmannova, V.; Menon, S.; Sedlackova, H.; Langhoff, J.; Surendranath, K.; Hühn, D.; et al. RECQ5 Helicase Cooperates with MUS81 Endonuclease in Processing Stalled Replication Forks at Common Fragile Sites during Mitosis. Mol. Cell 2017, 66, 658–671.e8. [Google Scholar] [CrossRef]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef]
- Chan, K.-L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Zhu, X.-D. Post-translational modifications of TRF1 and TRF2 and their roles in telomere maintenance. Mech. Ageing Dev. 2012, 133, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Pickett, H.A. Targeting telomeres: Advances in telomere maintenance mechanism-specific cancer therapies. Nat. Rev. Cancer 2022, 22, 515–532. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M. G-Quadruplexes at Telomeres: Friend or Foe? Molecules 2020, 25, 3686. [Google Scholar] [CrossRef]
- Liano, D.; Chowdhury, S.; Di Antonio, M. Cockayne Syndrome B Protein Selectively Resolves and Interact with Intermolecular DNA G-Quadruplex Structures. J. Am. Chem. Soc. 2021, 143, 20988–21002. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef]
- Wu, X.; Malkova, A. Break-induced replication mechanisms in yeast and mammals. Curr. Opin. Genet. Dev. 2021, 71, 163–170. [Google Scholar] [CrossRef]
- Poole, L.A.; Zhao, R.; Glick, G.G.; Lovejoy, C.A.; Eischen, C.M.; Cortez, D. SMARCAL1 maintains telomere integrity during DNA replication. Proc. Natl. Acad. Sci. USA 2015, 112, 14864–14869. [Google Scholar] [CrossRef]
- Balzerano, A.; Paccosi, E.; Proietti-De-Santis, L. Evolutionary Mechanisms of Cancer Suggest Rational Therapeutic Approaches. Cytogenet. Genome Res. 2021, 161, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Filippi, S.; Latini, P.; Frontini, M.; Palitti, F.; Egly, J.-M.; Proietti-De-Santis, L. CSB protein is (a direct target of HIF-1 and) a critical mediator of the hypoxic response. EMBO J. 2008, 27, 2545–2556. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, G.; Li, W. Elevated Expression of ERCC6 Confers Resistance to 5-Fluorouracil and Is Associated with Poor Patient Survival in Colorectal Cancer. DNA Cell Biol. 2017, 36, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, R.; Tsao, H.-S.; Zeinomar, N.; Stagnar, C.; Fitzpatrick, S.; Dzutsev, A. Integrative genomic analysis implicates ERCC6 and its interaction with ERCC8 in susceptibility to breast cancer. Sci. Rep. 2020, 10, 21276. [Google Scholar] [CrossRef] [PubMed]
- Orta, M.L.; Calderón-Montaño, J.M.; Domínguez, I.; Pastor, N.; Burgos-Morón, E.; López-Lázaro, M.; Cortés, F.; Mateos, S.; Helleday, T. 5-Aza-2′-deoxycytidine causes replication lesions that require Fanconi anemia-dependent homologous recombination for repair. Nucleic Acids Res. 2013, 41, 5827–5836. [Google Scholar] [CrossRef]
- Orta, M.L.; Höglund, A.; Calderón-Montaño, J.M.; Domínguez, I.; Burgos-Morón, E.; Visnes, T.; Pastor, N.; Ström, C.; López-Lázaro, M.; Helleday, T. The PARP inhibitor Olaparib disrupts base excision repair of 5-aza-2′-deoxycytidine lesions. Nucleic Acids Res. 2014, 42, 9108–9120. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.T.; Sengerová, B.; Cattell, E.; Inagawa, T.; Hartley, J.M.; Kiakos, K.; Burgess-Brown, N.A.; Swift, L.P.; Enzlin, J.H.; Schofield, C.J.; et al. Human SNM1A and XPF–ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 2011, 25, 1859–1870. [Google Scholar] [CrossRef]
- Yang, Q.; Li, Y.; Sun, R.; Li, J. Identification of a RAD52 Inhibitor Inducing Synthetic Lethality in BRCA2-Deficient Cancer Cells. Front. Pharmacol. 2021, 12, 637825. [Google Scholar] [CrossRef]
- Shadrick, W.R.; Ndjomou, J.; Kolli, R.; Mukherjee, S.; Hanson, A.M.; Frick, D.N. Discovering New Medicines Targeting Helicases: Challenges and Recent Progress. J. Biomol. Sreen. 2013, 18, 761–781. [Google Scholar] [CrossRef]
- Papillon, J.P.N.; Nakajima, K.; Adair, C.D.; Hempel, J.; Jouk, A.O.; Karki, R.G.; Mathieu, S.; Mobitz, H.; Ntaganda, R.; Smith, T.; et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)SMARCA4-Mutant Cancers. J. Med. Chem. 2018, 61, 10155–10172. [Google Scholar] [CrossRef] [PubMed]
- Cupido, T.; Pisa, R.; Kelley, M.E.; Kapoor, T.M. Designing a chemical inhibitor for the AAA protein spastin using active site mutations. Nat. Chem. Biol. 2019, 15, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Pisa, R.; Kapoor, T.M. Chemical strategies to overcome resistance against targeted anticancer therapeutics. Nat. Chem. Biol. 2020, 16, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Zhang, X.; Zhang, R.; Chai, X.; Zhao, Y.; Li, E.; Zhang, Q.; Yan, R.; Yang, J.; Liao, B. Blockage of ERCC6 Alleviates Spinal Cord Injury Through Weakening Apoptosis, Inflammation, Senescence, and Oxidative Stress. Front. Mol. Biosci. 2022, 9, 853654. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, G.T.; van Steeg, H.; Berg, R.J.; van Gool, A.J.; de Wit, J.; Weeda, G.; Morreau, H.; Beems, R.B.; van Kreijl, C.F.; de Gruijl, F.R.; et al. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell 1997, 89, 425–435. [Google Scholar] [CrossRef]
- Montané, X.; Bajek, A.; Roszkowski, K.; Montornés, J.M.; Giamberini, M.; Roszkowski, S.; Kowalczyk, O.; Garcia-Valls, R.; Tylkowski, B. Encapsulation for Cancer Therapy. Molecules 2020, 25, 1605. [Google Scholar] [CrossRef]
- Breen, D.M.; Kim, H.; Bennett, D.; Calle, R.A.; Collins, S.; Esquejo, R.M.; He, T.; Joaquim, S.; Joyce, A.; Lambert, M.; et al. GDF-15 Neutralization Alleviates Platinum-Based Chemotherapy-Induced Emesis, Anorexia, and Weight Loss in Mice and Nonhuman Primates. Cell Metab. 2020, 32, 938–950.e6. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Modification | Amino Acids | ID of Site | Modifying Enzymes | Function | Cellular Phenotype * | References | ||
|---|---|---|---|---|---|---|---|---|
| MS | MA | Ab | ||||||
| Phosphorylation | S10 | – | Yes | Yes | ATM | Relief of CSB’s N-terminal inhibition; regulation of CSB’s chromatin remodeling | Sensitivity to olaparib but not UV | [16,30] |
| S158 | Yes | Yes | Yes | CDK | Relief of CSB’s N-terminal inhibition; regulation of CSB’s chromatin remodeling | Sensitivity to olaparib but not UV | [16,30] | |
| Y932 | Yes | – | Yes | c-Abl | Regulation of CSB localization following oxidative damage | – | [31] | |
| S1013 | – | Yes | – | Possible ATM/ATR | Regulation of CSB’s DNA translocase activity; regulation of fork restart and MiDAS | Sensitivity to olaparib ** but not UV | [32] | |
| T1031 | – | Yes | Yes | CDK | Regulation of the CSB-MRE11 interaction; CSB recruitment to stalled forks; MRE11 recruitment to stalled forks in BRCA1/2-deficient cells; fork degradation in BRCA1/2-deficient cells | No sensitivity to olaparib | [22] | |
| S1276 | Yes | Yes | Yes | CDK | Regulation of MRE11/RAD50/NBS1- and CtIP-mediated end resection; recruitment of BRCA1-C to DSBs; cell survival in response to olaparib and CPT | Sensitivity to olaparib and CPT | [33,34,35] | |
| SUMOylation | K205 | – | Yes | Yes | SUMO-2/3 | TC-NER and UV resistance | Sensitivity to UV | [36] |
| Ubiquitylation | K991 | Yes | Yes | – | Oxidative damage repair; transcription regulation | Sensitivity to paraquat or potassium bromate but not UV or cisplatin | [37] | |
| PARylation | – | – | – | – | PARP1 | Mitochondria DNA damage sensing | – | [38] |
| Agents | Mechanisms | Approved Chemotherapy * | References |
|---|---|---|---|
| Radiation | |||
| UV radiation | Intrastrand crosslink | [7] | |
| Ionizing radiation | Oxidative damage, single-stranded breaks, double-stranded breaks | Broadly used | [15,54,61] |
| Enzyme inhibitors | |||
| Camptothecin (CPT) (Analogs include topotecan and irinotecan) | Topoisomerase I inhibitor | Analogs used against ovarian cancer, cervical cancer, colorectal cancer, small cell lung cancer; pancreatic cancer | [15,33,62] |
| Etoposide (ETP) | Topoisomerase II inhibitor | Small cell lung cancer, testicular cancer | [15] |
| Olaparib | PARP inhibitor | Breast cancer, ovarian epithelial, fallopian tube or primary peritoneal cancer, pancreatic cancer, prostate cancer | [15,22,33] |
| 3-aminobenzene (3-AB) | PARP inhibitor | [63] | |
| 3,4-dihydro-5[4(1-piperidinyl)butoxy]- 1(2H)-isoquinolinone (DPQ) | PARP inhibitor | [54] | |
| 5-AZA-2′-deoxycytidine (5-azadC) (also known as Decitabine) | Traps DNA methyltransferases (DNMTs) | Myelodysplastic syndromes (MDS) including chronic myelomonocytic leukemia | [64] |
| 5-fluorouracil (5-FU) | Thymidylate synthase inhibitor | Breast cancer, colorectal cancer, gastric cancer, pancreatic cancer | [65] |
| Crosslinking chemicals | |||
| Cisplatin | Intrastrand and interstrand crosslink | Bladder cancer, ovarian cancer, testicular cancer | [22,37,66,67,68,69] |
| Mitomycin C | Interstrand crosslink | Gastric and pancreatic adenocarcinoma, urothelial cancer | [65,68] |
| Formaldehyde | Interstrand crosslink | [14] | |
| Trioxsalen | Interstrand crosslink | [70] | |
| Carboplatin | Intrastrand and interstrand crosslink | Ovarian cancer | [69] |
| Oxaliplatin | Intrastrand and interstrand crosslink | Colorectal cancer, Stage III colon cancer | [65] |
| Methylating chemicals | |||
| Methyl methanesulfonate (MMS) | DNA methylation | [71] | |
| Oxidizing chemicals | |||
| Paraquot | Oxidative damage | [37,61] | |
| Hydrogen peroxide | Oxidative damage | [61] | |
| Potassium Bromate | Oxidative damage | [37] | |
| Methanedione | Oxidative damage | [56] | |
| Nucleotide analog | |||
| 5-hydroxymethyl-2′-deoxyuridine (HmdU) | Thymidine analog | [71] | |
| Other chemicals | |||
| N-acetoxy-2- acetylaminofluorene (NA-AAF) | DNA adducts | [72,73,74] | |
| Angelicin | Monoadducts | [70] | |
| 4-nitroquinoline-1-oxide (4-NQO) | Single-stranded breaks and bulky adducts | [73,75,76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walker, J.R.; Zhu, X.-D. Role of Cockayne Syndrome Group B Protein in Replication Stress: Implications for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 10212. https://doi.org/10.3390/ijms231810212
Walker JR, Zhu X-D. Role of Cockayne Syndrome Group B Protein in Replication Stress: Implications for Cancer Therapy. International Journal of Molecular Sciences. 2022; 23(18):10212. https://doi.org/10.3390/ijms231810212
Chicago/Turabian StyleWalker, John R., and Xu-Dong Zhu. 2022. "Role of Cockayne Syndrome Group B Protein in Replication Stress: Implications for Cancer Therapy" International Journal of Molecular Sciences 23, no. 18: 10212. https://doi.org/10.3390/ijms231810212