
Biological Mechanisms to Reduce Radioresistance and Increase the Efficacy of Radiotherapy: State of the Art


{kind=link}
{kind=link}
Abstract
:1. Introduction
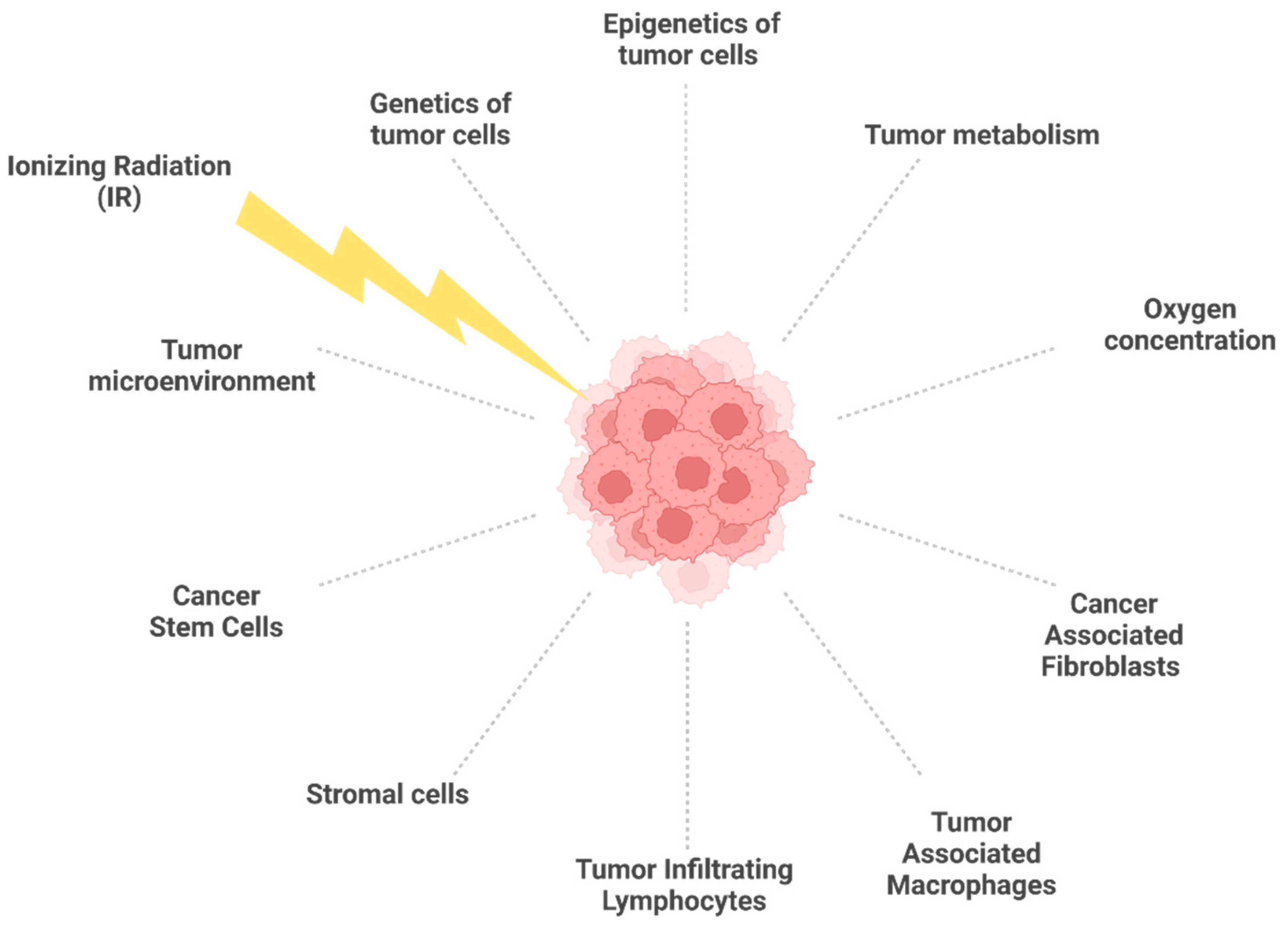
2. Molecular and Cellular Features Associated with Radioresistance
2.1. Basics of Radiobiology and Radiotherapy
2.2. Genetic and Epigenetic Features Associated with Radioresistance
2.2.1. Genetic Variants
2.2.2. DNA Methylation Changes
2.2.3. Histone Post-Translational Modifications
2.2.4. Transcriptomic Changes
2.3. Tumor Microenvironment and Tumor Metabolism in Radioresistant Tumors
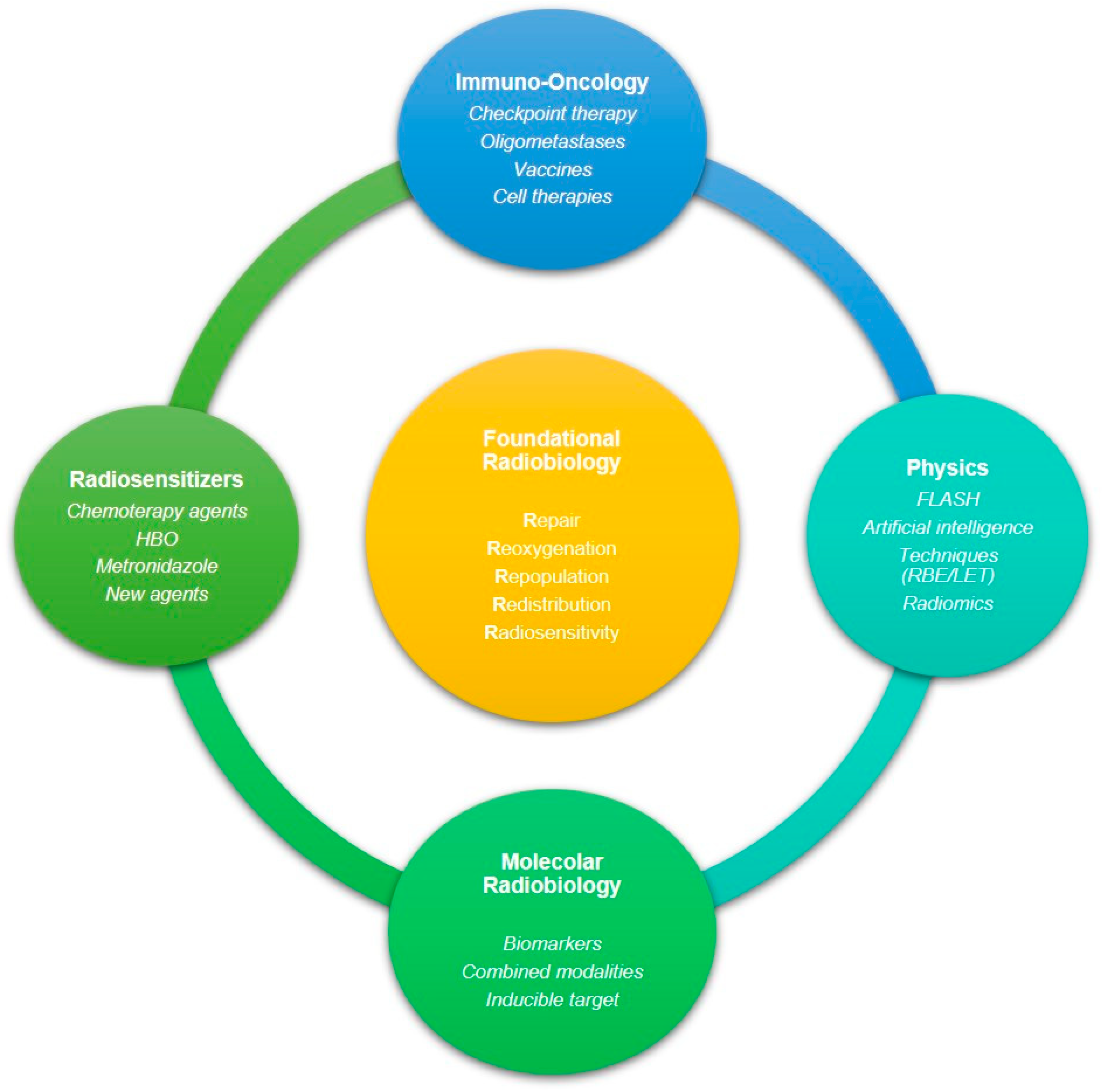
3. Targeting Strategies against Radioresistant Tumors
3.1. Hypoxic Cell Radiosensitizers
3.1.1. Hyperbaric Oxygen
3.1.2. Carbogen
3.1.3. Nicotinamide
3.1.4. Metronidazole and Its Analogs
3.1.5. Hypoxic Cell Cytotoxic Agents
3.2. Radiosensitizing Chemotherapy Agents
3.2.1. Fluoropyrimidines
3.2.2. Gemcitabine
3.2.3. Taxanes
3.2.4. Platinum-Based Drugs
3.2.5. Temozolomide
3.2.6. Histone Deacetylase Inhibitors (HDACi)
3.2.7. DNA Repair and Cell Cycle Inhibitors
3.3. Nanoparticles (NPs)
3.4. Immunomodulators
3.5. Radiation Therapy beyond Photons: Protons and Carbon Ions
3.5.1. SBRT
3.5.2. FLASH Radiotherapy
3.6. Diet
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Harper, J.W.; Elledge, S.J. The DNA Damage Response: Ten Years After. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological Consequences of Radiation-Induced DNA Damage: Relevance to Radiotherapy. Clin. Oncol. (R. Coll. Radiol.) 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical Mechanism of DSB End Resection and Its Regulation. DNA Repair (Amst.) 2015, 32, 66–74. [Google Scholar] [CrossRef]
- Dayal, R.; Singh, A.; Pandey, A.; Mishra, K.P. Reactive Oxygen Species as Mediator of Tumor Radiosensitivity. J. Cancer Res. Ther. 2014, 10, 811–818. [Google Scholar] [CrossRef]
- Panieri, E.; Gogvadze, V.; Norberg, E.; Venkatesh, R.; Orrenius, S.; Zhivotovsky, B. Reactive Oxygen Species Generated in Different Compartments Induce Cell Death, Survival, or Senescence. Free Radic. Biol. Med. 2013, 57, 176–187. [Google Scholar] [CrossRef]
- Beheshti, A.; McDonald, J.T.; Hada, M.; Takahashi, A.; Mason, C.E.; Mognato, M. Genomic Changes Driven by Radiation-Induced DNA Damage and Microgravity in Human Cells. Int. J. Mol. Sci. 2021, 22, 10507. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early Steps in the DNA Base Excision/Single-Strand Interruption Repair Pathway in Mammalian Cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef]
- Grifalconi, M.; Celotti, L.; Mognato, M. Bystander Response in Human Lymphoblastoid TK6 Cells. Mutat. Res. 2007, 625, 102–111. [Google Scholar] [CrossRef]
- Marín, I.; Boix, O.; García, A.; Sirois, I.; Caballe, A.; Zarzuela, E.; Ruano, I.; Attolini, C.S.-O.; Prats, N.; López-Domínguez, J.A.; et al. Induction of Senescence Renders Cancer Cells Highly Immunogenic. bioRxiv 2022. [Google Scholar] [CrossRef]
- Loaiza, N.; Demaria, M. Cellular Senescence and Tumor Promotion: Is Aging the Key? Biochim. Biophys. Acta 2016, 1865, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Brandenburg, S.; Demaria, M. Induction and Validation of Cellular Senescence in Primary Human Cells. J. Vis. Exp. 2018, 2018, 57782. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, D.T.; Nikjoo, H. Track Structure Analysis of Ultrasoft X-Rays Compared to High- and Low-LET Radiations. Int. J. Radiat. Biol. 1989, 55, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, F.A.; Durante, M. Cancer Risk from Exposure to Galactic Cosmic Rays: Implications for Space Exploration by Human Beings. Lancet Oncol. 2006, 7, 431–435. [Google Scholar] [CrossRef]
- Ray, S.; Cekanaviciute, E.; Lima, I.P.; Sørensen, B.S.; Costes, S.V. Comparing Photon and Charged Particle Therapy Using DNA Damage Biomarkers. Int. J. Part Ther. 2018, 5, 15–24. [Google Scholar] [CrossRef]
- Paganetti, H. Relative Biological Effectiveness (RBE) Values for Proton Beam Therapy. Variations as a Function of Biological Endpoint, Dose, and Linear Energy Transfer. Phys. Med. Biol. 2014, 59, R419–R472. [Google Scholar] [CrossRef]
- Palumbo, E.; Piotto, C.; Calura, E.; Fasanaro, E.; Groff, E.; Busato, F.; El Khouzai, B.; Rigo, M.; Baggio, L.; Romualdi, C.; et al. Individual Radiosensitivity in Oncological Patients: Linking Adverse Normal Tissue Reactions and Genetic Features. Front. Oncol. 2019, 9, 987. [Google Scholar] [CrossRef]
- Klement, R.J.; Champ, C.E. Calories, Carbohydrates, and Cancer Therapy with Radiation: Exploiting the Five R’s through Dietary Manipulation. Cancer Metast. Rev. 2014, 33, 217–229. [Google Scholar] [CrossRef]
- Shu, H.K.; Kim, M.M.; Chen, P.; Furman, F.; Julin, C.M.; Israel, M.A. The Intrinsic Radioresistance of Glioblastoma-Derived Cell Lines Is Associated with a Failure of P53 to Induce P21(BAX) Expression. Proc. Natl. Acad. Sci. USA 1998, 95, 14453–14458. [Google Scholar] [CrossRef]
- Sato, K.; Shimokawa, T.; Imai, T. Difference in Acquired Radioresistance Induction Between Repeated Photon and Particle Irradiation. Front. Oncol. 2019, 9, 1213. [Google Scholar] [CrossRef]
- Galeaz, C.; Totis, C.; Bisio, A. Radiation Resistance: A Matter of Transcription Factors. Front. Oncol. 2021, 11, 662840. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, Y.; Li, L.; Baba, T.; Nakagawa, H.; Shimura, T.; Yamamoto, Y.; Ohkubo, Y.; Fukumoto, M. Clinically Relevant Radioresistant Cells Efficiently Repair DNA Double-Strand Breaks Induced by X-Rays. Cancer Sci. 2009, 100, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, Y.; Roudkenar, M.H.; Urushihara, Y.; Saito, Y.; Tomita, K.; Roushandeh, A.M.; Sato, T.; Kurimasa, A.; Fukumoto, M. Clinically Relevant Radioresistant Cell Line: A Simple Model to Understand Cancer Radioresistance. Med. Mol. Morphol. 2017, 50, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.M.; Lynam-Lennon, N.; O’Neill, H.; O’Sullivan, J. Targeting Hallmarks of Cancer to Enhance Radiosensitivity in Gastrointestinal Cancers. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 298–313. [Google Scholar] [CrossRef]
- Bee, L.; Fabris, S.; Cherubini, R.; Mognato, M.; Celotti, L. The Efficiency of Homologous Recombination and Non-Homologous End Joining Systems in Repairing Double-Strand Breaks during Cell Cycle Progression. PLoS ONE 2013, 8, e69061. [Google Scholar] [CrossRef]
- Piotto, C.; Biscontin, A.; Millino, C.; Mognato, M. Functional Validation of MiRNAs Targeting Genes of DNA Double-Strand Break Repair to Radiosensitize Non-Small Lung Cancer Cells. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 1102–1118. [Google Scholar] [CrossRef]
- Szymonowicz, K.; Krysztofiak, A.; van der Linden, J.; Kern, A.; Deycmar, S.; Oeck, S.; Squire, A.; Koska, B.; Hlouschek, J.; Vüllings, M.; et al. Proton Irradiation Increases the Necessity for Homologous Recombination Repair Along with the Indispensability of Non-Homologous End Joining. Cells 2020, 9, 889. [Google Scholar] [CrossRef]
- Fontana, A.O.; Augsburger, M.A.; Grosse, N.; Guckenberger, M.; Lomax, A.J.; Sartori, A.A.; Pruschy, M.N. Differential DNA Repair Pathway Choice in Cancer Cells after Proton- and Photon-Irradiation. Radiother. Oncol. 2015, 116, 374–380. [Google Scholar] [CrossRef]
- Gerelchuluun, A.; Manabe, E.; Ishikawa, T.; Sun, L.; Itoh, K.; Sakae, T.; Suzuki, K.; Hirayama, R.; Asaithamby, A.; Chen, D.J.; et al. The Major DNA Repair Pathway after Both Proton and Carbon-Ion Radiation Is NHEJ, but the HR Pathway Is More Relevant in Carbon Ions. Radiat Res 2015, 183, 345–356. [Google Scholar] [CrossRef]
- Watanabe, R.; Wada, S.; Funayama, T.; Kobayashi, Y.; Saito, K.; Furusawa, Y. Monte Carlo Simulation of Radial Distribution of DNA Strand Breaks along the C and Ne Ion Paths. Radiat. Prot. Dosimetry 2011, 143, 186–190. [Google Scholar] [CrossRef]
- Qiao, G.-B.; Wu, Y.-L.; Yang, X.-N.; Zhong, W.-Z.; Xie, D.; Guan, X.-Y.; Fischer, D.; Kolberg, H.-C.; Kruger, S.; Stuerzbecher, H.-W. High-Level Expression of Rad51 Is an Independent Prognostic Marker of Survival in Non-Small-Cell Lung Cancer Patients. Br. J. Cancer 2005, 93, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhang, S.; Sun, Y.; Gu, J.; Ye, Z.; Sun, X.; Tang, Q. A Radioresponse-Related LncRNA Biomarker Signature for Risk Classification and Prognosis Prediction in Non-Small-Cell Lung Cancer. J. Oncol. 2021, 2021, 4338838. [Google Scholar] [CrossRef] [PubMed]
- Maacke, H.; Jost, K.; Opitz, S.; Miska, S.; Yuan, Y.; Hasselbach, L.; Lüttges, J.; Kalthoff, H.; Stürzbecher, H.W. DNA Repair and Recombination Factor Rad51 Is Over-Expressed in Human Pancreatic Adenocarcinoma. Oncogene 2000, 19, 2791–2795. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Z.; Zhao, L.; Li, L.; Zuo, W.; Han, L. High Expression of RAD51 Promotes DNA Damage Repair and Survival in KRAS-Mutant Lung Cancer Cells. BMB Rep. 2019, 52, 151–156. [Google Scholar] [CrossRef]
- Vaezi, A.; Feldman, C.H.; Niedernhofer, L.J. ERCC1 and XRCC1 as Biomarkers for Lung and Head and Neck Cancer. Pharmgenom. Pers. Med. 2011, 4, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Carlos-Reyes, A.; Muñiz-Lino, M.A.; Romero-Garcia, S.; López-Camarillo, C.; Hernández-de la Cruz, O.N. Biological Adaptations of Tumor Cells to Radiation Therapy. Front. Oncol. 2021, 11, 718636. [Google Scholar] [CrossRef]
- Todorovic, V.; Prevc, A.; Zakelj, M.N.; Savarin, M.; Brozic, A.; Groselj, B.; Strojan, P.; Cemazar, M.; Sersa, G. Mechanisms of Different Response to Ionizing Irradiation in Isogenic Head and Neck Cancer Cell Lines. Radiat. Oncol. 2019, 14, 214. [Google Scholar] [CrossRef]
- Li, X.; Zou, S.; Zhou, L.; Gao, A.; Xu, J.; He, C.; Zhou, J.; Wu, S.; Chen, Y. RAD18 Confers Radioresistance of Esophagus Squamous Cell Carcinoma through Regulating P-DNA-PKcs. Cancer Med. 2022. [Google Scholar] [CrossRef]
- Kokunai, T.; Tamaki, N. Relationship between Expression of P21WAF1/CIP1 and Radioresistance in Human Gliomas. Jpn. J. Cancer Res. 1999, 90, 638–646. [Google Scholar] [CrossRef]
- Nguyen, L.; Dobiasch, S.; Schneider, G.; Schmid, R.M.; Azimzadeh, O.; Kanev, K.; Buschmann, D.; Pfaffl, M.W.; Bartzsch, S.; Schmid, T.E.; et al. Impact of DNA Repair and Reactive Oxygen Species Levels on Radioresistance in Pancreatic Cancer. Radiother. Oncol. 2021, 159, 265–276. [Google Scholar] [CrossRef]
- Kuchur, O.A.; Kuzmina, D.O.; Dukhinova, M.S.; Shtil, A.A. The P53 Protein Family in the Response of Tumor Cells to Ionizing Radiation: Problem Development. Acta Nat. 2021, 13, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, N.; Grill, J.; Geoerger, B.; Lellouch-Tubiana, A.; Michalowski, M.B.; Vassal, G. P53 Pathway Dysfunction in Primary Childhood Ependymomas. Pediatr. Blood Cancer 2006, 46, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.H.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-Specific Prognostic Implications of TP53 Mutation in Medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef]
- Yogev, O.; Barker, K.; Sikka, A.; Almeida, G.S.; Hallsworth, A.; Smith, L.M.; Jamin, Y.; Ruddle, R.; Koers, A.; Webber, H.T.; et al. P53 Loss in MYC-Driven Neuroblastoma Leads to Metabolic Adaptations Supporting Radioresistance. Cancer Res. 2016, 76, 3025–3035. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, A.; Minaguchi, T.; Fujieda, K.; Hosokawa, Y.; Nishida, K.; Shikama, A.; Tasaka, N.; Sakurai, M.; Ochi, H.; Satoh, T. Abnormal Accumulation of P53 Predicts Radioresistance Leading to Poor Survival in Patients with Endometrial Carcinoma. Oncol. Lett. 2019, 18, 5952–5958. [Google Scholar] [CrossRef] [PubMed]
- Skinner, H.D.; Sandulache, V.C.; Ow, T.J.; Meyn, R.E.; Yordy, J.S.; Beadle, B.M.; Fitzgerald, A.L.; Giri, U.; Ang, K.K.; Myers, J.N. TP53 Disruptive Mutations Lead to Head and Neck Cancer Treatment Failure through Inhibition of Radiation-Induced Senescence. Clin. Cancer Res. 2012, 18, 290–300. [Google Scholar] [CrossRef]
- Casey, D.L.; Pitter, K.L.; Wexler, L.H.; Slotkin, E.K.; Gupta, G.P.; Wolden, S.L. TP53 Mutations Increase Radioresistance in Rhabdomyosarcoma and Ewing Sarcoma. Br. J. Cancer 2021, 125, 576–581. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Chiba, I.; Hirai, A.; Notani, K.; Kashiwazaki, H.; Tei, K.; Totsuka, Y.; Iizuka, T.; Kohgo, T.; Fukuda, H. Radioresistance in Oral Squamous Cell Carcinoma with P53 DNA Contact Mutation. Am. J. Clin. Oncol. 2003, 26, e124-9. [Google Scholar] [CrossRef]
- Singh, V.; Gupta, D.; Arora, R. NF-KB as a Key Player in Regulation of Cellular Radiation Responses and Identification of Radiation Countermeasures. Discoveries (Craiova) 2015, 3, e35. [Google Scholar] [CrossRef]
- Moussata, D.; Amara, S.; Siddeek, B.; Decaussin, M.; Hehlgans, S.; Paul-Bellon, R.; Mornex, F.; Gerard, J.-P.; Romestaing, P.; Rödel, F.; et al. XIAP as a Radioresistance Factor and Prognostic Marker for Radiotherapy in Human Rectal Adenocarcinoma. Am. J. Pathol. 2012, 181, 1271–1278. [Google Scholar] [CrossRef]
- Karki, R.; Pandya, D.; Elston, R.C.; Ferlini, C. Defining “Mutation” and “Polymorphism” in the Era of Personal Genomics. BMC Med. Genom. 2015, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Brookes, A.J. The Essence of SNPs. Gene 1999, 234, 177–186. [Google Scholar] [CrossRef]
- Werbrouck, C.; Evangelista, C.C.S.; Lobón-Iglesias, M.-J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788–6800. [Google Scholar] [CrossRef] [PubMed]
- Angèle, S.; Romestaing, P.; Moullan, N.; Vuillaume, M.; Chapot, B.; Friesen, M.; Jongmans, W.; Cox, D.G.; Pisani, P.; Gérard, J.-P.; et al. ATMHaplotypes and Cellular Response to DNA Damage: Association with Breast Cancer Risk and Clinical Radiosensitivity. Cancer Res. 2003, 63, 8717–8725. [Google Scholar]
- Qiu, L.; Feng, H.; Yu, H.; Li, M.; You, Y.; Zhu, S.; Yang, W.; Jiang, H.; Wu, X. Characterization of the Genomic Landscape in Cervical Cancer by Next Generation Sequencing. Genes 2022, 13, 287. [Google Scholar] [CrossRef]
- Bernichon, E.; Vallard, A.; Wang, Q.; Attignon, V.; Pissaloux, D.; Bachelot, T.; Heudel, P.E.; Ray-Coquard, I.; Bonnet, E.; de la Fouchardière, A.; et al. Genomic Alterations and Radioresistance in Breast Cancer: An Analysis of the ProfiLER Protocol. Ann. Oncol. 2017, 28, 2773–2779. [Google Scholar] [CrossRef]
- Jiang, C.; Guo, Y.; Li, Y.; Kang, J.; Sun, X.; Wu, H.; Feng, J.; Xu, Y. The Association between the ERCC1/2 Polymorphisms and Radiotherapy Efficacy in 87 Patients with Non-Small Cell Lung Cancer. J. Thorac. Dis. 2021, 13, 3126–3136. [Google Scholar] [CrossRef]
- Jin, J.-Y.; Wang, W.; Ten Haken, R.K.; Chen, J.; Bi, N.; Sadek, R.; Zhang, H.; Lawrence, T.S.; Kong, F.-M. (Spring) Use a Survival Model to Correlate Single-Nucleotide Polymorphisms of DNA Repair Genes with Radiation Dose–Response in Patients with Non-Small Cell Lung Cancer. Radiother. Oncol. 2015, 117, 77–82. [Google Scholar] [CrossRef]
- Gong, L.; Luo, M.; Sun, R.; Qiu, L.; Chen, C.; Luo, Z. Significant Association Between XRCC1 Expression and Its Rs25487 Polymorphism and Radiotherapy-Related Cancer Prognosis. Front. Oncol. 2021, 11, 654784. [Google Scholar] [CrossRef]
- Zhao, X.-M.; Xiang, Z.-L.; Chen, Y.-X.; Yang, P.; Hu, Y.; Zeng, Z.-C. A Sequence Polymorphism on 8q24 Is Associated with Survival in Hepatocellular Carcinoma Patients Who Received Radiation Therapy. Sci. Rep. 2018, 8, 2264. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miousse, I.R.; Kutanzi, K.R.; Koturbash, I. Effects of Ionizing Radiation on DNA Methylation: From Experimental Biology to Clinical Applications. Int. J. Radiat. Biol. 2017, 93, 457–469. [Google Scholar] [CrossRef]
- Zhai, G.; Li, J.; Zheng, J.; An, P.; Chen, X.; Wang, X.; Li, C. HTERT Promoter Methylation Promotes Small Cell Lung Cancer Progression and Radiotherapy Resistance. J. Radiat. Res. 2020, 61, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, R.; Wang, J. Characterization of a Prognostic Four-gene Methylation Signature Associated with Radiotherapy for Head and Neck Squamous Cell Carcinoma. Mol. Med. Rep. 2019, 20, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kwon, J.; Kim, D.; Park, M.; Kim, K.; Bae, I.; Kim, H.; Kong, J.; Kim, Y.; Shin, U.; et al. Gene Expression Profiles Associated with Radio-Responsiveness in Locally Advanced Rectal Cancer. Biology 2021, 10, 500. [Google Scholar] [CrossRef]
- Wu, C.; Guo, E.; Ming, J.; Sun, W.; Nie, X.; Sun, L.; Peng, S.; Luo, M.; Liu, D.; Zhang, L.; et al. Radiation-Induced DNMT3B Promotes Radioresistance in Nasopharyngeal Carcinoma through Methylation of P53 and P21. Mol. Ther. Oncolytics 2020, 17, 306–319. [Google Scholar] [CrossRef]
- Kim, S.-H.; Kang, B.-C.; Seong, D.; Lee, W.-H.; An, J.-H.; Je, H.-U.; Cha, H.-J.; Chang, H.-W.; Kim, S.-Y.; Kim, S.-W.; et al. EPHA3 Contributes to Epigenetic Suppression of PTEN in Radioresistant Head and Neck Cancer. Biomolecules 2021, 11, 599. [Google Scholar] [CrossRef]
- Hunt, C.R.; Ramnarain, D.; Horikoshi, N.; Iyengar, P.; Pandita, R.K.; Shay, J.W.; Pandita, T.K. Histone Modifications and DNA Double-Strand Break Repair after Exposure to Ionizing Radiations. Radiat. Res. 2013, 179, 383–392. [Google Scholar] [CrossRef]
- Tjeertes, J.V.; Miller, K.M.; Jackson, S.P. Screen for DNA-Damage-Responsive Histone Modifications Identifies H3K9Ac and H3K56Ac in Human Cells. EMBO J. 2009, 28, 1878–1889. [Google Scholar] [CrossRef]
- Sharma, A.K.; Bhattacharya, S.; Khan, S.A.; Khade, B.; Gupta, S. Dynamic Alteration in H3 Serine 10 Phosphorylation Is G1-Phase Specific during Ionization Radiation Induced DNA Damage Response in Human Cells. Mutat. Res. 2015, 773, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Sharda, A.; Rashid, M.; Shah, S.G.; Sharma, A.K.; Singh, S.R.; Gera, P.; Chilkapati, M.K.; Gupta, S. Elevated HDAC Activity and Altered Histone Phospho-Acetylation Confer Acquired Radio-Resistant Phenotype to Breast Cancer Cells. Clin. Epigenet. 2020, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Cojoc, M.; Hein, L.; Kurth, I.; Mäbert, K.; Trautmann, F.; Klink, B.; Schröck, E.; Wirth, M.P.; Krause, M.; et al. An Epigenetic Reprogramming Strategy to Resensitize Radioresistant Prostate Cancer Cells. Cancer Res. 2016, 76, 2637–2651. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde Dehydrogenase 1A1 in Stem Cells and Cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, N.; Balaji, K.; Jayachandran, K.; Inkman, M.; Zhang, J.; Dahiya, S.; Goldstein, M. Loss of H3K27 Trimethylation Promotes Radiotherapy Resistance in Medulloblastoma and Induces an Actionable Vulnerability to BET Inhibition. Cancer Res. 2022, 82, 2019–2030. [Google Scholar] [CrossRef]
- Mognato, M.; Burdak-Rothkamm, S.; Rothkamm, K. Interplay between DNA Replication Stress, Chromatin Dynamics and DNA-Damage Response for the Maintenance of Genome Stability. Mutat. Res./Rev. Mutat. Res. 2021, 787, 108346. [Google Scholar] [CrossRef]
- Storch, K.; Eke, I.; Borgmann, K.; Krause, M.; Richter, C.; Becker, K.; Schröck, E.; Cordes, N. Three-Dimensional Cell Growth Confers Radioresistance by Chromatin Density Modification. Cancer Res. 2010, 70, 3925–3934. [Google Scholar] [CrossRef]
- Chung, C.H.; Parker, J.S.; Ely, K.; Carter, J.; Yi, Y.; Murphy, B.A.; Ang, K.K.; El-Naggar, A.K.; Zanation, A.M.; Cmelak, A.J.; et al. Gene Expression Profiles Identify Epithelial-to-Mesenchymal Transition and Activation of Nuclear Factor-KappaB Signaling as Characteristics of a High-Risk Head and Neck Squamous Cell Carcinoma. Cancer Res. 2006, 66, 8210–8218. [Google Scholar] [CrossRef]
- Shen, S.; Bai, J.; Wei, Y.; Wang, G.; Li, Q.; Zhang, R.; Duan, W.; Yang, S.; Du, M.; Zhao, Y.; et al. A Seven-Gene Prognostic Signature for Rapid Determination of Head and Neck Squamous Cell Carcinoma Survival. Oncol. Rep. 2017, 38, 3403–3411. [Google Scholar] [CrossRef]
- Patil, S.; Tawk, B.; Grosser, M.; Lohaus, F.; Gudziol, V.; Kemper, M.; Nowak, A.; Haim, D.; Tinhofer, I.; Budach, V.; et al. Analyses of Molecular Subtypes and Their Association to Mechanisms of Radioresistance in Patients with HPV-Negative HNSCC Treated by Postoperative Radiochemotherapy. Radiother. Oncol. 2022, 167, 300–307. [Google Scholar] [CrossRef]
- Foy, J.-P.; Bazire, L.; Ortiz-Cuaran, S.; Deneuve, S.; Kielbassa, J.; Thomas, E.; Viari, A.; Puisieux, A.; Goudot, P.; Bertolus, C.; et al. A 13-Gene Expression-Based Radioresistance Score Highlights the Heterogeneity in the Response to Radiation Therapy across HPV-Negative HNSCC Molecular Subtypes. BMC Med. 2017, 15, 165. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, X.; Wang, J.; Wang, J.; Liu, X.; Huang, R.; Chen, C.; Deng, M.; Wang, H.; Han, F. Key Radioresistance Regulation Models and Marker Genes Identified by Integrated Transcriptome Analysis in Nasopharyngeal Carcinoma. Cancer Med. 2021, 10, 7404–7417. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear Long Noncoding RNAs: Key Regulators of Gene Expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Saitoh, N. Non-Coding RNAs and Chromatin Domains. Curr. Opin. Cell Biol. 2019, 58, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene Regulation by Long Non-Coding RNAs and Its Biological Functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Mognato, M.; Celotti, L. MicroRNAs Used in Combination with Anti-Cancer Treatments Can Enhance Therapy Efficacy. Mini Rev. Med. Chem. 2015, 15, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-C.; Du, L.-Q.; Tian, L.-L.; Wu, H.-L.; Jiang, X.-Y.; Zhang, H.; Li, D.-G.; Wang, Y.-Y.; Wu, H.-Y.; She, Y.; et al. Expression and Function of MiRNA in Postoperative Radiotherapy Sensitive and Resistant Patients of Non-Small Cell Lung Cancer. Lung Cancer 2011, 72, 92–99. [Google Scholar] [CrossRef]
- Aranza-Martínez, A.; Sánchez-Pérez, J.; Brito-Elias, L.; López-Camarillo, C.; Cantú de León, D.; Pérez-Plasencia, C.; López-Urrutia, E. Non-Coding RNAs Associated With Radioresistance in Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 752270. [Google Scholar] [CrossRef]
- de Mey, S.; Dufait, I.; De Ridder, M. Radioresistance of Human Cancers: Clinical Implications of Genetic Expression Signatures. Front. Oncol. 2021, 11, 761901. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Y.; Zhang, S.J.; Ma, Y.N.; He, Y. Comprehensive Analysis of Key Genes and MicroRNAs in Radioresistant Nasopharyngeal Carcinoma. BMC Med. Genom. 2019, 12, 73. [Google Scholar] [CrossRef]
- McDermott, N.; Meunier, A.; Wong, S.; Buchete, V.; Marignol, L. Profiling of a Panel of Radioresistant Prostate Cancer Cells Identifies Deregulation of Key MiRNAs. Clin. Transl. Radiat. Oncol. 2017, 2, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daemen, A.; Cooper, J.E.; Myrta, S.; Wongchenko, M.J.; Lin, E.; Long, J.E.; Foreman, O.; Modrusan, Z.; Tremayne, J.R.; de la Cruz, C.C.; et al. Transcriptional Subtypes Resolve Tumor Heterogeneity and Identify Vulnerabilities to MEK Inhibition in Lung Adenocarcinoma. Clin. Cancer Res. 2021, 27, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Niemira, M.; Collin, F.; Szalkowska, A.; Bielska, A.; Chwialkowska, K.; Reszec, J.; Niklinski, J.; Kwasniewski, M.; Kretowski, A. Molecular Signature of Subtypes of Non-Small-Cell Lung Cancer by Large-Scale Transcriptional Profiling: Identification of Key Modules and Genes by Weighted Gene Co-Expression Network Analysis (WGCNA). Cancers 2019, 12, 37. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, S.; He, J. The Mechanism of Long Non-Coding RNA in Cancer Radioresistance/Radiosensitivity: A Systematic Review. Front. Pharmacol. 2022, 13, 879704. [Google Scholar] [CrossRef] [PubMed]
- Matschke, J.; Larafa, S.; Jendrossek, V. Metabolic Reprograming of Antioxidant Defense: A Precision Medicine Perspective for Radiotherapy of Lung Cancer? Biochem. Soc. Trans. 2021, 49, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A Peek into Cancer-Associated Fibroblasts: Origins, Functions and Translational Impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef]
- Ansems, M.; Span, P.N. The Tumor Microenvironment and Radiotherapy Response; a Central Role for Cancer-Associated Fibroblasts. Clin. Transl. Radiat. Oncol. 2020, 22, 90–97. [Google Scholar] [CrossRef]
- Li, H.; Fan, X.; Houghton, J. Tumor Microenvironment: The Role of the Tumor Stroma in Cancer. J. Cell Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef]
- Brewer, G.; Fortier, A.-M.; Park, M.; Moraes, C. The Case for Cancer-Associated Fibroblasts: Essential Elements in Cancer Drug Discovery? Future Drug Discov. 2022, 4, FDD71. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Lindström, A.; Midtbö, K.; Arnesson, L.-G.; Garvin, S.; Shabo, I. Fusion between M2-Macrophages and Cancer Cells Results in a Subpopulation of Radioresistant Cells with Enhanced DNA-Repair Capacity. Oncotarget 2017, 8, 51370–51386. [Google Scholar] [CrossRef] [PubMed]
- Jang, B.-S.; Kim, I.A. Relationship between Macrophage and Radiosensitivity in Human Primary and Recurrent Glioblastoma: In Silico Analysis with Publicly Available Datasets. Biomedicines 2022, 10, 292. [Google Scholar] [CrossRef] [PubMed]
- Fu, E.; Liu, T.; Yu, S.; Chen, X.; Song, L.; Lou, H.; Ma, F.; Zhang, S.; Hussain, S.; Guo, J.; et al. M2 Macrophages Reduce the Radiosensitivity of Head and Neck Cancer by Releasing HB-EGF. Oncol. Rep. 2020, 44, 698–710. [Google Scholar] [CrossRef]
- Monjazeb, A.M.; Schalper, K.A.; Villarroel-Espindola, F.; Nguyen, A.; Shiao, S.L.; Young, K. Effects of Radiation on the Tumor Microenvironment. Semin. Radiat. Oncol. 2020, 30, 145–157. [Google Scholar] [CrossRef]
- Guo, S.; Chen, X.; Guo, C.; Wang, W. Tumour-Associated Macrophages Heterogeneity Drives Resistance to Clinical Therapy. Expert Rev. Mol. Med. 2022, 24, e17. [Google Scholar] [CrossRef]
- Ragunathan, K.; Upfold, N.L.E.; Oksenych, V. Interaction between Fibroblasts and Immune Cells Following DNA Damage Induced by Ionizing Radiation. Int. J. Mol. Sci. 2020, 21, 8635. [Google Scholar] [CrossRef]
- Abad, E.; Graifer, D.; Lyakhovich, A. DNA Damage Response and Resistance of Cancer Stem Cells. Cancer Lett. 2020, 474, 106–117. [Google Scholar] [CrossRef]
- Skvortsov, S.; Debbage, P.; Lukas, P.; Skvortsova, I. Crosstalk between DNA Repair and Cancer Stem Cell (CSC) Associated Intracellular Pathways. Semin. Cancer Biol. 2015, 31, 36–42. [Google Scholar] [CrossRef]
- Arnold, C.R.; Mangesius, J.; Skvortsova, I.-I.; Ganswindt, U. The Role of Cancer Stem Cells in Radiation Resistance. Front. Oncol. 2020, 10, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Cancer Stem Cells: Role in Tumor Growth, Recurrence, Metastasis, and Treatment Resistance. Medicine 2016, 95, S20–S25. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Begg, K.; Tavassoli, M. Inside the Hypoxic Tumour: Reprogramming of the DDR and Radioresistance. Cell Death Discov. 2020, 6, 77. [Google Scholar] [CrossRef]
- Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules 2019, 9, 339. [Google Scholar] [CrossRef]
- Rakotomalala, A.; Escande, A.; Furlan, A.; Meignan, S.; Lartigau, E. Hypoxia in Solid Tumors: How Low Oxygenation Impacts the “Six Rs” of Radiotherapy. Front. Endocrinol. 2021, 12, 742215. [Google Scholar] [CrossRef]
- Bader, S.B.; Dewhirst, M.W.; Hammond, E.M. Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer. Cancers 2020, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting Tumour Hypoxia in Cancer Treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Grimes, D.R.; Partridge, M. A Mechanistic Investigation of the Oxygen Fixation Hypothesis and Oxygen Enhancement Ratio. Biomed. Phys. Eng. Express 2015, 1, 045209. [Google Scholar] [CrossRef]
- Olcina, M.M.; Grand, R.J.; Hammond, E.M. ATM Activation in Hypoxia-Causes and Consequences. Mol. Cell Oncol. 2014, 1, e29903. [Google Scholar] [CrossRef] [Green Version]
- Hauth, F.; Toulany, M.; Zips, D.; Menegakis, A. Cell-Line Dependent Effects of Hypoxia Prior to Irradiation in Squamous Cell Carcinoma Lines. Clin. Transl. Radiat. Oncol. 2017, 5, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Bian, Y.; Liu, L.; Liu, L.; Liu, X.; Ma, S. Molecular Pathways Associated with Oxidative Stress and Their Potential Applications in Radiotherapy (Review). Int. J. Mol. Med. 2022, 49, 65. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and Glucose Metabolism in the Solid Tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Harada, H. Hypoxia-Inducible Factor 1-Mediated Characteristic Features of Cancer Cells for Tumor Radioresistance. J. Radiat. Res. 2016, 57 (Suppl. 1), i99–i105. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in Hypoxia-Mediated Apoptosis, Cell Proliferation and Tumour Angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Jin, X.; Kuang, Y.; Li, L.; Li, H.; Zhao, T.; He, Y.; Di, C.; Kang, J.; Yuan, L.; Yu, B.; et al. A Positive Feedback Circuit Comprising P21 and HIF-1α Aggravates Hypoxia-Induced Radioresistance of Glioblastoma by Promoting Glut1/LDHA-Mediated Glycolysis. FASEB J. 2022, 36, e22229. [Google Scholar] [CrossRef]
- Wu, Q.; You, L.; Nepovimova, E.; Heger, Z.; Wu, W.; Kuca, K.; Adam, V. Hypoxia-Inducible Factors: Master Regulators of Hypoxic Tumor Immune Escape. J. Hematol. Oncol. 2022, 15, 77. [Google Scholar] [CrossRef]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 Regulatory Pathway and Its Potential for Therapeutic Intervention in Malignancy and Ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar]
- McCann, E.; O’Sullivan, J.; Marcone, S. Targeting Cancer-Cell Mitochondria and Metabolism to Improve Radiotherapy Response. Transl. Oncol. 2021, 14, 100905. [Google Scholar] [CrossRef]
- Tang, L.; Wei, F.; Wu, Y.; He, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Deng, H.; et al. Role of Metabolism in Cancer Cell Radioresistance and Radiosensitization Methods. J. Exp. Clin. Cancer Res. 2018, 37, 87. [Google Scholar] [CrossRef] [PubMed]
- Krysztofiak, A.; Szymonowicz, K.; Hlouschek, J.; Xiang, K.; Waterkamp, C.; Larafa, S.; Goetting, I.; Vega-Rubin-de-Celis, S.; Theiss, C.; Matschke, V.; et al. Metabolism of Cancer Cells Commonly Responds to Irradiation by a Transient Early Mitochondrial Shutdown. iScience 2021, 24, 103366. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Jiang, H.; Sun, X.; Xu, H.; Wei, X.; Wei, Y.; Xiao, G.; Song, Z.; Zhou, F. Mitochondrial Dysfunction Induces Radioresistance in Colorectal Cancer by Activating [Ca2+]m-PDP1-PDH-Histone Acetylation Retrograde Signaling. Cell Death Dis. 2021, 12, 837. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chen, L.; Xu, H.; Xie, C.; Zhou, Y.; Zhou, F. Mitochondrial Dysfunctions Regulated Radioresistance through Mitochondria-to-Nucleus Retrograde Signaling Pathway of NF-ΚB/PI3K/AKT2/MTOR. Radiat. Res. 2018, 190, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Grasso, D.; Medeiros, H.C.D.; Zampieri, L.X.; Bol, V.; Danhier, P.; van Gisbergen, M.W.; Bouzin, C.; Brusa, D.; Grégoire, V.; Smeets, H.; et al. Fitter Mitochondria Are Associated With Radioresistance in Human Head and Neck SQD9 Cancer Cells. Front. Pharmacol. 2020, 11, 263. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Wozny, A.-S.; Alphonse, G.; Cassard, A.; Malésys, C.; Louati, S.; Beuve, M.; Lalle, P.; Ardail, D.; Nakajima, T.; Rodriguez-Lafrasse, C. Impact of Hypoxia on the Double-Strand Break Repair after Photon and Carbon Ion Irradiation of Radioresistant HNSCC Cells. Sci. Rep. 2020, 10, 21357. [Google Scholar] [CrossRef]
- Ruba, T.; Tamilselvi, D.R. Radiosensitizers and Radioprotectors for Effective Radiation Therapy—A Review. Asian J. Appl. Sci. Technol. 2018, 2, 77–86. [Google Scholar]
- Lesueur, P.; Chevalier, F.; Austry, J.-B.; Waissi, W.; Burckel, H.; Noël, G.; Habrand, J.-L.; Saintigny, Y.; Joly, F. Poly-(ADP-Ribose)-Polymerase Inhibitors as Radiosensitizers: A Systematic Review of Pre-Clinical and Clinical Human Studies. Oncotarget 2017, 8, 69105–69124. [Google Scholar] [CrossRef]
- Al-Waili, N.S.; Butler, G.J.; Beale, J.; Hamilton, R.W.; Lee, B.Y.; Lucas, P. Hyperbaric Oxygen and Malignancies: A Potential Role in Radiotherapy, Chemotherapy, Tumor Surgery and Phototherapy. Med. Sci. Monit. 2005, 11, RA279–RA289. [Google Scholar]
- Churchill-Davidson, I.; Foster, C.A.; Wiernik, G.; Collins, C.D.; Pizey, N.C.; Skeggs, D.B.; Purser, P.R. The Place of Oxygen in Radiotherapy. Br. J. Radiol. 1966, 39, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.; Cox, J. Physical and Biological Basis of Radiation Therapy. In Radiation Oncology: Rationale, Technique, Results; Cox, J.D., Ang, K.K., Eds.; Mosby: St. Louis, MO, USA, 2003; pp. 3–62. [Google Scholar]
- Hoskin, P.J.; Saunders, M.I.; Phillips, H.; Cladd, H.; Powell, M.E.; Goodchild, K.; Stratford, M.R.; Rojas, A. Carbogen and Nicotinamide in the Treatment of Bladder Cancer with Radical Radiotherapy. Br. J. Cancer 1997, 76, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.E.; Dewey, D.L. Hydrated electrons and radiobiological sensitisation. Biochem. Biophys. Res. Commun. 1963, 12, 473–477. [Google Scholar] [CrossRef]
- Asquith, J.C.; Foster, J.L.; Willson, R.L.; Ings, R.; McFadzean, J.A. Metronidazole (“Flagyl”). A Radiosensitizer of Hypoxic Cells. Br. J. Radiol. 1974, 47, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Alper, T. The Modification of Damage Caused by Primary Ionization of Biological Targets. Radiat. Res. 1956, 5, 573–586. [Google Scholar] [CrossRef]
- Guichard, M.; Malaise, E.P. Radiosensitizing Effects of Misonidazole and SR 2508 on a Human Melanoma Transplanted in Nude Mice: Influence on Repair of Potentially Lethal Damage. Int. J. Radiat. Oncol. Biol. Phys. 1982, 8, 465–468. [Google Scholar] [CrossRef]
- Overgaard, J.; Hansen, H.S.; Overgaard, M.; Bastholt, L.; Berthelsen, A.; Specht, L.; Lindeløv, B.; Jørgensen, K. A Randomized Double-Blind Phase III Study of Nimorazole as a Hypoxic Radiosensitizer of Primary Radiotherapy in Supraglottic Larynx and Pharynx Carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother. Oncol. 1998, 46, 135–146. [Google Scholar] [CrossRef]
- Kovacs, M.S.; Hocking, D.J.; Evans, J.W.; Siim, B.G.; Wouters, B.G.; Brown, J.M. Cisplatin Anti-Tumour Potentiation by Tirapazamine Results from a Hypoxia-Dependent Cellular Sensitization to Cisplatin. Br. J. Cancer 1999, 80, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Pu, A.T.; Robertson, J.M.; Lawrence, T.S. Current Status of Radiation Sensitization by Fluoropyrimidines. Oncology (Williston Park) 1995, 9, 707–714; discussion 714, 717–718, 721. [Google Scholar]
- Pinedo, H.M.; Peters, G.F. Fluorouracil: Biochemistry and Pharmacology. J. Clin. Oncol. 1988, 6, 1653–1664. [Google Scholar] [CrossRef]
- Hasegawa, K.; Okamoto, H.; Kawamura, K.; Kato, R.; Kobayashi, Y.; Sekiya, T.; Udagawa, Y. The Effect of Chemotherapy or Radiotherapy on Thymidine Phosphorylase and Dihydropyrimidine Dehydrogenase Expression in Cancer of the Uterine Cervix. Eur. J. Obstet. Gynecol. Reprod. Biol. 2012, 163, 67–70. [Google Scholar] [CrossRef] [PubMed]
- de Sousa Cavalcante, L.; Monteiro, G. Gemcitabine: Metabolism and Molecular Mechanisms of Action, Sensitivity and Chemoresistance in Pancreatic Cancer. Eur. J. Pharmacol. 2014, 741, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Plunkett, W. Fludarabine- and Gemcitabine-Induced Apoptosis: Incorporation of Analogs into DNA Is a Critical Event. Cancer Chemother. Pharmacol. 1995, 36, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Abal, M.; Andreu, J.M.; Barasoain, I. Taxanes: Microtubule and Centrosome Targets, and Cell Cycle Dependent Mechanisms of Action. Curr. Cancer Drug Targets 2003, 3, 193–203. [Google Scholar] [CrossRef]
- Rosenthal, D.I.; Carbone, D.P. Taxol plus Radiation for Head and Neck Cancer. J. Infus. Chemother. 1995, 5, 46–54. [Google Scholar] [PubMed]
- Rosenberg, B.; VanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum Compounds: A New Class of Potent Antitumour Agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef]
- Szumiel, I.; Nias, A.H. The Effect of Combined Treatment with a Platinum Complex and Ionizing Radiation on Chinese Hamster Ovary Cells in Vitro. Br. J. Cancer 1976, 33, 450–458. [Google Scholar] [CrossRef]
- Stratford, I.J.; Williamson, C.; Adams, G.E. Combination Studies with Misonidazole and a Cis-Platinum Complex: Cytotoxicity and Radiosensitization in Vitro. Br. J. Cancer 1980, 41, 517–522. [Google Scholar] [CrossRef]
- O’Hara, J.A.; Douple, E.B.; Richmond, R.C. Enhancement of Radiation-Induced Cell Kill by Platinum Complexes (Carboplatin and Iproplatin) in V79 Cells. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1419–1422. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Camphausen, K.; Tofilon, P.J. Inhibition of Histone Deacetylation: A Strategy for Tumor Radiosensitization. J. Clin. Oncol. 2007, 25, 4051–4056. [Google Scholar] [CrossRef]
- Johnson, A.M.; Bennett, P.V.; Sanidad, K.Z.; Hoang, A.; Jardine, J.H.; Keszenman, D.J.; Wilson, P.F. Evaluation of Histone Deacetylase Inhibitors as Radiosensitizers for Proton and Light Ion Radiotherapy. Front. Oncol. 2021, 11, 735940. [Google Scholar] [CrossRef]
- Cappellacci, L.; Perinelli, D.R.; Maggi, F.; Grifantini, M.; Petrelli, R. Recent Progress in Histone Deacetylase Inhibitors as Anticancer Agents. Curr. Med. Chem. 2020, 27, 2449–2493. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent Developments of HDAC Inhibitors: Emerging Indications and Novel Molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef] [PubMed]
- Antrobus, J.; Parsons, J.L. Histone Deacetylases and Their Potential as Targets to Enhance Tumour Radiosensitisation. Radiation 2022, 2, 149–167. [Google Scholar] [CrossRef]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous End Joining Drives Poly(ADP-Ribose) Polymerase (PARP) Inhibitor Lethality in Homologous Recombination-Deficient Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef]
- Barcellini, A.; Loap, P.; Murata, K.; Villa, R.; Kirova, Y.; Okonogi, N.; Orlandi, E. PARP Inhibitors in Combination with Radiotherapy: To Do or Not to Do? Cancers 2021, 13, 5380. [Google Scholar] [CrossRef]
- Césaire, M.; Thariat, J.; Candéias, S.M.; Stefan, D.; Saintigny, Y.; Chevalier, F. Combining PARP Inhibition, Radiation, and Immunotherapy: A Possible Strategy to Improve the Treatment of Cancer? Int. J. Mol. Sci. 2018, 19, 3793. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, Q.; Nagasawa, H.; Okayasu, R.; Liber, H.L.; Bedford, J.S. Silencing Expression of the Catalytic Subunit of DNA-Dependent Protein Kinase by Small Interfering RNA Sensitizes Human Cells for Radiation-Induced Chromosome Damage, Cell Killing, and Mutation. Cancer Res. 2002, 62, 6400–6404. [Google Scholar] [PubMed]
- Toulany, M.; Kehlbach, R.; Florczak, U.; Sak, A.; Wang, S.; Chen, J.; Lobrich, M.; Rodemann, H.P. Targeting of AKT1 Enhances Radiation Toxicity of Human Tumor Cells by Inhibiting DNA-PKcs-Dependent DNA Double-Strand Break Repair. Mol. Cancer Ther. 2008, 7, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Geng, W.; Tian, D.; Wang, Q.; Shan, S.; Zhou, J.; Xu, W.; Shan, H. DNA-PKcs Inhibitor Increases the Sensitivity of Gastric Cancer Cells to Radiotherapy. Oncol. Rep. 2019, 42, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, C.E.; Jiang, Y.; Thomas, H.D.; Willmore, E.; Kyle, S.; Wittner, A.; Phillips, N.; Zhao, Y.; Tudhope, S.J.; Prendergast, L.; et al. Selective DNA-PKcs Inhibition Extends the Therapeutic Index of Localized Radiotherapy and Chemotherapy. J. Clin. Investig. 2020, 130, 258–271. [Google Scholar] [CrossRef]
- Waqar, S.N.; Robinson, C.; Olszanski, A.J.; Spira, A.; Hackmaster, M.; Lucas, L.; Sponton, L.; Jin, H.; Hering, U.; Cronier, D.; et al. Phase I Trial of ATM Inhibitor M3541 in Combination with Palliative Radiotherapy in Patients with Solid Tumors. Investig. New Drugs 2022, 40, 596–605. [Google Scholar] [CrossRef]
- Hafsi, H.; Dillon, M.T.; Barker, H.E.; Kyula, J.N.; Schick, U.; Paget, J.T.; Smith, H.G.; Pedersen, M.; McLaughlin, M.; Harrington, K.J. Combined ATR and DNA-PK Inhibition Radiosensitizes Tumor Cells Independently of Their P53 Status. Front. Oncol. 2018, 8, 245. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 Inhibitors and Cancer Therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Fukumori, Y.; Ichikawa, H. Nanoparticles for Cancer Therapy and Diagnosis. Adv. Powder Technol. 2006, 17, 173. [Google Scholar] [CrossRef]
- Ukleja, J.; Kusaka, E.; Miyamoto, D.T. Immunotherapy Combined With Radiation Therapy for Genitourinary Malignancies. Front. Oncol. 2021, 11, 663852. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation Modulates the Peptide Repertoire, Enhances MHC Class I Expression, and Induces Successful Antitumor Immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Farhood, B.; Eleojo Musa, A.; Taeb, S.; Najafi, M. Damage-Associated Molecular Patterns in Tumor Radiotherapy. Int. Immunopharmacol. 2020, 86, 106761. [Google Scholar] [CrossRef] [PubMed]
- Abuodeh, Y.; Venkat, P.; Kim, S. Systematic Review of Case Reports on the Abscopal Effect. Curr. Probl. Cancer 2016, 40, 25–37. [Google Scholar] [CrossRef]
- Craig, D.J.; Nanavaty, N.S.; Devanaboyina, M.; Stanbery, L.; Hamouda, D.; Edelman, G.; Dworkin, L.; Nemunaitis, J.J. The Abscopal Effect of Radiation Therapy. Future Oncol. 2021, 17, 1683–1694. [Google Scholar] [CrossRef]
- Mole, R.H. Whole Body Irradiation; Radiobiology or Medicine? Br. J. Radiol. 1953, 26, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Dagoglu, N.; Karaman, S.; Caglar, H.B.; Oral, E.N. Abscopal Effect of Radiotherapy in the Immunotherapy Era: Systematic Review of Reported Cases. Cureus 2019, 11, e4103. [Google Scholar] [CrossRef]
- Blanchard, P.; Gunn, G.B.; Lin, A.; Foote, R.L.; Lee, N.Y.; Frank, S.J. Proton Therapy for Head and Neck Cancers. Semin. Radiat. Oncol. 2018, 28, 53–63. [Google Scholar] [CrossRef]
- Malouff, T.D.; Mahajan, A.; Krishnan, S.; Beltran, C.; Seneviratne, D.S.; Trifiletti, D.M. Carbon Ion Therapy: A Modern Review of an Emerging Technology. Front. Oncol. 2020, 10, 82. [Google Scholar] [CrossRef]
- Mohamad, O.; Sishc, B.J.; Saha, J.; Pompos, A.; Rahimi, A.; Story, M.D.; Davis, A.J.; Kim, D.W.N. Carbon Ion Radiotherapy: A Review of Clinical Experiences and Preclinical Research, with an Emphasis on DNA Damage/Repair. Cancers 2017, 9, 66. [Google Scholar] [CrossRef]
- Yang, J.; Gao, J.; Wu, X.; Hu, J.; Hu, W.; Kong, L.; Lu, J.J. Salvage Carbon Ion Radiation Therapy for Locally Recurrent or Radiation-Induced Second Primary Sarcoma of the Head and Neck. J. Cancer 2018, 9, 2215–2223. [Google Scholar] [CrossRef]
- Hayashi, K.; Koto, M.; Ikawa, H.; Hagiwara, Y.; Tsuji, H.; Ogawa, K.; Kamada, T. Feasibility of Re-Irradiation Using Carbon Ions for Recurrent Head and Neck Malignancies after Carbon-Ion Radiotherapy. Radiother. Oncol. 2019, 136, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, B.L.T.; Pijls-Johannesma, M.; Joore, M.A.; van den Ende, P.; Langendijk, J.A.; Lambin, P.; Kessels, A.G.H.; Grutters, J.P.C. Systematic Review and Meta-Analysis of Radiotherapy in Various Head and Neck Cancers: Comparing Photons, Carbon-Ions and Protons. Cancer Treat. Rev. 2011, 37, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Schlaff, C.D.; Krauze, A.; Belard, A.; O’Connell, J.J.; Camphausen, K.A. Bringing the Heavy: Carbon Ion Therapy in the Radiobiological and Clinical Context. Radiat. Oncol. 2014, 9, 88. [Google Scholar] [CrossRef]
- Helm, A.; Ebner, D.K.; Tinganelli, W.; Simoniello, P.; Bisio, A.; Marchesano, V.; Durante, M.; Yamada, S.; Shimokawa, T. Combining Heavy-Ion Therapy with Immunotherapy: An Update on Recent Developments. Int. J. Part Ther. 2018, 5, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, A.; Ueda, Y.; Yamada, S.; Harada, Y.; Shimada, H.; Hasegawa, M.; Tsujii, H.; Ochiai, T.; Yonemitsu, Y. Carbon-Ion Beam Treatment Induces Systemic Antitumor Immunity against Murine Squamous Cell Carcinoma. Cancer 2010, 116, 3740–3748. [Google Scholar] [CrossRef]
- Ando, K.; Fujita, H.; Hosoi, A.; Ma, L.; Wakatsuki, M.; Seino, K.-I.; Kakimi, K.; Imai, T.; Shimokawa, T.; Nakano, T. Intravenous Dendritic Cell Administration Enhances Suppression of Lung Metastasis Induced by Carbon-Ion Irradiation. J. Radiat. Res. 2017, 58, 446–455. [Google Scholar] [CrossRef]
- De Meerleer, G.; Khoo, V.; Escudier, B.; Joniau, S.; Bossi, A.; Ost, P.; Briganti, A.; Fonteyne, V.; Van Vulpen, M.; Lumen, N.; et al. Radiotherapy for Renal-Cell Carcinoma. Lancet Oncol. 2014, 15, e170–e177. [Google Scholar] [CrossRef]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor Response to Radiotherapy Regulated by Endothelial Cell Apoptosis. Science 2003, 300, 1155–1159. [Google Scholar] [CrossRef]
- Sathishkumar, S.; Boyanovsky, B.; Karakashian, A.A.; Rozenova, K.; Giltiay, N.V.; Kudrimoti, M.; Mohiuddin, M.; Ahmed, M.M.; Nikolova-Karakashian, M. Elevated Sphingomyelinase Activity and Ceramide Concentration in Serum of Patients Undergoing High Dose Spatially Fractionated Radiation Treatment: Implications for Endothelial Apoptosis. Cancer Biol. Ther. 2005, 4, 979–986. [Google Scholar] [CrossRef]
- Siva, S.; Pham, D.; Gill, S.; Corcoran, N.M.; Foroudi, F. A Systematic Review of Stereotactic Radiotherapy Ablation for Primary Renal Cell Carcinoma. BJU Int. 2012, 110, E737–E743. [Google Scholar] [CrossRef]
- Favaudon, V.; Caplier, L.; Monceau, V.; Pouzoulet, F.; Sayarath, M.; Fouillade, C.; Poupon, M.-F.; Brito, I.; Hupé, P.; Bourhis, J.; et al. Ultrahigh Dose-Rate FLASH Irradiation Increases the Differential Response between Normal and Tumor Tissue in Mice. Sci. Transl. Med. 2014, 6, 245ra93. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Gao, F.; Yang, Y.; Wu, D.; Zhang, Y.; Feng, G.; Dai, T.; Du, X. FLASH Radiotherapy: History and Future. Front. Oncol. 2021, 11, 644400. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.A.; Moran, J.M.; Jaffray, D.A.; Buchsbaum, J.C. A Roadmap to Clinical Trials for FLASH. Med. Phys. 2022, 49, 4099–4108. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.D.; Simone, B.A.; Palazzo, J.; Savage, J.E.; Sano, Y.; Dan, T.; Jin, L.; Champ, C.E.; Zhao, S.; Lim, M.; et al. Caloric Restriction Augments Radiation Efficacy in Breast Cancer. Cell Cycle 2013, 12, 1955–1963. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Raffaghello, L.; Brandhorst, S.; Safdie, F.M.; Bianchi, G.; Martin-Montalvo, A.; Pistoia, V.; Wei, M.; Hwang, S.; Merlino, A.; et al. Fasting Cycles Retard Growth of Tumors and Sensitize a Range of Cancer Cell Types to Chemotherapy. Sci. Transl. Med. 2012, 4, 124ra27. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, P.; Abate, L.E.; Seyfried, T.N. Antiangiogenic and Proapoptotic Effects of Dietary Restriction on Experimental Mouse and Human Brain Tumors. Clin. Cancer Res. 2004, 10, 5622–5629. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Busato, F.; Khouzai, B.E.; Mognato, M. Biological Mechanisms to Reduce Radioresistance and Increase the Efficacy of Radiotherapy: State of the Art. Int. J. Mol. Sci. 2022, 23, 10211. https://doi.org/10.3390/ijms231810211
Busato F, Khouzai BE, Mognato M. Biological Mechanisms to Reduce Radioresistance and Increase the Efficacy of Radiotherapy: State of the Art. International Journal of Molecular Sciences. 2022; 23(18):10211. https://doi.org/10.3390/ijms231810211
Chicago/Turabian StyleBusato, Fabio, Badr El Khouzai, and Maddalena Mognato. 2022. "Biological Mechanisms to Reduce Radioresistance and Increase the Efficacy of Radiotherapy: State of the Art" International Journal of Molecular Sciences 23, no. 18: 10211. https://doi.org/10.3390/ijms231810211