
Genomic Insights of First ermB-Positive ST338-SCCmecVT/CC59 Taiwan Clone of Community-Associated Methicillin-Resistant Staphylococcus aureus in Poland


Abstract
:1. Introduction
2. Results
2.1. Preliminary Phenotype and Genotype-Based Results
2.2. Genomic-Based Results
2.2.1. Genome-Assembly Features
2.2.2. Genome Annotation and Taxonomy Confirmation
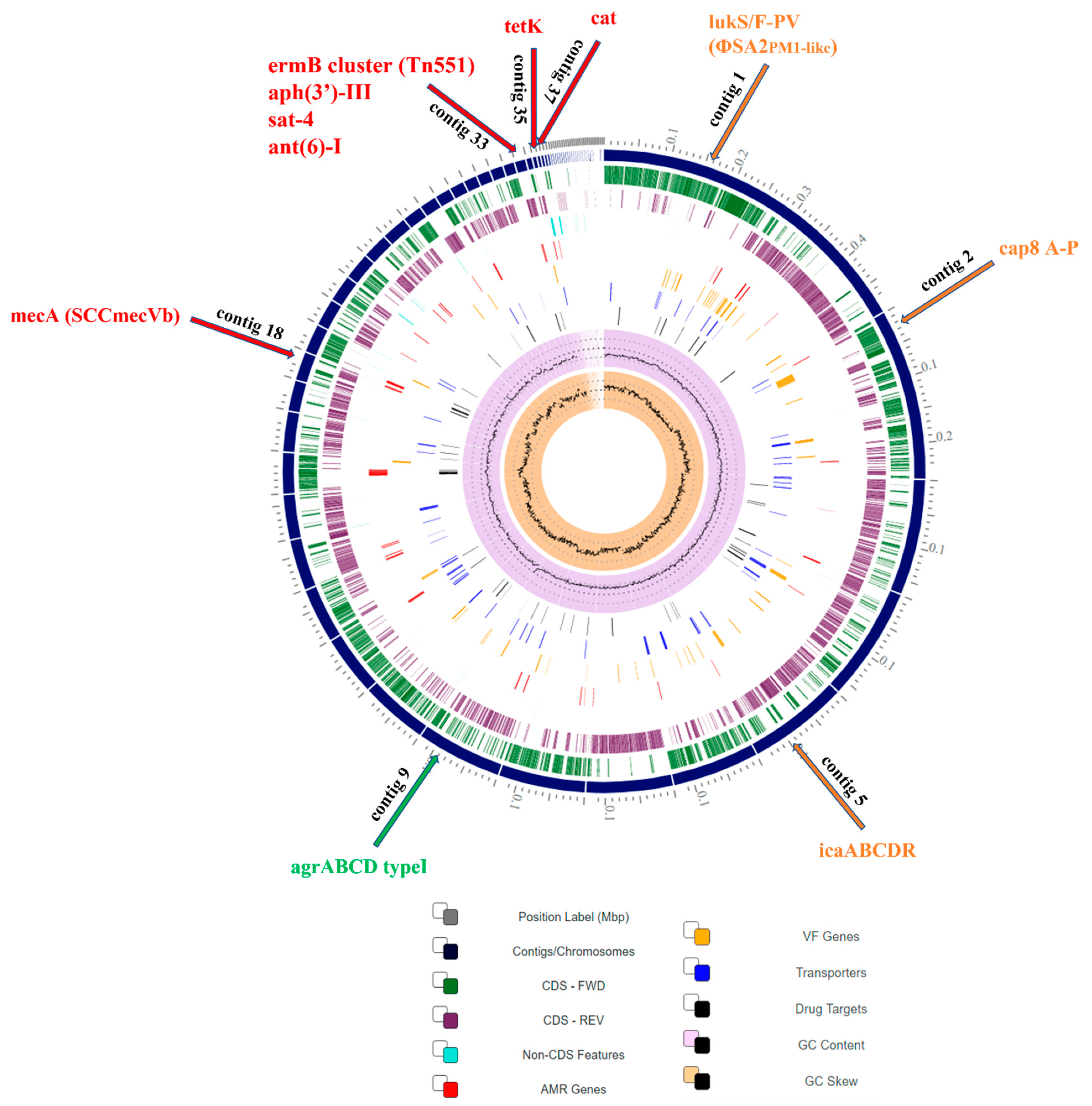
2.2.3. Comparative Genomics of CA-MRSA SO574/12
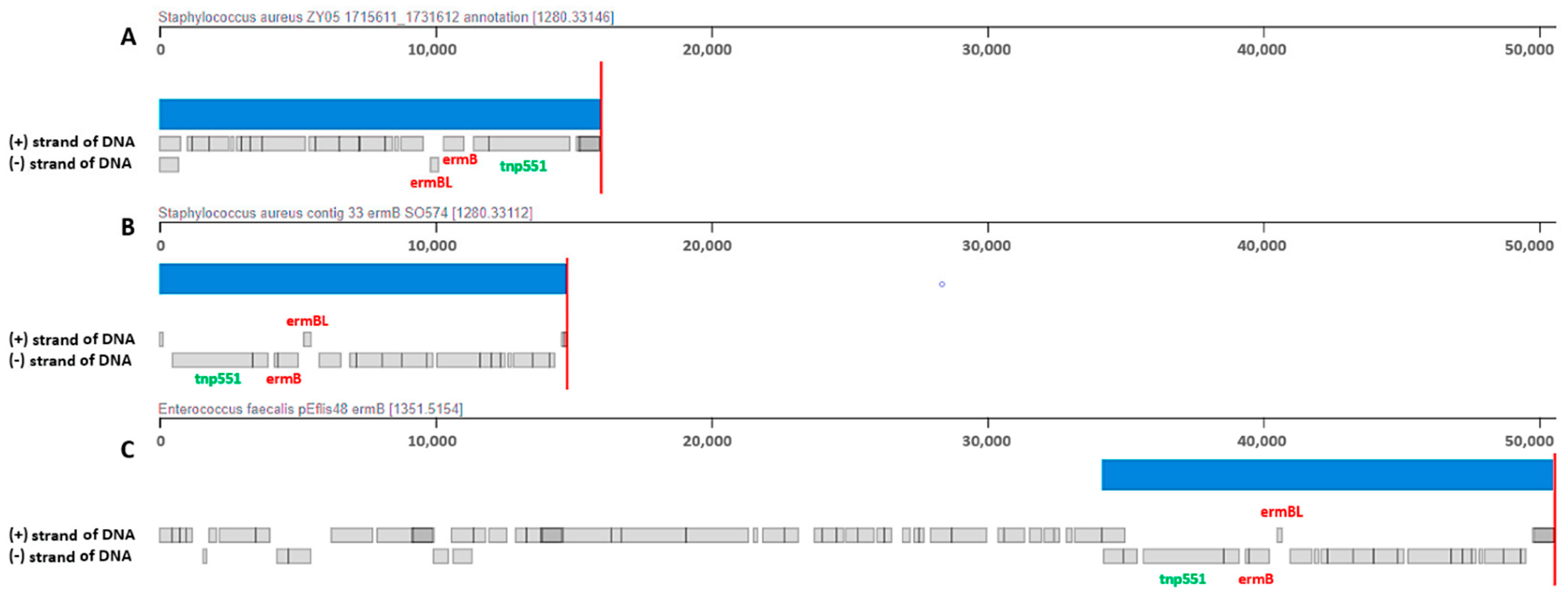
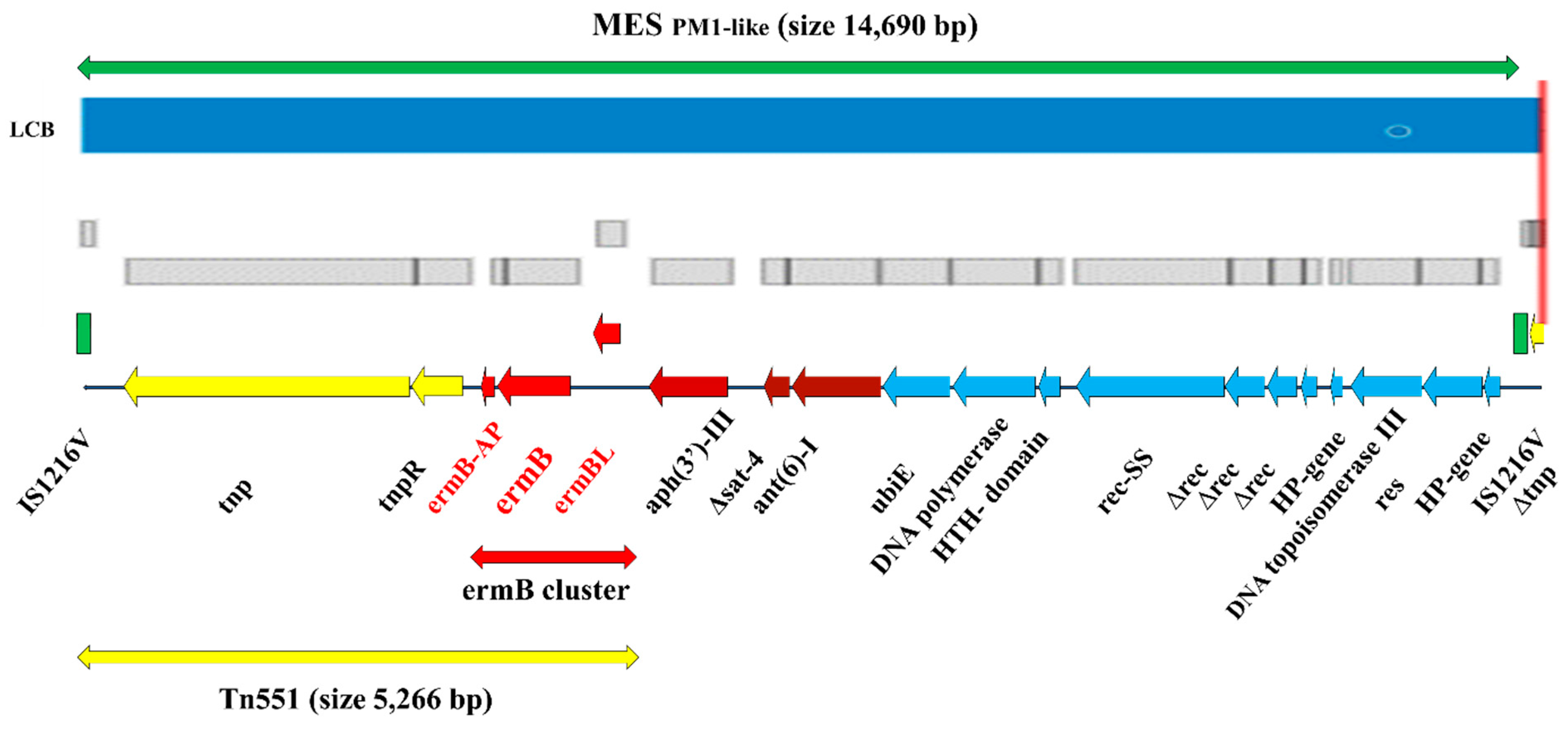
2.2.4. Genetic Structure and Organization of ermB Carrying MESPM1-like Structure in CA-MRSA SO574/12
2.2.5. Genomic-Based Antimicrobial Resistance Analysis
2.2.6. Genomic-Based Virulence Factors Analysis
3. Discussion
4. Materials and Methods
4.1. Bacterial Strain
4.2. Phenotype Characteristics
4.2.1. Confirmation of Resistance to Methicillin
4.2.2. Detecting Resistance to Other Antibiotics
4.2.3. Type of Regulation of Resistance to Macrolide, Lincosamide, and Streptogramin B (MLS-B) Antibiotics (Qualitative Method)
4.2.4. Resistance to MLS-B Antibiotics (Quantitative Method)
4.3. Genotype and Genomic Characteristics
4.3.1. Genomic DNA Extraction
4.3.2. Detection of Antibiotic-Resistance Genetic Determinants—Targeted PCR Amplification
4.3.3. SCCmec (staphylococcal chromosome cassettes mec) Assignment
4.3.4. Multilocus Sequence Typing (MLST)
4.3.5. Whole-Genome Library Preparation and Sequencing
4.3.6. Sequence Quality Verification, Trimming, and Assembling
4.3.7. Genome Annotation and Genomic Features Assignments
4.3.8. Comparative Genomics and Mobile ermB-Carrying Genetic-Structure Analysis
4.3.9. Reference Genome, Plasmid, and Mobile Genetic-Structure Sequences
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bogut, A.; Koziol-Montewka, M.; Baranowicz, I.; Jozwiak, L.; Al-Doori, Z.; Morrison, D.; Kaczor, D.; Ksiazek, A. Community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) in Poland: Further evidence for the changing epidemiology of MRSA. New Microbiol. 2008, 31, 229–234. [Google Scholar] [PubMed]
- Otto, M. Community-associated MRSA: What makes them special? Int. J. Med. Microbiol. 2013, 303, 324–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Luo, Y.; Zhang, S.; Liang, Z.; Wang, Y.; Zhang, Y.; Zhou, G.; Jia, Y.; Chen, L.; She, D. Community-acquired necrotizing pneumonia caused by methicillin-resistant Staphylococcus aureus producing Panton-Valentine leukocidin in a Chinese teenager: Case report and literature review. Int. J. Infect. Dis. 2014, 26, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolo, J.; Miragaia, M.; Turlej-Rogacka, A.; Empel, J.; Bouchami, O.; Faria, N.A.; Tavares, A.; Hryniewicz, W.; Fluit, A.C.; de Lencastre, H.; et al. High genetic diversity among community-associated Staphylococcus aureus in Europe: Results from a multicenter study. PLoS ONE 2012, 7, e34768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, K.; Chung, D.R. Changing epidemiology of community-associated methicillin-resistant Staphylococcus aureus in the Asia-Pacific region. Expert Rev. Anti-Infect. Ther. 2016, 14, 1007–1022. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Jena, S.; Panda, S.; Sharma, S.; Dhawan, B.; Nath, G.; Singh, N.P.; Nayak, K.C.; Singh, D.V. Antibiotic Susceptibility, Virulence Pattern, and Typing of Staphylococcus aureus Strains Isolated from Variety of Infections in India. Front. Microbiol. 2019, 10, 2763. [Google Scholar] [CrossRef]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31, e00020-18. [Google Scholar] [CrossRef] [Green Version]
- Boyle-Vavra, S.; Ereshefsky, B.; Wang, C.C.; Daum, R.S. Successful multiresistant community-associated methicillin-resistant Staphylococcus aureus lineage from Taipei, Taiwan, that carries either the novel staphylococcal chromosome cassette mec (SCCmec) type VT or SCCmec type IV. J. Clin. Microbiol. 2005, 43, 4719–4730. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Ma, X.X.; Takeuchi, F.; Okuma, K.; Yuzawa, H.; Hiramatsu, K. Novel type V staphylococcal cassette chromosome mec driven by a novel cassette chromosome recombinase, ccrC. Antimicrob. Agents Chemother. 2004, 48, 2637–2651. [Google Scholar] [CrossRef] [Green Version]
- Takano, T.; Higuchi, W.; Zaraket, H.; Otsuka, T.; Baranovich, T.; Enany, S.; Saito, K.; Isobe, H.; Dohmae, S.; Ozaki, K.; et al. Novel characteristics of community-acquired methicillin-resistant Staphylococcus aureus strains belonging to multilocus sequence type 59 in Taiwan. Antimicrob. Agents Chemother. 2008, 52, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Panton, P.N.; Valentine, F.C.O. Staphylococcal toxin. Lancet 1932, 219, 506–508. [Google Scholar] [CrossRef]
- Boyle-Vavra, S.; Daum, R.S. Community-acquired methicillin-resistant Staphylococcus aureus: The role of Panton-Valentine leukocidin. Lab. Investig. 2007, 87, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tromp, A.T.; van Strijp, J.A.G. Studying Staphylococcal Leukocidins: A Challenging Endeavor. Front. Microbiol. 2020, 11, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClure, J.A.; Lakhundi, S.; Niazy, A.; Dong, G.; Obasuyi, O.; Gordon, P.; Chen, S.; Conly, J.M.; Zhang, K. Staphylococcus aureus ST59: Concurrent but Separate Evolution of North American and East Asian Lineages. Front. Microbiol. 2021, 12, 631845. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Lauderdale, T.L.Y.; Huang, Y.C. Evolution and Population Structures of Prevalent Methicillin-Resistant Staphylococcus aureus in Taiwan. Front. Microbiol. 2021, 12, 725340. [Google Scholar] [CrossRef]
- Ma, X.X.; Ito, T.; Tiensasitorn, C.; Jamklang, M.; Chongtrakool, P.; Boyle-Vavra, S.; Daum, R.S.; Hiramatsu, K. Novel type of staphylococcal cassette chromosome mec identified in community-acquired methicillin resistant Staphylococcus aureus strains. Antimicrob. Agents Chemother. 2002, 46, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Hung, W.C.; Takano, T.; Higuchi, W.; Iwao, Y.; Khokhlova, O.; Teng, L.J.; Yamamoto, T. Comparative genomics of community-acquired ST59 methicillin-resistant Staphylococcus aureus in Taiwan: Novel mobile resistance structures with IS1216V. PLoS ONE 2012, 7, e46987. [Google Scholar] [CrossRef]
- Su, Y.C.; Hung, W.W.; Lin, J.M.; Chang, C.C.; Chen, Y.H.; Lai, Y.L.; Tseng, S.P.; Lu, P.L.; Yamamoto, T.; Teng, L.J.; et al. Tracking the evolution of the two successful CC59 methicillin-resistant Staphylococcus aureus clones in Taiwan: The divergence time of the two clades is estimated to be the 1980s. Int. J. Antimicrob. Agents 2020, 56, 106047. [Google Scholar] [CrossRef]
- Vázquez-Laslop, N.; Mankin, A.S. How Macrolide Antibiotics Work. Trends Biochem. Sci. 2018, 43, 668–684. [Google Scholar] [CrossRef]
- Mlynarczyk-Bonikowska, B.; Kowalewski, C.; Krolak-Ulinska, A.; Marusza, W. Molecular Mechanisms of Drug Resistance in Staphylococcus aureus. Int. J. Mol. Sci. 2022, 23, 8088. [Google Scholar] [CrossRef]
- Mlynarczyk, A.; Szymanek-Majchrzak, K.; Grzybowska, W.; Durlik, M.; Deborska-Materkowska, D.; Paczek, L.; Chmura, A.; Swoboda-Kopec, E.; Tyski, S.; Mlynarczyk, G. Molecular and phenotypic characteristics of methicillin-resistant Staphylococcus aureus strains isolated from hospitalized patients in transplantation wards. Transplant. Proc. 2014, 46, 2579–2582. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.H.; Clewell, D.B. Complete nucleotide sequence of macrolide-lincosamide-streptogramin B-resistance transposon Tn917 in Streptococcus faecalis. J. Bacteriol. 1985, 164, 782–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Młynarczyk, A.; Młynarczyk, G.; Jeljaszewicz, J. The genome of Staphylococcus aureus: A review. Zentralbl. Bakteriol. 1998, 287, 277–314. [Google Scholar] [CrossRef]
- Wu, S.W.; de Lencastre, H.; Tomasz, A. The Staphylococcus aureus transposon Tn551: Complete nucleotide sequence and transcriptional analysis of the expression of the erythromycin resistance gene. Microb. Drug Resist. 1999, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Szymanek-Majchrzak, K.; Mlynarczyk, A.; Dobrzaniecka, K.; Majchrzak, K.; Mierzwinska-Nastalska, E.; Chmura, A.; Kwiatkowski, A.; Durlik, M.; Deborska-Materkowska, D.; Paczek, L.; et al. Epidemiological and drug-resistance types of methicillin-resistant Staphylococcus aureus strains isolated from surgical and transplantation ward patients during 2010 to 2011. Transplant. Proc. 2016, 48, 1414–1417. [Google Scholar] [CrossRef]
- Luczak-Kadlubowska, A.; Sulikowska, A.; Empel, J.; Piasecka, A.; Orczykowska, M.; Kozinska, A.; Hryniewicz, W. Countrywide molecular survey of methicillin-resistant Staphylococcus aureus strains in Poland. J. Clin. Microbiol. 2008, 46, 2930–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kot, B.; Wierzchowska, K.; Piechota, M.; Grużewska, A. Antimicrobial Resistance Patterns in Methicillin-Resistant Staphylococcus aureus from Patients Hospitalized during 2015–2017 in Hospitals in Poland. Med. Princ. Pract. 2020, 29, 61–68. [Google Scholar] [CrossRef]
- European Committee on Antimicrobial Susceptibility Testing, EUCAST Recommendations (2022b). Available online: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_12.0_Breakpoint_Tables.pdf (accessed on 1 January 2022).
- Szymanek, K.; Mlynarczyk, A.; Mlynarczyk, G. Regulatory systems of gene expression in Staphylococcus aureus. Post. Mikrobiol. 2009, 48, 7–22. [Google Scholar]
- Chen, Y.; Hong, J.; Chen, Y.; Wang, H.; Yu, Y.; Qu, T. Characterization of a community-acquired methicillin-resistant sequence type 338 Staphylococcus aureus strain containing a staphylococcal cassette chromosome mec type VT. Int. J. Infect. Dis. 2020, 90, 181–187. [Google Scholar] [CrossRef]
- Chen, C.J.; Huang, Y.C. New epidemiology of Staphylococcus aureus infection in Asia. Clin. Microbiol. Infect. 2014, 20, 605–623. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Mai, J.; Liu, Y.; Huang, Y.; Zhong, H.; Xie, Y.; Deng, Q.; Huang, L.; Yao, S.; He, Y.; et al. Prevalence and Characterization of Staphylococcus aureus Isolated from Women and Children in Guangzhou, China. Front. Microbiol. 2018, 9, 2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundmann, H.; Aanensen, D.M.; van den Wijngaard, C.C.; Spratt, B.G.; Harmsen, D.; Friedrich, A.W. Geographic Distribution of Staphylococcus aureus Causing Invasive Infections in Europe: A Molecular-Epidemiological Analysis. PLoS Med. 2010, 7, e1000215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundmann, H.; Schouls, L.M.; Aanensen, D.M.; Pluister, G.N.; Tami, A.; Chlebowicz, M.; Glasner, C.; Sabat, A.J.; Weist, K.; Heuer, O.; et al. The dynamic changes of dominant clones of Staphylococcus aureus causing bloodstream infections in the European region: Results of a second structured survey. Euro Surveill. 2014, 19, 20987. [Google Scholar] [CrossRef] [PubMed]
- Bletz, S.; Mellmann, A.; Rothgänger, J.; Harmsen, D. Ensuring backwards compatibility: Traditional genotyping efforts in the era of whole genome sequencing. Clin. Microbiol. Infect. 2015, 21, 347.e1–347.e4. [Google Scholar] [CrossRef] [Green Version]
- Glasner, C.; Pluister, G.; Westh, H.; Arends, J.P.; Empel, J.; Giles, E.; Laurent, F.; Layer, F.; Marstein, L.; Matussek, A.; et al. Staphylococcus aureus spa type t437: Identification of the most dominant community-associated clone from Asia across Europe. Clin. Microbiol. Infect. 2015, 21, 163.e1–163.e8. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Zhou, W.; Yin, Z.; Zhang, S.; Chen, Y.; Shen, P.; Ji, J.; Chen, W.; Zheng, B.; Xiao, Y. The genetic feature and virulence determinant of Highly Virulent Community-Associated MRSA ST338-SCCmec Vb in China. Emerg. Microbes Infect. 2021, 7, 1052–1064. [Google Scholar] [CrossRef]
- Hernando-Amado, S.; Sanz-García, F.; Blanco, P.; Martínez, J.L. Fitness costs associated with the acquisition of antibiotic resistance. Essays Biochem. 2017, 61, 37–48. [Google Scholar] [CrossRef]
- Touati, A.; Bellil, Z.; Barache, D.; Mairi, A. Fitness Cost of Antibiotic Resistance in Staphylococcus aureus: A Systematic Review. Microb. Drug Resist. 2021, 27, 1218–1231. [Google Scholar] [CrossRef]
- Mlynarczyk, A.; Mlynarczyk, B.; Kmera-Muszynska, M.; Majewski, S.; Mlynarczyk, G. Mechanisms of the resistance and tolerance to beta-lactam and glycopeptide antibiotics in pathogenic Gram-positive cocci. Mini Rev. Med. Chem. 2009, 9, 1527–1537. [Google Scholar] [CrossRef]
- Min, Y.H.; Kwon, A.R.; Yoon, E.J.; Shim, M.J.; Choi, E.C. Translational attenuation and mRNA stabilization as mechanisms of erm(B) induction by erythromycin. Antimicrob. Agents Chemother. 2008, 52, 1782–1789. [Google Scholar] [CrossRef] [Green Version]
- Arenz, S.; Ramu, H.; Gupta, P.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N. Molecular basis for erythromycin-dependent ribosome stalling during translation of the ErmBL leader peptide. Nat. Commun. 2014, 5, 3501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.T.; Tseng, S.P.; Hung, W.W.; Chang, C.C.; Chen, Y.H.; Jao, Y.T.; Chen, Y.H.; Teng, L.J.; Hung, W.C. A Possible Role of Insertion Sequence IS1216V in Dissemination of Multidrug-Resistant Elements MESPM1 and MES6272-2 between Enterococcus and ST59 Staphylococcus aureus. Microorganisms 2020, 8, 1905. [Google Scholar] [CrossRef] [PubMed]
- Boerlin, P.; Burnens, A.P.; Frey, J.; Kuhnert, P.; Nicolet, J. Molecular epidemiology and genetic linkage of macrolide and aminoglycoside resistance in Staphylococcus intermedius of canine origin. Vet. Microbiol. 2001, 79, 155–169. [Google Scholar] [CrossRef]
- Oh, T.G.; Kwon, A.R.; Choi, E.C. Induction of ermAMR from a clinical strain of Enterococcus faecalis by 16-membered-ring macrolide antibiotics. J. Bacteriol. 1998, 180, 5788–5791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.; Kannan, K.; Mankin, A.S.; Vázquez-Laslop, N. Regulation of gene expression by macrolide-induced ribosomal frameshifting. Mol. Cell 2013, 52, 629–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzyubak, E.; Yap, M.N. The Expression of Antibiotic Resistance Methyltransferase Correlates with mRNA Stability Independently of Ribosome Stalling. Antimicrob. Agents Chemother. 2016, 60, 7178–7188. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Jiang, K.; Du, X.; Lu, Y.; Liao, L.; He, Z.; He, W. Translational Attenuation Mechanism of ErmB Induction by Erythromycin Is Dependent on Two Leader Peptides. Front. Microbiol. 2021, 12, 690744. [Google Scholar] [CrossRef]
- Dabul, A.N.G.; Avaca-Crusca, J.S.; Van Tyne, D.; Gilmore, M.S.; Camargo, I.L.B.C. Resistance in In Vitro Selected Tigecycline-Resistant Methicillin-Resistant Staphylococcus aureus Sequence Type 5 Is Driven by Mutations in mepR and mepA Genes. Microb. Drug Resist. 2018, 24, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Khoshnood, S.; Heidary, M.; Asadi, A.; Soleimani, S.; Motahar, M.; Savari, M.; Saki, M.; Abdi, M. A review on mechanism of action, resistance, synergism, and clinical implications of mupirocin against Staphylococcus aureus. Biomed. Pharmacother. 2019, 109, 1809–1818. [Google Scholar] [CrossRef]
- European Committee on Antimicrobial Susceptibility Testing, EUCAST Recommendations (2022a). Available online: https://www.eucast.org/ast_of_bacteria/ (accessed on 1 January 2022).
- Szymanek-Majchrzak, K.; Kosiński, J.; Żak, K.; Sułek, K.; Młynarczyk, A.; Młynarczyk, G. Prevalence of methicillin-resistant and mupirocin resistant Staphylococcus aureus strains among medical students of Medical University of Warsaw. Epidemiol. Rev. 2019, 73, 39–48. [Google Scholar] [CrossRef]
- Szymanek-Majchrzak, K.; Mlynarczyk, A.; Mlynarczyk, G. Characteristics of glycopeptide-resistant Staphylococcus aureus strains isolated from inpatients of three teaching hospitals in Warsaw, Poland. Antimicrob. Resist. Infect. Control 2018, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Okuma, K.; Iwakawa, K.; Turnidge, J.D.; Grubb, W.B.; Bell, J.M.; O’Brien, F.G.; Coombs, G.W.; Pearman, J.W.; Tenover, F.C.; Kapi, M.; et al. Dissemination of new methicillin resistant Staphylococcus aureus clones in the community. J. Clin. Microbiol. 2002, 40, 4289–4294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, D.C.; de Lencastre, H. Multiplex PCR strategy for rapid identification of structural types and variants of the mec element in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2002, 46, 2155–2161. [Google Scholar] [CrossRef] [Green Version]
- Enright, M.C.; Day, N.P.; Davies, C.E.; Peacock, S.J.; Spratt, B.G. Multilocus sequence typing for characterization of methicillin-resistant and methicillin susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 2000, 38, 1008–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, S. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 21 August 2021).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.; Zobel, J.; Holt, K. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [Green Version]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Brettin, T.; Davis, J.J.; Gerdes, S.; Kenyon, R.; Machi, D.; Mao, C.; Olson, R.; Overbeek, R.; Pusch, G.D.; et al. Assembly, Annotation, and Comparative Genomics in PATRIC, the All Bacterial Bioinformatics Resource Center. Methods Mol. Biol. 2018, 1704, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Ito, T.; Ma, X.X.; Watanabe, S.; Kreiswirth, B.N.; Etienne, J.; Hiramatsu, K. Combination of multiplex PCRs for staphylococcal cassette chromosome mec type assignment: Rapid identification system for mec, ccr and major difference in junkyard regions. Antimicrob. Agents Chemother. 2007, 51, 264–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Working Group on the Classification of Staphylococcal Cassette Chromosome Elements (IWG-SCC). Classification of staphylococcal cassette chromosome mec (SCCmec): Guidelines for reporting novel SCCmec elements. Antimicrob. Agents Chemother. 2009, 56, 4961–4967. [Google Scholar] [CrossRef] [Green Version]
- Bartels, M.D.; Petersen, A.; Worning, P.; Nielsen, J.B.; Larner-Svensson, H.; Johansen, H.K.; Andersen, L.P.; Jarløv, J.O.; Boye, K.; Larsen, A.R.; et al. Comparing whole-genome sequencing with Sanger sequencing for spa typing of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 2014, 52, 4305–4308. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total Genome Sequenced Bacteria. J. Clin. Micobiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [Green Version]
- Zankari, E.; Allesoe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 2764–2768. [Google Scholar] [CrossRef] [Green Version]
- Bortolaia, V.; Kaas, R.F.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Clausen, P.T.L.C.; Aarestrup, F.M.; Lund, O. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinform. 2018, 19, 307. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garcia-Fernandez, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. PlasmidFinder and pMLST: In Silico detection and typing of plasmids. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 2020, 76, 101–109. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 8, D606–D612. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Strain Characteristic |
|---|---|
| MRSA (gene) | Yes, low-level resistance (mecA) |
| MRSA phenotype | CA-MRSA |
| SCCmec type (subtype) | Composite Vb, also named VT (5C2&5) |
| ccr genes complex type | ccrC1-allel-2, ccrC1-allel-8 |
| mec gene complex class | C2 |
| spa type (profile) | t437 (04-20-17-20-17-25-34) |
| MLST type (profile) | ST338 (19-23-15-48-19-20-15) |
| Clonal complex | CC59 |
| International epidemic clone | Taiwan |
| MLS-B phenotype (gene) | cMLS-B (ermB) |
| AMR profile | E, AZ, CL, DA, MY, FOX, P, AK, TET |
| Main virulence factor (gene) | PVL (lukS/F-PV) |
| AGR TCSTS | agr ABCD type I |
| ACME | ND |
| Biofilm determination genes | icaABCDR |
| Capsule determination genes | cap8 A-P |
| MGE (main carried gene): | |
| Plasmids | rep7a (tetK), rep7b (cat), rep19 (blaZ) |
| Tn | Tn551 (ermB) |
| IS | IS1216V, IS3-like family, IS200-like family |
| MES | MESPM1-like (ermB, aph(3′)-III, Δsat-4, ant(6)-I), SCCmec (mecA) |
| Prophages | ΦSA2PM1-like (lukS/F-PV) |
| (A) | |||||||||||||
| Antimicrobial Resistance Profile—Strain MRSA SO574/12 | |||||||||||||
| ATB results | Antibiotics, MIC (mg/L) or Diameter (mm) | ||||||||||||
| Macrolide, lincosamide, streptogramin B group (MLS-B) | β-lactams | Aminoglycosides | Quinolones | ||||||||||
| E MIC >256 | AZ MIC >256 | CL MIC >256 | DA MIC >256 | MY 6 | MLS-B test | FOX 16 | P 17 | CPT MIC 0.5 | AK 17 | CN 30 | CIP 22 | LEV 29 | |
| HL-R | HL-R | HL-R | HL-R | HL-R | cMLS-B | LL-R | LL-R | S | LL-R | S | I | I | |
| Gene | ermB | mecA | blaZ, blaR1, blaI | ND | aph(3′)-III | ND | gyrA, gyrB, norA | gyrA, gyrB, parF | |||||
| Phenotype expression | + constitutive resistance | + | + | − | + | − | + | + | |||||
| (B) | |||||||||||||
| ATB results | Antibiotics, MIC (mg/L) or Diameter (mm) | ||||||||||||
| Glycopeptides | Oxazolidinone | Lipopeptide | Tetracyclines | Miscellaneous agent | |||||||||
| VA MIC 1.5 | TP MIC 0.75 | LZD MIC 1.5 | DPC MIC 0.25 | TET 17 | TGC MIC 0.094 | MUP 35 | FUS 30 | SC MIC 48 | |||||
| S | S | S | S | LL-R | S | S | S | S | |||||
| Gene | ND | tcaA, tcaB, tcaR | ND | clsA, gdpD, mprF, liaF, liaR, liaS | tetK, tet38 | rpsJ (S10p), mepA, mepR | iso-tRNA | fusA | ND | ||||
| Phenotype expression | − | NEx | − | NEx | + | NEx | NEx | NEx | − | ||||
| Genome Feature | MRSA SO574/12 Characteristic |
|---|---|
| Size (bp) | 2,792,694 |
| G+C content (%) | 32.74 |
| Coarse consistency (%) | 100 |
| Fine consistency (%) | 99.8 |
| Completeness (%) | 100 |
| Contamination (%) | 0 |
| Number of raw reads | 683,054 |
| Total contigs count | 67 |
| Largest contig (bp) | 494,917 |
| Contigs N50 (bp) | 127,953 |
| Contigs N75 (bp) | 65,061 |
| Contigs L50 | 7 |
| Contigs L75 | 14 |
| Quast quality genome | good |
| Genome Feature | Clinical Isolate Genome of MRSA SO574/12 | Reference Genome CA-MRSA ZY05 SCCmecVb/ST338/CC59 |
|---|---|---|
| Size (bp) | 2,792,694 | 2,822,516 |
| G+C content (%) | 32.74 | 32.9 |
| Protein encoding genes with functional assignment | 1854 | 1844 |
| Proteins encoding without functional assignment | 789 | 778 |
| tRNA genes | 60 | 60 |
| Protein coding sequences | 2643 | 2622 |
| Proteins with functional assignments | 2131 | 2121 |
| Hypothetical proteins | 512 | 501 |
| Total number of specialty genes: | 330 | 314 |
| Virulence factor genes (VFG) | 98 | 95 |
| Antimicrobial resistance genes (AMR) | 80 | 72 |
| Transporter genes (TG) | 98 | 96 |
| Drug target genes (DTG) | 54 | 51 |
| Mechanism of Resistance | Gene | Gene Product/Function/KEGG Code | Class of Antibiotic | Antibiotics | PubMed ID |
|---|---|---|---|---|---|
| Antibiotic inactivation enzyme (transferases, hydrolases) and/or regulator modulating expression of antibiotic resistance genes | ant(6)-I | Aminoglycoside 6-nucleotidyl-transferase (EC 2.7.7.-), ANT(6)-I | Aminoglycoside | streptomycin | 19603075; 2168151; 8293959 |
| aph(3′)-III/aph(3′)-IV/aph(3′)-VI/aph(3′)-VII | Aminoglycoside 3′-phosphotransferase (EC 2.7.1.95), APH(3′)-III/APH(3′)-IV/APH(3′)-VI/APH(3′)-VII | Aminoglycoside | butirosin, neomycin, kanamycin, amikacin, kanamycin, lividomycin, isepamicin, ribostamycin, paromomycin | 6313476; 2846986; 2848443; 2550983 | |
| blaI blaR1 blaZ | β-lactamase repressor BlaI β-lactamase regulatory sensor-transducer BlaR1 Class A β-lactamase (EC 3.5.2.6), BlaZ | β-lactams, Penicillins | penicillin, ampicillin, amoxicillin, piperacillin | 9220009; 12591921; 6793593; 2555777 | |
| catA8 | Chloramphenicol O-acetyltransferase (EC 2.3.1.28), CatA8 family | Phenicols | choramphenicol | 15150221 | |
| sat-4 | Streptothricin acetyltransferase Sat-4 | Streptothricins | streptothricin | 31605529 | |
| Antibiotic resistance gene cluster, cassette, or operon, antibiotic target replacement protein | mecA | Penicillin-binding protein PBP2a, methicillin-resistance determinant MecA, transpeptidase | β-lactams | almost all β-lactams, except V generation of cephalosporins, e.g., amoxicillin, cefoxitin, ceftazidime, amoxicillin/clavulanic acid, meropenem | 1507425; 1691614; 1544435; 30209034 |
| folA/dfrC | Dihydrofolate reductase (EC 1.5.1.3) | Diaminopyrimidines | trimetoprim | 8540692 | |
| Antibiotic target-modifying enzyme | ermB | 23S rRNA (adenine(2058)-N(6))-dimethyl- transferase (EC 2.1.1.184), ErmB | Macrolides, Linosamides, Streptogramin B | erythromycin, azithromycin, clarithromycin, clindamycin, lincomycin, quinupristin, virginiamycin S, pristinamycin IA | 11959553 |
| Efflux pump conferring antibiotic resistance and/or the gene modulating antibiotic efflux | bceA bceB bceR bceS | Bacitracin export ATP-binding protein BceA Bacitracin export permease protein BceB Two-component response regulator BceR Two-component sensor histidine kinase BceS | Peptide antibiotics | bacitracin | 25118291 |
| sav1866 | Efflux ABC transporter, permease/ATP-binding protein YgaD | Ansamycins | rifampicin, rifaximin | 18690712 | |
| tet38 | Tetracycline resistance, MFS efflux pump Tet38 | Tetracyclines | tetracycline | 26324534; 33619028 | |
| tetK | Tetracycline resistance, MFS efflux pump TetK | Tetracyclines | tetracycline, doxycycline | 7877638 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymanek-Majchrzak, K.; Młynarczyk, G. Genomic Insights of First ermB-Positive ST338-SCCmecVT/CC59 Taiwan Clone of Community-Associated Methicillin-Resistant Staphylococcus aureus in Poland. Int. J. Mol. Sci. 2022, 23, 8755. https://doi.org/10.3390/ijms23158755
Szymanek-Majchrzak K, Młynarczyk G. Genomic Insights of First ermB-Positive ST338-SCCmecVT/CC59 Taiwan Clone of Community-Associated Methicillin-Resistant Staphylococcus aureus in Poland. International Journal of Molecular Sciences. 2022; 23(15):8755. https://doi.org/10.3390/ijms23158755
Chicago/Turabian StyleSzymanek-Majchrzak, Ksenia, and Grażyna Młynarczyk. 2022. "Genomic Insights of First ermB-Positive ST338-SCCmecVT/CC59 Taiwan Clone of Community-Associated Methicillin-Resistant Staphylococcus aureus in Poland" International Journal of Molecular Sciences 23, no. 15: 8755. https://doi.org/10.3390/ijms23158755