
The NEL Family of Bacterial E3 Ubiquitin Ligases


Abstract
:1. Introduction
1.1. Type III Secretion Systems
1.2. Ubiquitination Systems
2. Ubiquitination in Bacterial Pathogenesis
2.1. Ubiquitination and Bacterial Invasion and Adhesion
2.2. Manipulation of the Innate Immune Response (NF-KB)
2.3. Manipulation of Defense-Associated Ubiquitination Machinery of Host Plants
2.4. Subversion of Xenophagy
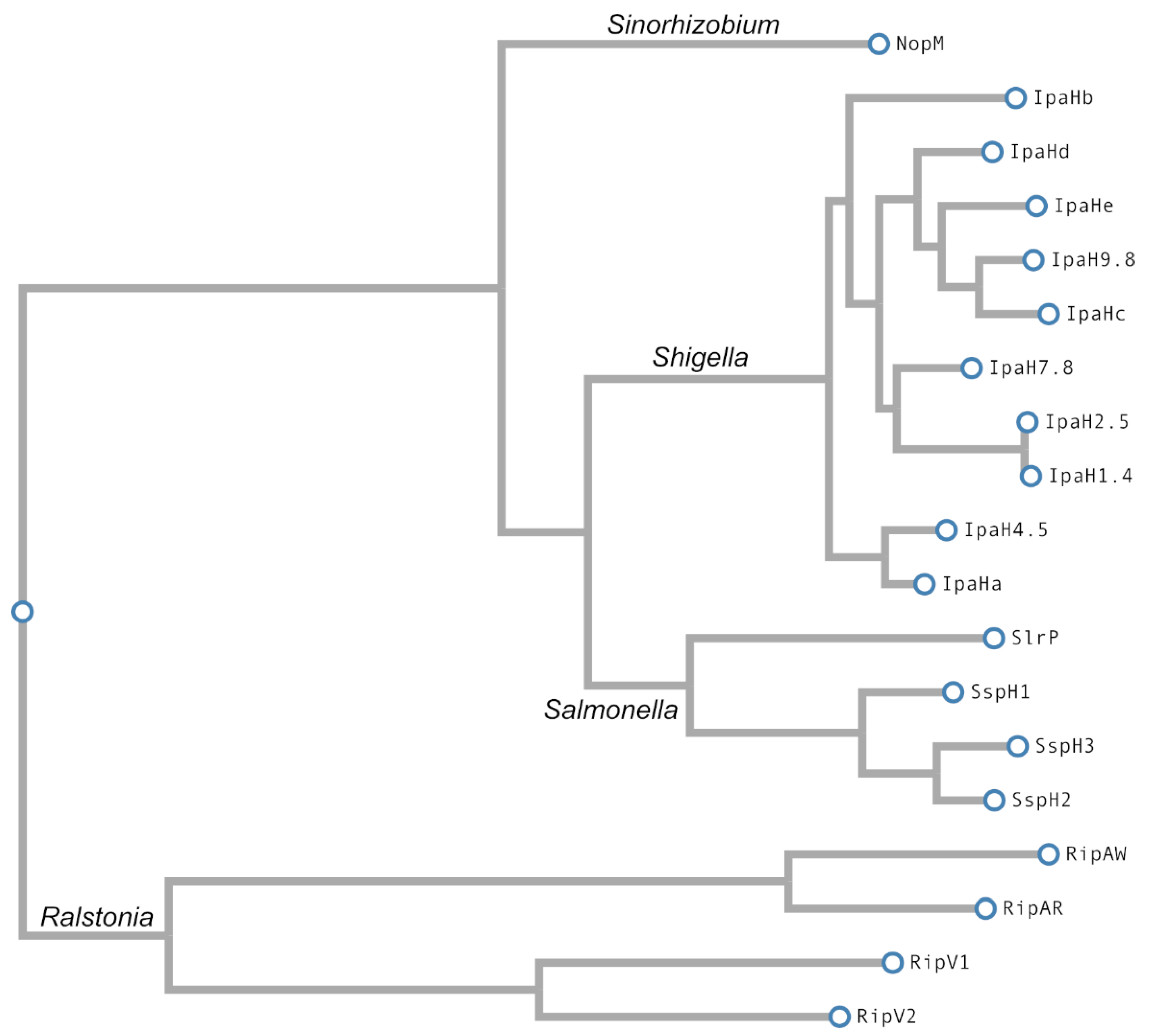
3. E3 Ubiquitin Ligases of the NEL Family
3.1. Members of the Family

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Hosts | Effector Name | Domains | References |
|---|---|---|---|---|
| Salmonella enterica | Mammals | SlrP | LRR-NEL | [104,105] |
| SspH1 | LRR-NEL | [106,107] | ||
| SspH2 | LRR-NEL | [106,108] | ||
| SspH3 | LRR-NEL | [109] | ||
| Shigella flexneri | Primates | IpaH proteins (1.4, 2.5, 4.5, 7.8, 9.8, a, b, c, d, e) | LRR-NEL | [107,110] |
| Sinorhizobium fredii | Plants | NopM | LRR-NEL | [111,112] |
| Ralstonia solanacearum | Plants | RipAR | NEL | [113] |
| RipAW | NEL | [113] | ||
| RipV1 | NEL | [113,114,115] | ||
| RipV2 | NEL | [116] |
3.2. Expression, Translocation, and Subcellular Localization
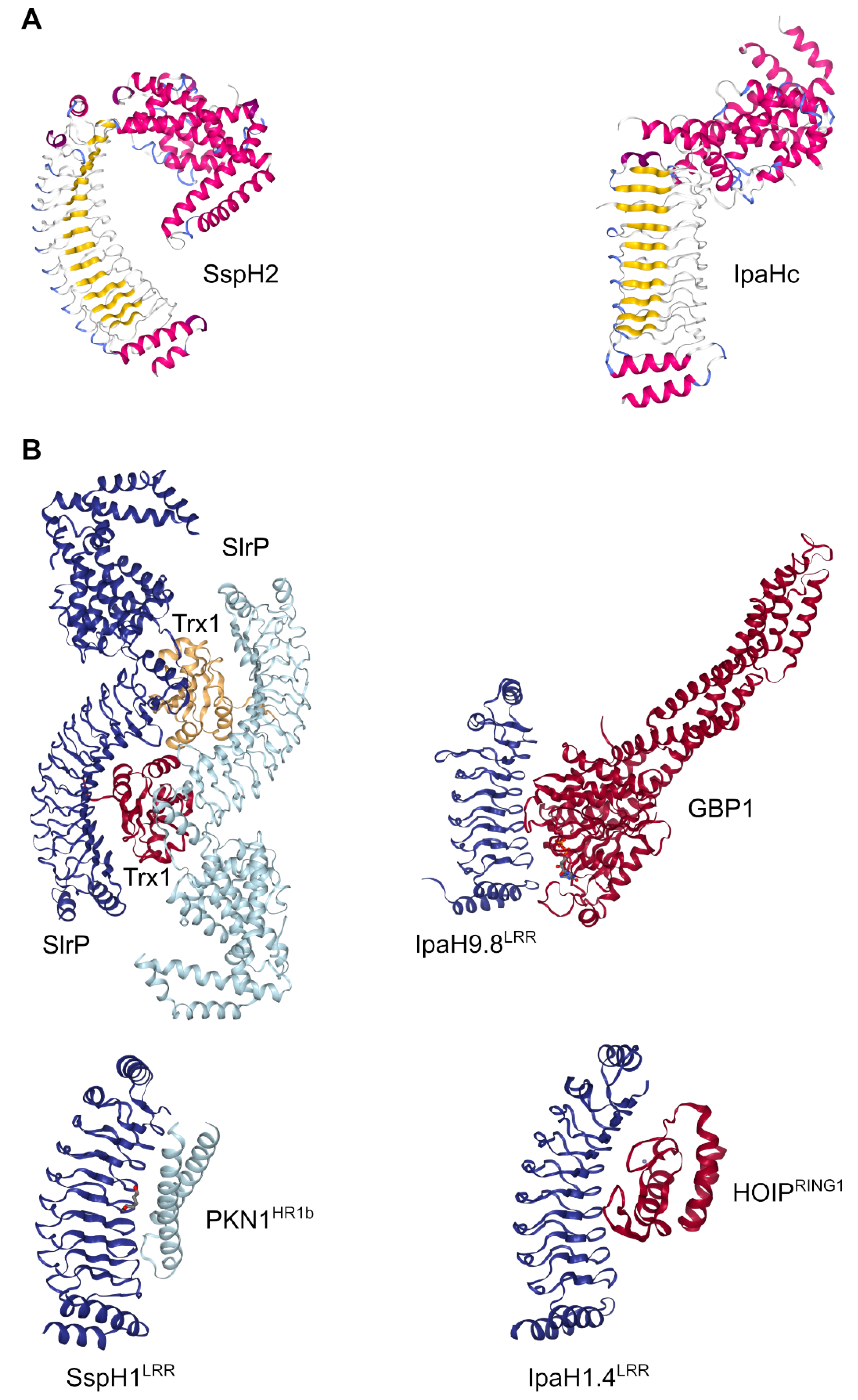
3.3. Enzymatic Activity and Structural Studies

3.4. Role in Virulence or Symbiosis
3.5. Targets of NEL Effectors
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hajra, D.; Nair, A.V.; Chakravortty, D. An elegant nano-injection machinery for sabotaging the host: Role of Type III secretion system in virulence of different human and animal pathogenic bacteria. Phys. Life Rev. 2021, 38, 25–54. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.J.; Chau-Ly, I.J.; Lewis, J.D. What the Wild Things Do: Mechanisms of Plant Host Manipulation by Bacterial Type III-Secreted Effector Proteins. Microorganisms 2021, 9, 1029. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, J.; Worrall, L.J.; Strynadka, N.C.J. Recent structural advances towards understanding of the bacterial type III secretion injectisome. Trends Biochem. Sci. 2022, in press. [CrossRef] [PubMed]
- Hu, Y.; Huang, H.; Cheng, X.; Shu, X.; White, A.P.; Stavrinides, J.; Köster, W.; Zhu, G.; Zhao, Z.; Wang, Y. A global survey of bacterial type III secretion systems and their effectors. Environ. Microbiol. 2017, 19, 3879–3895. [Google Scholar] [CrossRef] [PubMed]
- Gazi, A.D.; Kokkinidis, M.; Fadouloglou, V.E. α-Helices in the Type III Secretion Effectors: A Prevalent Feature with Versatile Roles. Int. J. Mol. Sci. 2021, 22, 5412. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.; Xin, X.; Mecey, C.; He, S.Y. Subcellular Localization of Pseudomonas syringae pv. tomato Effector Proteins in Plants. Methods Mol. Biol. 2017, 1531, 141–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, P. Functional domains and motifs of bacterial type III effector proteins and their roles in infection. FEMS Microbiol. Rev. 2011, 35, 1100–1125. [Google Scholar] [CrossRef] [Green Version]
- Gilzer, D.; Schreiner, M.; Niemann, H.H. Direct interaction of a chaperone-bound type III secretion substrate with the export gate. Nat. Commun. 2022, 13, 2858. [Google Scholar] [CrossRef]
- Kim, B.H.; Kim, H.G.; Kim, J.S.; Jang, J.I.; Park, Y.K. Analysis of functional domains present in the N-terminus of the SipB protein. Microbiology 2007, 153, 2998–3008. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Chen, J.; Zhou, D. Delineation and characterization of the actin nucleation and effector translocation activities of Salmonella SipC. Mol. Microbiol. 2005, 55, 1379–1389. [Google Scholar] [CrossRef]
- Brown, N.F.; Szeto, J.; Jiang, X.; Coombes, B.K.; Brett Finlay, B.; Brumell, J.H. Mutational analysis of Salmonella translocated effector members SifA and SopD2 reveals domains implicated in translocation, subcellular localization and function. Microbiology 2006, 152, 2323–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen-Vercoe, E.; Toh, M.C.W.; Waddell, B.; Ho, H.; DeVinney, R. A carboxy-terminal domain of Tir from enterohemorrhagic Escherichia coli O157:H7 (EHEC O157:H7) required for efficient type III secretion. FEMS Microbiol. Lett. 2005, 243, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavrinides, J.; Ma, W.; Guttman, D.S. Terminal reassortment drives the quantum evolution of type III effectors in bacterial pathogens. PLoS Pathog. 2006, 2, e104. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.P.; Kingsley, R.A. Salmonella pathogenesis and host-adaptation in farmed animals. Curr. Opin. Microbiol. 2021, 63, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Lou, L.; Zhang, P.; Piao, R.; Wang, Y. Salmonella Pathogenicity Island 1 (SPI-1) and Its Complex Regulatory Network. Front. Cell. Infect. Microbiol. 2019, 9, 270. [Google Scholar] [CrossRef] [Green Version]
- Jennings, E.; Thurston, T.L.M.; Holden, D.W. Salmonella SPI-2 Type III Secretion System Effectors: Molecular Mechanisms And Physiological Consequences. Cell Host Microbe 2017, 22, 217–231. [Google Scholar] [CrossRef]
- Bao, H.; Wang, S.; Zhao, J.H.; Liu, S.L. Salmonella secretion systems: Differential roles in pathogen-host interactions. Microbiol. Res. 2020, 241, 126591. [Google Scholar] [CrossRef]
- Schnupf, P.; Sansonetti, P.J. Shigella Pathogenesis: New Insights through Advanced Methodologies. Microbiol. Spectr. 2019, 7, 1–24. [Google Scholar] [CrossRef]
- Belotserkovsky, I.; Sansonetti, P.J. Shigella and enteroinvasive Escherichia coli. Curr. Top. Microbiol. Immunol. 2018, 416, 1–26. [Google Scholar] [CrossRef]
- Bajunaid, W.; Haidar-Ahmad, N.; Kottarampatel, A.H.; Manigat, F.O.; Silué, N.; Tchagang, C.F.; Tomaro, K.; Campbell-Valois, F.X. The T3SS of Shigella: Expression, Structure, Function, and Role in Vacuole Escape. Microorganisms 2020, 8, 1933. [Google Scholar] [CrossRef]
- Muthuramalingam, M.; Whittier, S.K.; Picking, W.L.; Picking, W.D. The Shigella Type III Secretion System: An Overview from Top to Bottom. Microorganisms 2021, 9, 451. [Google Scholar] [CrossRef] [PubMed]
- Nasser, A.; Mosadegh, M.; Azimi, T.; Shariati, A. Molecular mechanisms of Shigella effector proteins: A common pathogen among diarrheic pediatric population. Mol. Cell. Pediatr. 2022, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef] [PubMed]
- Pueppke, S.G.; Broughton, W.J. Rhizobium sp. strain NGR234 and R. fredii USDA257 share exceptionally broad, nested host ranges. Mol. Plant Microbe Interact. 1999, 12, 293–318. [Google Scholar] [CrossRef] [Green Version]
- Vinardell, J.M.; Acosta-Jurado, S.; Zehner, S.; Göttfert, M.; Becker, A.; Baena, I.; Blom, J.; Crespo-Rivas, J.C.; Goesmann, A.; Jaenicke, S.; et al. The Sinorhizobium fredii HH103 Genome: A Comparative Analysis With S. fredii Strains Differing in Their Symbiotic Behavior With Soybean. Mol. Plant Microbe Interact. 2015, 28, 811–824. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Krishnan, H.B. Sinorhizobium fredii USDA257, a cultivar-specific soybean symbiont, carries two copies of y4yA and y4yB, two open reading frames that are located in a region that encodes the type III protein secretion system. Mol. Plant Microbe Interact. 2000, 13, 1010–1014. [Google Scholar] [CrossRef]
- Viprey, V.; Del Greco, A.; Golinowski, W.; Broughton, W.J.; Perret, X. Symbiotic implications of type III protein secretion machinery in Rhizobium. Mol. Microbiol. 1998, 28, 1381–1389. [Google Scholar] [CrossRef]
- Staehelin, C.; Krishnan, H.B. Nodulation outer proteins: Double-edged swords of symbiotic rhizobia. Biochem. J. 2015, 470, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Paudel, S.; Dobhal, S.; Alvarez, A.M.; Arif, M. Taxonomy and Phylogenetic Research on Ralstonia solanacearum Species Complex: A Complex Pathogen with Extraordinary Economic Consequences. Pathogens 2020, 9, 886. [Google Scholar] [CrossRef]
- Salanoubat, M.; Genin, S.; Artiguenave, F.; Gouzy, J.; Mangenot, S.; Arlat, M.; Billault, A.; Brottiert, P.; Camus, J.C.; Cattolico, L.; et al. Genome sequence of the plant pathogen Ralstonia solanacearum. Nature 2002, 415, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Boucher, C.; Martinel, A.; Barberis, P.; Alloing, G.; Zischek, C. Virulence genes are carried by a megaplasmid of the plant pathogen Pseudomonas solanacearum. Mol. Gen. Genet. 1986, 205, 270–275. [Google Scholar] [CrossRef]
- Landry, D.; González-Fuente, M.; Deslandes, L.; Peeters, N. The large, diverse, and robust arsenal of Ralstonia solanacearum type III effectors and their in planta functions. Mol. Plant Pathol. 2020, 21, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Suleman, M.; Ma, M.; Ge, G.; Hua, D.; Li, H. The role of alternative oxidase in plant hypersensitive response. Plant Biol. 2021, 23, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, C.R.R.; Carrere, S.; Lonjon, F.; Vailleau, F.; Macho, A.P.; Genin, S.; Peeters, N. Pangenomic type III effector database of the plant pathogenic Ralstonia spp. PeerJ 2019, 7, e7346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Maupin-Furlow, J.A. Ubiquitin-like proteins and their roles in archaea. Trends Microbiol. 2013, 21, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Toma-Fukai, S.; Shimizu, T. Structural Diversity of Ubiquitin E3 Ligase. Molecules 2021, 26, 6682. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Wilkinson, K.D.; Urban, M.K.; Haas, A.L. Ubiquitin is the ATP-dependent proteolysis factor I of rabbit reticulocytes. J. Biol. Chem. 1980, 255, 7529–7532. [Google Scholar] [CrossRef]
- Salas-Lloret, D.; González-Prieto, R. Insights in Post-Translational Modifications: Ubiquitin and SUMO. Int. J. Mol. Sci. 2022, 23, 3281. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Wakatsuki, S.; Walters, K.J. Ubiquitin-binding domains from structures to functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Chau, V.; Tobias, J.W.; Bachmair, A.; Marriott, D.; Ecker, D.J.; Gonda, D.K.; Varshavsky, A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 1989, 243, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Braten, O.; Livneh, I.; Ziv, T.; Admon, A.; Kehat, I.; Caspi, L.H.; Gonen, H.; Bercovich, B.; Godzik, A.; Jahandideh, S.; et al. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc. Natl. Acad. Sci. USA 2016, 113, E4639–E4647. [Google Scholar] [CrossRef] [Green Version]
- Chiu, R.K.; Brun, J.; Ramaekers, C.; Theys, J.; Weng, L.; Lambin, P.; Gray, D.A.; Wouters, B.G. Lysine 63-polyubiquitination guards against translesion synthesis-induced mutations. PLoS Genet. 2006, 2, e116. [Google Scholar] [CrossRef]
- Chen, Z.J.; Sun, L.J. Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 2009, 33, 275–286. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Haas, A.L.; Rose, I.A. The mechanism of ubiquitin activating enzyme. A kinetic and equilibrium analysis. J. Biol. Chem. 1982, 257, 10329–10337. [Google Scholar] [CrossRef]
- Vozandychova, V.; Stojkova, P.; Hercik, K.; Rehulka, P.; Stulik, J. The Ubiquitination System within Bacterial Host-Pathogen Interactions. Microorganisms 2021, 9, 638. [Google Scholar] [CrossRef] [PubMed]
- Cotton, T.R.; Lechtenberg, B.C. Chain reactions: Molecular mechanisms of RBR ubiquitin ligases. Biochem. Soc. Trans. 2020, 48, 1737–1750. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Zhang, Y.; Huibregtse, J.M.; Zhou, D.; Chen, J. Crystal structure of SopA, a Salmonella effector protein mimicking a eukaryotic ubiquitin ligase. Nat. Struct. Mol. Biol. 2008, 15, 65–70. [Google Scholar] [CrossRef]
- Ashida, H.; Kim, M.; Sasakawa, C. Exploitation of the host ubiquitin system by human bacterial pathogens. Nat. Rev. Microbiol. 2014, 12, 399–413. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, H.; Hu, L.; Wang, J.; Zhou, Y.; Pang, Z.; Liu, L.; Shao, F. Structure of a Shigella effector reveals a new class of ubiquitin ligases. Nat. Struct. Mol. Biol. 2008, 15, 1302–1308. [Google Scholar] [CrossRef]
- Singer, A.U.; Rohde, J.R.; Lam, R.; Skarina, T.; Kagan, O.; Dileo, R.; Chirgadze, N.Y.; Cuff, M.E.; Joachimiak, A.; Tyers, M.; et al. Structure of the Shigella T3SS effector IpaH defines a new class of E3 ubiquitin ligases. Nat. Struct. Mol. Biol. 2008, 15, 1293–1301. [Google Scholar] [CrossRef]
- Levin, I.; Eakin, C.; Blanc, M.; Klevit, R.E.; Miller, S.I.; Brzovic, P.S. Identification of an unconventional E3 binding surface on the UbcH5 ~ Ub conjugate recognized by a pathogenic bacterial E3 ligase. Proc. Natl. Acad. Sci. USA 2010, 107, 2848–2853. [Google Scholar] [CrossRef] [Green Version]
- Keszei, A.F.A.; Sicheri, F. Mechanism of catalysis, E2 recognition, and autoinhibition for the IpaH family of bacterial E3 ubiquitin ligases. Proc. Natl. Acad. Sci. USA 2017, 114, 1311–1316. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Sheedlo, M.J.; Yu, K.; Tan, Y.; Nakayasu, E.S.; Das, C.; Liu, X.; Luo, Z.-Q. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 2016, 533, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the immune system. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Wu, L.; Tang, C.; Wang, H.; Wei, Y. Autophagy-Inflammation Interplay During Infection: Balancing Pathogen Clearance and Host Inflammation. Front. Pharmacol. 2022, 13, 832750. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.G.; Pruneda, J.N. Bacteria make surgical strikes on host ubiquitin signaling. PLoS Pathog. 2021, 17, e1009341. [Google Scholar] [CrossRef] [PubMed]
- Bassères, E.; Coppotelli, G.; Pfirrmann, T.; Andersen, J.B.; Masucci, M.; Frisan, T. The ubiquitin C-terminal hydrolase UCH-L1 promotes bacterial invasion by altering the dynamics of the actin cytoskeleton. Cell. Microbiol. 2010, 12, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Fiskin, E.; Bionda, T.; Dikic, I.; Behrends, C. Global Analysis of Host and Bacterial Ubiquitinome in Response to Salmonella Typhimurium Infection. Mol. Cell 2016, 62, 967–981. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; You, Q.; Zhu, H.; Chang, Z.; Li, Q.; Wang, H.; Wang, C.; Wang, H.; Hui, L.; Du, C.; et al. Bacterial effector NleL promotes enterohemorrhagic E. coli-induced attaching and effacing lesions by ubiquitylating and inactivating JNK. PLoS Pathog. 2017, 13, e1006534. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Sebban, H.; Yamaoka, S.; Courtois, G. Posttranslational modifications of NEMO and its partners in NF-kappaB signaling. Trends Cell Biol. 2006, 16, 569–577. [Google Scholar] [CrossRef]
- Kamanova, J.; Sun, H.; Lara-Tejero, M.; Galán, J.E. The Salmonella Effector Protein SopA Modulates Innate Immune Responses by Targeting TRIM E3 Ligase Family Members. PLoS Pathog. 2016, 12, e1005552. [Google Scholar] [CrossRef]
- Ye, Z.; Petrof, E.O.; Boone, D.; Claud, E.C.; Sun, J. Salmonella Effector AvrA Regulation of Colonic Epithelial Cell Inflammation by Deubiquitination. Am. J. Pathol. 2007, 171, 882–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Negrate, G.; Faustin, B.; Welsh, K.; Loeffler, M.; Krajewska, M.; Hasegawa, P.; Mukherjee, S.; Orth, K.; Krajewski, S.; Godzik, A.; et al. Salmonella Secreted Factor L Deubiquitinase of Salmonella typhimurium Inhibits NF-κB, Suppresses IκBα Ubiquitination and Modulates Innate Immune Responses. J. Immunol. 2008, 180, 5045–5056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; El Qaidi, S.; Hardwidge, P.R. SseL Deubiquitinates RPS3 to Inhibit Its Nuclear Translocation. Pathogens 2018, 7, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesquita, F.S.; Holden, D.W.; Rolhion, N. Lack of effect of the Salmonella deubiquitinase SseL on the NF-κB pathway. PLoS ONE 2013, 8, e53064. [Google Scholar] [CrossRef]
- Kim, D.W.; Lenzen, G.; Page, A.L.; Legrain, P.; Sansonetti, P.J.; Parsot, C. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. USA 2005, 102, 14046–14051. [Google Scholar] [CrossRef] [Green Version]
- Sanada, T.; Kim, M.; Mimuro, H.; Suzuki, M.; Ogawa, M.; Oyama, A.; Ashida, H.; Kobayashi, T.; Koyama, T.; Nagai, S.; et al. The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature 2012, 483, 623–626. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, H.; Chae, W.B.; Oh, M.H. Pattern recognition receptors and their interactions with bacterial type III effectors in plants. Genes Genom. 2019, 41, 499–506. [Google Scholar] [CrossRef]
- Fick, A.; Swart, V.; van den Berg, N. The Ups and Downs of Plant NLR Expression During Pathogen Infection. Front. Plant Sci. 2022, 13, 921148. [Google Scholar] [CrossRef]
- Balint-Kurti, P. The plant hypersensitive response: Concepts, control and consequences. Mol. Plant Pathol. 2019, 20, 1163–1178. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, P.; Joshi, B.J.; Maupin-Furlow, J.A.; Uthandi, S. Bacterial effectors mimicking ubiquitin-proteasome pathway tweak plant immunity. Microbiol. Res. 2021, 250, 126810. [Google Scholar] [CrossRef]
- Abramovitch, R.B.; Janjusevic, R.; Stebbins, C.E.; Martin, G.B. Type III effector AvrPtoB requires intrinsic E3 ubiquitin ligase activity to suppress plant cell death and immunity. Proc. Natl. Acad. Sci. USA 2006, 103, 2851–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janjusevic, R.; Abramovitch, R.B.; Martin, G.B.; Stebbins, C.E. A bacterial inhibitor of host programmed cell death defenses is an E3 ubiquitin ligase. Science 2006, 311, 222–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Zhou, X.; Sun, L.; Wang, K.; Yang, F.; Liao, H.; Rong, W.; Yin, J.; Chen, H.; Chen, X.; et al. The Xanthomonas effector XopK harbours E3 ubiquitin-ligase activity that is required for virulence. New Phytol. 2018, 220, 219–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, J.W.; Schulman, B.A. Cullin-RING Ubiquitin Ligase Regulatory Circuits: A Quarter Century Beyond the F-Box Hypothesis. Annu. Rev. Biochem. 2021, 90, 403–429. [Google Scholar] [CrossRef]
- Schrammeijer, B.; Risseeuw, E.; Pansegrau, W.; Regensburg-Tuïnk, T.J.G.; Crosby, W.L.; Hooykaas, P.J.J. Interaction of the virulence protein VirF of Agrobacterium tumefaciens with plant homologs of the yeast Skp1 protein. Curr. Biol. 2001, 11, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Vergunst, A.C.; Schrammeijer, B.; Den Dulk-Ras, A.; De Vlaam, C.M.T.; Regensburg-Tuink, T.J.G.; Hooykaas, P.J.J. VirB/D4-dependent protein translocation from Agrobacterium into plant cells. Science 2000, 290, 979–982. [Google Scholar] [CrossRef]
- Magori, S.; Citovsky, V. Hijacking of the Host SCF Ubiquitin Ligase Machinery by Plant Pathogens. Front. Plant Sci. 2011, 2, 87. [Google Scholar] [CrossRef] [Green Version]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Chan, H.; Lu, L.; Wong, K.T.; Wong, S.H.; Li, M.X.; Xiao, Z.G.; Cho, C.H.; Gin, T.; Chan, M.T.V.; et al. Autophagy in intracellular bacterial infection. Semin. Cell Dev. Biol. 2020, 101, 41–50. [Google Scholar] [CrossRef]
- Wu, S.; Shen, Y.; Zhang, S.; Xiao, Y.; Shi, S. Salmonella Interacts With Autophagy to Offense or Defense. Front. Microbiol. 2020, 11, 721. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Cai, W. Autophagy and Bacterial Infection. Adv. Exp. Med. Biol. 2020, 1207, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, F.S.; Thomas, M.; Sachse, M.; Santos, A.J.M.; Figueira, R.; Holden, D.W. The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PLoS Pathog. 2012, 8, e1002743. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zhang, Y.-G.; Lin, Z.; Lu, R.; Xia, Y.; Meng, C.; Pan, Z.; Xu, X.; Jiao, X.; Sun, J. Salmonella Enteritidis Effector AvrA Suppresses Autophagy by Reducing Beclin-1 Protein. Front. Immunol. 2020, 11, 686. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Gao, S.; Wang, T.; Yan, J.; Xu, G.; Li, Y.; Niu, H.; Huang, R.; Wu, S. A novel contribution of spvB to pathogenesis of Salmonella Typhimurium by inhibiting autophagy in host cells. Oncotarget 2016, 7, 8295–8309. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhou, P.; Cheng, S.; Lu, Q.; Nowak, K.; Hopp, A.-K.; Li, L.; Shi, X.; Zhou, Z.; Gao, W.; et al. A Bacterial Effector Reveals the V-ATPase-ATG16L1 Axis that Initiates Xenophagy. Cell 2019, 178, 552–566.e20. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; Gao, S.; Yuan, H.; Zuo, L.; Wu, C.; Huang, R.; Wu, S. Salmonella spvC Gene Inhibits Autophagy of Host Cells and Suppresses NLRP3 as Well as NLRC4. Front. Immunol. 2021, 12, 639019. [Google Scholar] [CrossRef]
- Lal, N.K.; Thanasuwat, B.; Huang, P.-J.; Cavanaugh, K.A.; Carter, A.; Michelmore, R.W.; Dinesh-Kumar, S.P. Phytopathogen Effectors Use Multiple Mechanisms to Manipulate Plant Autophagy. Cell Host Microbe 2020, 28, 558–571.e6. [Google Scholar] [CrossRef]
- Leong, J.X.; Raffeiner, M.; Spinti, D.; Langin, G.; Franz-Wachtel, M.; Guzman, A.R.; Kim, J.-G.; Pandey, P.; Minina, A.E.; Macek, B.; et al. A bacterial effector counteracts host autophagy by promoting degradation of an autophagy component. EMBO J. 2022, e110352. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Norkowski, S.; Schmidt, M.A.; Rüter, C. The species-spanning family of LPX-motif harbouring effector proteins. Cell. Microbiol. 2018, 20, e12945. [Google Scholar] [CrossRef] [Green Version]
- Soundararajan, V.; Patel, N.; Subramanian, V.; Sasisekharan, V.; Sasisekharan, R. The many faces of the YopM effector from plague causative bacterium Yersinia pestis and its implications for host immune modulation. Innate Immun. 2011, 17, 548–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPhee, J.B.; Bliska, J.B. Letter to the Editor and response. Innate Immun. 2011, 17, 558–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Wang, Y.; Du, Z.; Guan, K.; Cao, Y.; Yang, H.; Zhou, P.; Wu, F.; Chen, J.; Wang, P.; et al. The Yersinia Type III secretion effector YopM Is an E3 ubiquitin ligase that induced necrotic cell death by targeting NLRP3. Cell Death Dis. 2016, 7, e2519. [Google Scholar] [CrossRef] [PubMed]
- Tsolis, R.M.; Townsend, S.M.; Miao, E.A.; Miller, S.I.; Ficht, T.A.; Adams, L.G.; Bäumler, A.J. Identification of a putative Salmonella enterica serotype typhimurium host range factor with homology to IpaH and YopM by signature-tagged mutagenesis. Infect. Immun. 1999, 67, 6385–6393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal-Bayard, J.; Ramos-Morales, F. Salmonella type III secretion effector SlrP is an E3 ubiquitin ligase for mammalian thioredoxin. J. Biol. Chem. 2009, 284, 27587–27595. [Google Scholar] [CrossRef] [Green Version]
- Miao, E.A.; Scherer, C.A.; Tsolis, R.M.; Kingsley, R.A.; Adams, L.G.; Baumler, A.J.; Miller, S.I. Salmonella typhimurium leucine-rich repeat proteins are targeted to the SPI1 and SPI2 type III secretion systems. Mol. Microbiol. 1999, 34, 850–864. [Google Scholar] [CrossRef] [Green Version]
- Rohde, J.R.; Breitkreutz, A.; Chenal, A.; Sansonetti, P.J.; Parsot, C. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe 2007, 1, 77–83. [Google Scholar] [CrossRef]
- Quezada, C.M.; Hicks, S.W.; Galán, J.E.; Stebbins, C.E. A family of Salmonella virulence factors functions as a distinct class of autoregulated E3 ubiquitin ligases. Proc. Natl. Acad. Sci. USA 2009, 106, 4864–4869. [Google Scholar] [CrossRef] [Green Version]
- Herod, A.; Emond-Rheault, J.; Tamber, S.; Goodridge, L.; Lévesque, R.C.; Rohde, J. Genomic and phenotypic analysis of SspH1 identifies a new Salmonella effector, SspH3. Mol. Microbiol. 2021, 117, 770–789. [Google Scholar] [CrossRef]
- Ashida, H.; Toyotome, T.; Nagai, T.; Sasakawa, C. Shigella chromosomal IpaH proteins are secreted via the type III secretion system and act as effectors. Mol. Microbiol. 2007, 63, 680–693. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.A.; López-Baena, F.J.; Ollero, F.J.; Vinardell, J.M.; Espuny, M.D.R.; Bellogín, R.A.; Ruiz-Sainz, J.E.; Thomas, J.R.; Sumpton, D.; Ault, J.; et al. NopM and NopD are rhizobial nodulation outer proteins: Identification using LC-MALDI and LC-ESI with a monolithic capillary column. J. Proteome Res. 2007, 6, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.C.; Zhang, D.; Hann, D.R.; Xie, Z.P.; Staehelin, C. Biochemical properties and in planta effects of NopM, a rhizobial E3 ubiquitin ligase. J. Biol. Chem. 2018, 293, 15304–15315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, M.; Oda, K.; Mukaihara, T. Ralstonia solanacearum novel E3 ubiquitin ligase (NEL) effectors RipAW and RipAR suppress pattern-triggered immunity in plants. Microbiology 2017, 163, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Mukaihara, T.; Tamura, N.; Iwabuchi, M. Genome-wide identification of a large repertoire of Ralstonia solanacearum type III effector proteins by a new functional screen. Mol. Plant Microbe Interact. 2010, 23, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Peeters, N.; Carrère, S.; Anisimova, M.; Plener, L.; Cazalé, A.C.; Genin, S. Repertoire, unified nomenclature and evolution of the Type III effector gene set in the Ralstonia solanacearum species complex. BMC Genom. 2013, 14, 859. [Google Scholar] [CrossRef]
- Cheng, D.; Zhou, D.; Wang, Y.; Wang, B.; He, Q.; Song, B.; Chen, H. Ralstonia solanacearum type III effector RipV2 encoding a novel E3 ubiquitin ligase (NEL) is required for full virulence by suppressing plant PAMP-triggered immunity. Biochem. Biophys. Res. Commun. 2021, 550, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Bossi, N.; Uzzau, S.; Maloriol, D.; Bossi, L. Variable assortment of prophages provides a transferable repertoire of pathogenic determinants in Salmonella. Mol. Microbiol. 2001, 39, 260–272. [Google Scholar] [CrossRef]
- Buysse, J.M.; Stover, C.K.; Oaks, E.V.; Venkatesan, M.; Kopecko, D.J. Molecular cloning of invasion plasmid antigen (ipa) genes from Shigella flexneri: Analysis of ipa gene products and genetic mapping. J. Bacteriol. 1987, 169, 2561–2569. [Google Scholar] [CrossRef] [Green Version]
- Hartman, A.B.; Venkatesan, M.; Oaks, E.V.; Buysse, J.M. Sequence and molecular characterization of a multicopy invasion plasmid antigen gene, ipaH, of Shigella flexneri. J. Bacteriol. 1990, 172, 1905–1915. [Google Scholar] [CrossRef] [Green Version]
- Bongrand, C.; Sansonetti, P.J.; Parsot, C. Characterization of the promoter, MxiE box and 5’ UTR of genes controlled by the activity of the type III secretion apparatus in Shigella flexneri. PLoS ONE 2012, 7, e32862. [Google Scholar] [CrossRef] [Green Version]
- Jin, Q.; Yuan, Z.; Xu, J.; Wang, Y.; Shen, Y.; Lu, W.; Wang, J.; Liu, H.; Yang, J.; Yang, F.; et al. Genome sequence of Shigella flexneri 2a: Insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 2002, 30, 4432–4441. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Goldberg, M.B.; Burland, V.; Venkatesan, M.M.; Deng, W.; Fournier, G.; Mayhew, G.F.; Plunkett, G.; Rose, D.J.; Darling, A.; et al. Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect. Immun. 2003, 71, 2775–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambara, K.; Ardissone, S.; Kobayashi, H.; Saad, M.M.; Schumpp, O.; Broughton, W.J.; Deakin, W.J. Rhizobia utilize pathogen-like effector proteins during symbiosis. Mol. Microbiol. 2009, 71, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Mukaihara, T.; Tamura, N.; Murata, Y.; Iwabuchi, M. Genetic screening of Hrp type III-related pathogenicity genes controlled by the HrpB transcriptional activator in Ralstonia solanacearum. Mol. Microbiol. 2004, 54, 863–875. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Higgins, D.G. Clustal omega. Curr. Protoc. Bioinforma. 2014, 48, 3.13.1–3.13.16. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Narm, K.E.; Kalafatis, M.; Slauch, J.M. HilD, HilC, and RtsA Form Homodimers and Heterodimers To Regulate Expression of the Salmonella Pathogenicity Island I Type III Secretion System. J. Bacteriol. 2020, 202, e00012-20. [Google Scholar] [CrossRef]
- Kalafatis, M.; Slauch, J.M. Long-Distance Effects of H-NS Binding in the Control of hilD Expression in the Salmonella SPI1 Locus. J. Bacteriol. 2021, 203, e0030821. [Google Scholar] [CrossRef]
- Fass, E.; Groisman, E.A. Control of Salmonella pathogenicity island-2 gene expression. Curr. Opin. Microbiol. 2009, 12, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.D.; Kim, K.; Slauch, J.M. PhoP-mediated repression of the SPI1 T3SS in Salmonella enterica serovar Typhimurium. J. Bacteriol. 2019, 201, e00264-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante, V.H.; Martínez, L.C.; Santana, F.J.; Knodler, L.A.; Steele-mortimer, O.; Puente, J.L. HilD-mediated transcriptional cross-talk between SPI-1 and SPI-2. Proc. Natl. Acad. Sci. USA 2008, 105, 14591–14596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, L.C.; Banda, M.M.; Fernández-Mora, M.; Santana, F.J.; Bustamante, V.H. HilD induces expression of Salmonella pathogenicity island 2 genes by displacing the global negative regulator H-NS from ssrAB. J. Bacteriol. 2014, 196, 3746–3755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordero-Alba, M.; Ramos-Morales, F. Patterns of expression and translocation of the ubiquitin ligase SlrP in Salmonella enterica serovar Typhimurium. J. Bacteriol. 2014, 196, 3912–3922. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Hensel, M. Systematic analysis of the SsrAB virulon of Salmonella enterica. Infect. Immun. 2010, 78, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Miao, E.A.; Miller, S.I. A conserved amino acid sequence directing intracellular type III secretion by Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 2000, 97, 7539–7544. [Google Scholar] [CrossRef] [Green Version]
- Haraga, A.; Miller, S.I. A Salmonella enterica serovar Typhimurium translocated leucine-rich repeat effector protein inhibits NF-kappa B-dependent gene expression. Infect. Immun. 2003, 71, 4052–4058. [Google Scholar] [CrossRef] [Green Version]
- Bernal-Bayard, J.; Cardenal-Muñoz, E.; Ramos-Morales, F. The Salmonella type III secretion effector, Salmonella leucine-rich repeat protein (SlrP), targets the human chaperone ERdj3. J. Biol. Chem. 2010, 285, 16360–16368. [Google Scholar] [CrossRef] [Green Version]
- Miao, E.A.; Brittnacher, M.; Haraga, A.; Jeng, R.L.; Welch, M.D.; Miller, S.I. Salmonella effectors translocated across the vacuolar membrane interact with the actin cytoskeleton. Mol. Microbiol. 2003, 48, 401–415. [Google Scholar] [CrossRef]
- Hicks, S.W.; Charron, G.; Hang, H.C.; Galán, J.E. Subcellular targeting of Salmonella virulence proteins by host-mediated S-palmitoylation. Cell Host Microbe 2011, 10, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Dorman, M.J.; Dorman, C.J. Regulatory Hierarchies Controlling Virulence Gene Expression in Shigella flexneri and Vibrio cholerae. Front. Microbiol. 2018, 9, 2686. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, C.; Glaser, P.; Rusniok, C.; Nedjari, H.; D’Hauteville, H.; Kunst, F.; Sansonetti, P.; Parsot, C. The virulence plasmid pWR100 and the repertoire of proteins secreted by the type III secretion apparatus of Shigella flexneri. Mol. Microbiol. 2000, 38, 760–771. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.L.; Falconi, M.; Micheli, G.; Colonna, B.; Prosseda, G. The Multifaceted Activity of the VirF Regulatory Protein in the Shigella Lifestyle. Front. Mol. Biosci. 2016, 3, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardini, M.L.; Mounier, J.; D’Hauteville, H.; Coquis-Rondon, M.; Sansonetti, P.J. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc. Natl. Acad. Sci. USA 1989, 86, 3867–3871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, B.; Sasakawa, C.; Tobe, T.; Makino, S.; Komatsu, K.; Yoshikawa, M. A dual transcriptional activation system for the 230 kb plasmid genes coding for virulence-associated antigens of Shigella flexneri. Mol. Microbiol. 1989, 3, 627–635. [Google Scholar] [CrossRef]
- Mavris, M.; Page, A.L.; Tournebize, R.; Demers, B.; Sansonetti, P.; Parsot, C. Regulation of transcription by the activity of the Shigella flexneri type III secretion apparatus. Mol. Microbiol. 2002, 43, 1543–1553. [Google Scholar] [CrossRef]
- Le Gall, T.; Mavris, M.; Martino, M.C.; Bernardini, M.L.; Denamur, E.; Parsot, C.; Le Gall, T.; Mavris, M.; Martino, M.C.; Bernardini, M.L.; et al. Analysis of virulence plasmid gene expression defines three classes of effectors in the type III secretion system of Shigella flexneri. Microbiology 2005, 151, 951–962. [Google Scholar] [CrossRef] [Green Version]
- Campbell-Valois, F.X.; Pontier, S.M. Implications of Spatiotemporal Regulation of Shigella flexneri Type Three Secretion Activity on Effector Functions: Think Globally, Act Locally. Front. Cell. Infect. Microbiol. 2016, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Toyotome, T.; Suzuki, T.; Kuwae, A.; Nonaka, T.; Fukuda, H.; Imajoh-Ohmi, S.; Toyofuku, T.; Hori, M.; Sasakawa, C. Shigella protein IpaH(9.8) is secreted from bacteria within mammalian cells and transported to the nucleus. J. Biol. Chem. 2001, 276, 32071–32079. [Google Scholar] [CrossRef] [Green Version]
- Bierne, H.; Pourpre, R. Bacterial Factors Targeting the Nucleus: The Growing Family of Nucleomodulins. Toxins 2020, 12, 220. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Mimuro, H.; Kim, M.; Ogawa, M.; Ashida, H.; Toyotome, T.; Franchi, L.; Suzuki, M.; Sanada, T.; Suzuki, T.; et al. Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to Demolish macrophages. Proc. Natl. Acad. Sci. USA 2014, 111, E4254–E4263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashida, H.; Nakano, H.; Sasakawa, C. Shigella IpaH0722 E3 ubiquitin ligase effector targets TRAF2 to inhibit PKC-NF-κB activity in invaded epithelial cells. PLoS Pathog. 2013, 9, e1003409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Lu, L.; Liu, X.; Liu, X.; Pan, C.; Feng, E.; Wang, D.; Niu, C.; Zhu, L.; Wang, H. Proteomic Analysis of Shigella Virulence Effectors Secreted under Different Conditions. J. Microbiol. Biotechnol. 2017, 27, 171–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teulet, A.; Camuel, A.; Perret, X.; Giraud, E. The Versatile Roles of Type III Secretion Systems in Rhizobia-Legume Symbioses. Annu. Rev. Microbiol. 2022, 76, 45–65. [Google Scholar] [CrossRef]
- Kobayashi, H.; Naciri-Graven, Y.; Broughton, W.J.; Perret, X. Flavonoids induce temporal shifts in gene-expression of nod-box controlled loci in Rhizobium sp. NGR234. Mol. Microbiol. 2004, 51, 335–347. [Google Scholar] [CrossRef]
- Krishnan, H.B.; Kuo, C.I.; Pueppke, S.G. Elaboration of flavonoid-induced proteins by the nitrogen-fixing soybean symbiont Rhizobium fredii is regulated by both nodD1 and nodD2, and is dependent on the cultivar-specificity locus, nolXWBTUV. Microbiology 1995, 141, 2245–2251. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.F.; Long, S.R. Interactions of NodD at the nod Box: NodD binds to two distinct sites on the same face of the helix and induces a bend in the DNA. J. Mol. Biol. 1993, 233, 336–348. [Google Scholar] [CrossRef]
- López-Baena, F.J.; Vinardell, J.M.; Pérez-Montaño, F.; Crespo-Rivas, J.C.; Bellogín, R.A.; Espuny, M.d.R.; Ollero, F.J. Regulation and symbiotic significance of nodulation outer proteins secretion in Sinorhizobium fredii HH103. Microbiology 2008, 154, 1825–1836. [Google Scholar] [CrossRef] [Green Version]
- Marie, C.; Deakin, W.J.; Ojanen-Reuhs, T.; Diallo, E.; Reuhs, B.; Broughton, W.J.; Perret, X. TtsI, a key regulator of Rhizobium species NGR234 is required for type III-dependent protein secretion and synthesis of rhamnose-rich polysaccharides. Mol. Plant Microbe Interact. 2004, 17, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Krause, A.; Doerfel, A.; Göttfert, M. Mutational and transcriptional analysis of the type III secretion system of Bradyrhizobium japonicum. Mol. Plant Microbe Interact. 2002, 15, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Wassem, R.; Kobayashi, H.; Kambara, K.; Le Quéré, A.; Walker, G.C.; Broughton, W.J.; Deakin, W.J. TtsI regulates symbiotic genes in Rhizobium species NGR234 by binding to tts boxes. Mol. Microbiol. 2008, 68, 736–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Xiang, Q.W.; Ge, Y.Y.; Huang, Q.Y.; Liang, Y.; Xu, C.C.; Zhang, D.; Zhang, M.X.; Zhu, P.F.; Xie, Z.P.; et al. Use of a Xanthomonas/pepper translocation system for characterization of rhizobial type 3 effectors. In Recent Trends in PGPR Research for Sustainable Crop Productivity; Sayyed, R.Z., Reddy, M.S., Al-Turki, A.I., Eds.; Scientific Publishers: Jodhpurt, India, 2016; pp. 174–178. ISBN 9788172339906. [Google Scholar]
- Van Gijsegem, F.; Vasse, J.; De Rycke, R.; Castello, P.; Boucher, C. Genetic dissection of Ralstonia solanacearum hrp gene cluster reveals that the HrpV and HrpX proteins are required for Hrp pilus assembly. Mol. Microbiol. 2002, 44, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Cunnac, S.; Boucher, C.; Genin, S. Characterization of the cis-acting regulatory element controlling HrpB-mediated activation of the type III secretion system and effector genes in Ralstonia solanacearum. J. Bacteriol. 2004, 186, 2309–2318. [Google Scholar] [CrossRef] [Green Version]
- Yoshimochi, T.; Zhang, Y.; Kiba, A.; Hikichi, Y.; Ohnishi, K. Expression of hrpG and activation of response regulator HrpG are controlled by distinct signal cascades in Ralstonia solanacearum. J. Gen. Plant Pathol. 2009, 75, 196–204. [Google Scholar] [CrossRef]
- Aldon, D.; Brito, B.; Boucher, C.; Genin, S. A bacterial sensor of plant cell contact controls the transcriptional induction of Ralstonia solanacearum pathogenicity genes. EMBO J. 2000, 19, 2304–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marenda, M.; Brito, B.; Callard, D.; Genin, S.; Barberis, P.; Boucher, C.; Arlat, M. PrhA controls a novel regulatory pathway required for the specific induction of Ralstonia solanacearum hrp genes in the presence of plant cells. Mol. Microbiol. 1998, 27, 437–453. [Google Scholar] [CrossRef] [Green Version]
- Brito, B.; Aldon, D.; Barberis, P.; Boucher, C.; Genin, S. A signal transfer system through three compartments transduces the plant cell contact-dependent signal controlling Ralstonia solanacearum hrp genes. Mol. Plant Microbe Interact. 2002, 15, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Zuluaga, A.P.; Puigvert, M.; Valls, M. Novel plant inputs influencing Ralstonia solanacearum during infection. Front. Microbiol. 2013, 4, 349. [Google Scholar] [CrossRef] [Green Version]
- Plener, L.; Manfredi, P.; Valls, M.; Genin, S. PrhG, a transcriptional regulator responding to growth conditions, is involved in the control of the type III secretion system regulon in Ralstonia solanacearum. J. Bacteriol. 2010, 192, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, L.; Yoshimochi, T.; Kiba, A.; Hikichi, Y.; Ohnishi, K. Functional analysis of Ralstonia solanacearum PrhG regulating the hrp regulon in host plants. Microbiology 2013, 159, 1695–1704. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Zhang, W.; Han, L.; Ru, X.; Cao, Y.; Hikichi, Y.; Ohnishi, K.; Pan, G.; Zhang, Y. A CysB regulator positively regulates cysteine synthesis, expression of type III secretion system genes, and pathogenicity in Ralstonia solanacearum. Mol. Plant Pathol. 2022, 23, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, F.; Wu, D.; Hikichi, Y.; Kiba, A.; Igarashi, Y.; Ding, W.; Ohnishi, K. PrhN, a putative marR family transcriptional regulator, is involved in positive regulation of type III secretion system and full virulence of Ralstonia solanacearum. Front. Microbiol. 2015, 6, 357. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Zhang, W.; Shi, H.; Luo, F.; Hikichi, Y.; Shi, X.; Ohnishi, K. A putative LysR-type transcriptional regulator PrhO positively regulates the type III secretion system and contributes to the virulence of Ralstonia solanacearum. Mol. Plant Pathol. 2018, 19, 1808–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, W.; Han, L.; Li, J.; Shi, X.; Hikichi, Y.; Ohnishi, K. Involvement of a PadR regulator PrhP on virulence of Ralstonia solanacearum by controlling detoxification of phenolic acids and type III secretion system. Mol. Plant Pathol. 2019, 20, 1477–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonjon, F.; Lohou, D.; Cazalé, A.-C.; Büttner, D.; Ribeiro, B.G.; Péanne, C.; Genin, S.; Vailleau, F. HpaB-Dependent Secretion of Type III Effectors in the Plant Pathogens Ralstonia solanacearum and Xanthomonas campestris pv. vesicatoria. Sci. Rep. 2017, 7, 4879. [Google Scholar] [CrossRef]
- Norkowski, S.; Körner, B.; Greune, L.; Stolle, A.-S.; Lubos, M.-L.; Hardwidge, P.R.; Schmidt, M.A.; Rüter, C. Bacterial LPX motif-harboring virulence factors constitute a species-spanning family of cell-penetrating effectors. Cell. Mol. Life Sci. 2017, 75, 2273–2289. [Google Scholar] [CrossRef]
- Takagi, K.; Kim, M.; Sasakawa, C.; Mizushima, T. Crystal structure of the substrate-recognition domain of the Shigella E3 ligase IpaH9.8. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Zouhir, S.; Bernal-bayard, J.; Cordero-alba, M.; Cardenal-muñoz, E.; Guimaraes, B.; Lazar, N.; Ramos-morales, F.; Nessler, S. The Structure of the SlrP-hTrx1 Complex Sheds Light on the Autoinhibition Mechanism of the Type III Secretion System Effectors of the NEL Family. Biochem. J. 2014, 144, 2014. [Google Scholar]
- Haraga, A.; Miller, S.I.; Science, B.; Article, O.; Haraga, A.; Miller, S.I. A Salmonella type III secretion effector interacts with the mammalian serine/threonine protein kinase PKN1. Cell. Microbiol. 2006, 8, 837–846. [Google Scholar] [CrossRef]
- Scheffner, M.; Staub, O. HECT E3s and human disease. BMC Biochem. 2007, 8, S6. [Google Scholar] [CrossRef] [Green Version]
- Budhidarmo, R.; Nakatani, Y.; Day, C.L. RINGs hold the key to ubiquitin transfer. Trends Biochem. Sci. 2012, 37, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Kinnucan, E.; Wang, G.; Beaudenon, S.; Howley, P.M.; Huibregtse, J.M.; Pavletich, N.P. Structure of an E6AP-UbcH7 complex: Insights into ubiquitination by the E2-E3 enzyme cascade. Science 1999, 286, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.-C.; Keszei, A.F.A.; Rohde, J.R.; Tyers, M.; Sicheri, F. Conserved structural mechanisms for autoinhibition in IpaH ubiquitin ligases. J. Biol. Chem. 2012, 287, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Xiong, Y.; Huang, H. Substrate-binding destabilizes the hydrophobic cluster to relieve the autoinhibition of bacterial ubiquitin ligase IpaH9.8. Commun. Biol. 2020, 3, 752. [Google Scholar] [CrossRef]
- Keszei, A.F.A.; Tang, X.; McCormick, C.; Zeqiraj, E.; Rohde, J.R.; Tyers, M.; Sicheri, F. Structure of an SspH1-PKN1 complex reveals the basis for host substrate recognition and mechanism of activation for a bacterial E3 ubiquitin ligase. Mol. Cell. Biol. 2014, 34, 362–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, Y.; Wang, D.; Wang, Y.; Xu, X.; Zhang, Y.; Li, Y.; Zhang, M.; Gong, X.; Tang, Y.; et al. Mechanistic insights into the subversion of the linear ubiquitin chain assembly complex by the E3 ligase IpaH1.4 of Shigella flexneri. Proc. Natl. Acad. Sci. USA 2022, 119, e2116776119. [Google Scholar] [CrossRef]
- Ji, C.; Du, S.; Li, P.; Zhu, Q.; Yang, X.; Long, C.; Yu, J.; Shao, F.; Xiao, J. Structural mechanism for guanylate-binding proteins (GBPs) targeting by the Shigella E3 ligase IpaH9.8. PLoS Pathog. 2019, 15, e1007876. [Google Scholar] [CrossRef]
- Edwards, D.J.; Streich, F.C.; Ronchi, V.P.; Todaro, D.R.; Haas, A.L. Convergent evolution in the assembly of polyubiquitin degradation signals by the Shigella flexneri IpaH9.8 ligase. J. Biol. Chem. 2014, 289, 34114–34128. [Google Scholar] [CrossRef] [Green Version]
- Cook, M.; Delbecq, S.P.; Schweppe, T.P.; Guttman, M.; Klevit, R.E.; Brzovic, P.S. The ubiquitin ligase SspH1 from Salmonella uses a modular and dynamic E3 domain to catalyze substrate ubiquitylation. J. Biol. Chem. 2019, 294, 783–793. [Google Scholar] [CrossRef] [Green Version]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlic, A.; Rose, P.W. NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Santos, R.L.; Tsolis, R.M.; Stender, S.; Hardt, W.-D.; Bäumler, A.J.; Adams, L.G. The Salmonella enterica serotype Typhimurium effector proteins SipA, SopA, SopB, SopD, and SopE2 act in concert to induce diarrhea in calves. Infect. Immun. 2002, 70, 3843–3855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shappo, M.O.E.; Li, Q.; Lin, Z.; Hu, M.; Ren, J.; Xu, Z.; Pan, Z.; Jiao, X. SspH2 as anti-inflammatory candidate effector and its contribution in Salmonella Enteritidis virulence. Microb. Pathog. 2020, 142, 104041. [Google Scholar] [CrossRef] [PubMed]
- Kidwai, A.S.; Mushamiri, I.; Niemann, G.S.; Brown, R.N.; Adkins, J.N.; Heffron, F. Diverse secreted effectors are required for Salmonella persistence in a mouse infection model. PLoS ONE 2013, 8, e70753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordero-Alba, M.; García-Gómez, J.J.; Aguilera-Herce, J.; Ramos-Morales, F. Proteomic insight into the effects of the Salmonella ubiquitin ligase SlrP on host cells. Biochem. Biophys. Res. Commun. 2016, 472, 539–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhavsar, A.P.; Brown, N.F.; Stoepel, J.; Wiermer, M.; Martin, D.D.O.; Hsu, K.J.; Imami, K.; Ross, C.J.; Hayden, M.R.; Foster, L.J.; et al. The Salmonella type III effector SspH2 specifically exploits the NLR co-chaperone activity of SGT1 to subvert immunity. PLoS Pathog. 2013, 9, e1003518. [Google Scholar] [CrossRef] [Green Version]
- Halici, S.; Zenk, S.F.; Jantsch, J.; Hensel, M. Functional analysis of the Salmonella pathogenicity island 2-mediated inhibition of antigen presentation in dendritic cells. Infect. Immun. 2008, 76, 4924–4933. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, L.M.; Xu, H.; Carden, S.E.; Fisher, S.; Reyes, M.; Heilshorn, S.C.; Monack, D.M. A microfluidic-based genetic screen to identify microbial virulence factors that inhibit dendritic cell migration. Integr. Biol. 2014, 6, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Sereny, B. Experimental Shigella keratoconjunctivitis; a preliminary report. Acta Microbiol. Acad. Sci. Hung. 1955, 2, 293–296. [Google Scholar]
- Voino-Yasenetsky, M.V.; Voino-Yasenetskaya, M.K. Experimental pneumonia caused by bacteria of the Shigella group. Acta Morphol. Acad. Sci. Hung. 1962, 11, 439–454. [Google Scholar]
- Fernandez-Prada, C.M.; Hoover, D.L.; Tall, B.D.; Hartman, A.B.; Kopelowitz, J.; Venkatesan, M.M. Shigella flexneri IpaH(7.8) facilitates escape of virulent bacteria from the endocytic vacuoles of mouse and human macrophages. Infect. Immun. 2000, 68, 3608–3619. [Google Scholar] [CrossRef] [Green Version]
- Paetzold, S.; Lourido, S.; Raupach, B.; Zychlinsky, A. Shigella flexneri phagosomal escape is independent of invasion. Infect. Immun. 2007, 75, 4826–4830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Sun, J.; Wan, L.; Yang, X.; Lin, H.; Zhang, Y.; He, X.; Zhong, H.; Guan, K.; Min, M.; et al. The Shigella Type III Secretion Effector IpaH4.5 Targets NLRP3 to Activate Inflammasome Signaling. Front. Cell. Infect. Microbiol. 2020, 10, 511798. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, G.; Roncaioli, J.L.; Chavez, R.A.; Schubert, A.F.; Kofoed, E.M.; Reja, R.; Cheung, T.K.; Liang, Y.; Webster, J.D.; Lehoux, I.; et al. Shigella ubiquitin ligase IpaH7.8 targets gasdermin D for degradation to prevent pyroptosis and enable infection. Cell Host Microbe 2021, 29, 1521–1530.e10. [Google Scholar] [CrossRef] [PubMed]
- Mou, X.; Souter, S.; Du, J.; Reeves, A.Z.; Lesser, C.F. Synthetic bottom-up approach reveals the complex interplay of Shigella effectors in regulation of epithelial cell death. Proc. Natl. Acad. Sci. USA 2018, 115, 6452–6457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, J.; Toyotome, T.; Kataoka, N.; Ohno, M.; Abe, H.; Shimura, Y.; Seyedarabi, A.; Pickersgill, R.; Sasakawa, C. Shigella effector IpaH9.8 binds to a splicing factor U2AF(35) to modulate host immune responses. Biochem. Biophys. Res. Commun. 2005, 333, 531–539. [Google Scholar] [CrossRef]
- Wang, F.; Jiang, Z.; Li, Y.; He, X.; Zhao, J.; Yang, X.; Zhu, L.; Yin, Z.; Li, X.; Wang, X.; et al. Shigella flexneri T3SS effector IpaH4.5 modulates the host inflammatory response via interaction with NF-κB p65 protein. Cell. Microbiol. 2013, 15, 474–485. [Google Scholar] [CrossRef]
- Otsubo, R.; Mimuro, H.; Ashida, H.; Hamazaki, J.; Murata, S.; Sasakawa, C. Shigella effector IpaH4.5 targets 19S regulatory particle subunit RPN13 in the 26S proteasome to dampen cytotoxic T lymphocyte activation. Cell. Microbiol. 2019, 21, e12974. [Google Scholar] [CrossRef]
- Niu, Y.; Fu, S.; Chen, G.; Wang, H.; Wang, Y.; Hu, J.; Jin, X.; Zhang, M.; Lu, M.; He, Y.; et al. Different epitopes of Ralstonia solanacearum effector RipAW are recognized by two Nicotiana species and trigger immune responses. Mol. Plant Pathol. 2022, 23, 188–203. [Google Scholar] [CrossRef]
- Pensec, F.; Lebeau, A.; Daunay, M.C.; Chiroleu, F.; Guidot, A.; Wicker, E. Towards the Identification of Type III Effectors Associated with Ralstonia solanacearum Virulence on Tomato and Eggplant. Phytopathology 2015, 105, 1529–1544. [Google Scholar] [CrossRef] [Green Version]
- Xin, D.W.; Liao, S.; Xie, Z.P.; Hann, D.R.; Steinle, L.; Boller, T.; Staehelin, C. Functional analysis of NopM, a novel E3 ubiquitin ligase (NEL) domain effector of Rhizobium sp. strain NGR234. PLoS Pathog. 2012, 8, e1002707. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.S.; Sadowsky, M.J. Secretion systems and signal exchange between nitrogen-fixing rhizobia and legumes. Front. Plant Sci. 2015, 6, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walch, P.; Selkrig, J.; Knodler, L.A.; Rettel, M.; Stein, F.; Fernandez, K.; Viéitez, C.; Potel, C.M.; Scholzen, K.; Geyer, M.; et al. Global mapping of Salmonella enterica-host protein-protein interactions during infection. Cell Host Microbe 2021, 29, 1316–1332.e12. [Google Scholar] [CrossRef]
- Auweter, S.D.; Bhavsar, A.P.; De Hoog, C.L.; Li, Y.; Chan, Y.A.; van der Heijden, J.; Lowden, M.J.; Coombes, B.K.; Rogers, L.D.; Stoynov, N.; et al. Quantitative mass spectrometry catalogues Salmonella pathogenicity island-2 effectors and identifies their cognate host binding partners. J. Biol. Chem. 2011, 286, 24023–24035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meyer, M.; Fijalkowski, I.; Jonckheere, V.; De Sutter, D.; Eyckerman, S.; Van Damme, P. Capturing Salmonella SspH2 Host Targets in Virus-Like Particles. Front. Med. 2021, 8, 725072. [Google Scholar] [CrossRef] [PubMed]
- Wandel, M.P.; Pathe, C.; Werner, E.I.; Ellison, C.J.; Boyle, K.B.; von der Malsburg, A.; Rohde, J.; Randow, F. GBPs Inhibit Motility of Shigella flexneri but Are Targeted for Degradation by the Bacterial Ubiquitin Ligase IpaH9.8. Cell Host Microbe 2017, 22, 507–518.e5. [Google Scholar] [CrossRef] [Green Version]
- Seyedarabi, A.; Sullivan, J.A.; Sasakawa, C.; Pickersgill, R.W. A disulfide driven domain swap switches off the activity of Shigella IpaH9.8 E3 ligase. FEBS Lett. 2010, 584, 4163–4168. [Google Scholar] [CrossRef] [Green Version]
- Ashida, H.; Kim, M.; Schmidt-Supprian, M.; Ma, A.; Ogawa, M.; Sasakawa, C. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat. Cell Biol. 2010, 12, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Jiang, W.; Yu, Q.; Liu, W.; Zhou, P.; Li, J.; Xu, J.; Xu, B.; Wang, F.; Shao, F. Ubiquitination and degradation of GBPs by a Shigella effector to suppress host defence. Nature 2017, 551, 378–383. [Google Scholar] [CrossRef]
- Piro, A.S.; Hernandez, D.; Luoma, S.; Feeley, E.M.; Finethy, R.; Yirga, A.; Frickel, E.M.; Lesser, C.F.; Coers, J. Detection of Cytosolic Shigella flexneri via a C-Terminal Triple-Arginine Motif of GBP1 Inhibits Actin-Based Motility. MBio 2017, 8, e01979-17. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Wei, C.; Guan, K.; Yuan, Y.; Zhang, Y.; Ma, S.; Cao, Y.; Wang, F.; Zhong, H.; He, X. Bacterial E3 Ubiquitin Ligase IpaH4.5 of Shigella flexneri Targets TBK1 To Dampen the Host Antibacterial Response. J. Immunol. 2016, 196, 1199–1208. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Wang, X.; Lin, H.; Wan, L.; Chen, J.; Yang, X.; Li, D.; Zhang, Y.; He, X.; Wang, B.; et al. Shigella escapes lysosomal degradation through inactivation of Rab31 by IpaH4.5. J. Med. Microbiol. 2021, 70, 001382. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.F.; Liu, Z.; Chen, D.; Alto, N.M. Shigella flexneri suppresses NF-κB activation by inhibiting linear ubiquitin chain ligation. Nat. Microbiol. 2016, 1, 16084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak, H.; Iwai, K.; Dikic, I. Generation and physiological roles of linear ubiquitin chains. BMC Biol. 2012, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, F.; Deribe, Y.L.; Skånland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; van Wijk, S.J.L.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.L.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Noad, J.; von der Malsburg, A.; Pathe, C.; Michel, M.A.; Komander, D.; Randow, F. LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat. Microbiol. 2017, 2, 17063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Suzuki, T.; Mimuro, H.; Mizushima, T.; Sasakawa, C. Shigella hijacks the glomulin-cIAPs-inflammasome axis to promote inflammation. EMBO Rep. 2018, 19, 89–101. [Google Scholar] [CrossRef]
- Hansen, J.M.; de Jong, M.F.; Wu, Q.; Zhang, L.-S.; Heisler, D.B.; Alto, L.T.; Alto, N.M. Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions. Cell 2021, 184, 3178–3191.e18. [Google Scholar] [CrossRef]
- O’Connor, H.F.; Lyon, N.; Leung, J.W.; Agarwal, P.; Swaim, C.D.; Miller, K.M.; Huibregtse, J.M. Ubiquitin-Activated Interaction Traps (UBAITs) identify E3 ligase binding partners. EMBO Rep. 2015, 16, 1699–1712. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.C.; Na, C.H.; Peng, J. Quantitative proteomics to decipher ubiquitin signaling. Amino Acids 2012, 43, 1049–1060. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.K.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Walton, A.; Stes, E.; Cybulski, N.; Bel, M.; Iñigo, S.; Durand, A.N.; Timmerman, E.; Heyman, J.; Pauwels, L.; De Veylder, L.; et al. It’s Time for Some “Site”-Seeing: Novel Tools to Monitor the Ubiquitin Landscape in Arabidopsis thaliana. Plant Cell 2016, 28, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Stes, E.; Laga, M.; Walton, A.; Samyn, N.; Timmerman, E.; De Smet, I.; Goormachtig, S.; Gevaert, K. A COFRADIC protocol to study protein ubiquitination. J. Proteome Res. 2014, 13, 3107–3113. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; González-Prieto, R.; Xiao, Z.; Verlaan-De Vries, M.; Vertegaal, A.C.O. The STUbL RNF4 regulates protein group SUMOylation by targeting the SUMO conjugation machinery. Nat. Commun. 2017, 8, 1809. [Google Scholar] [CrossRef] [Green Version]
- Salas-Lloret, D.; Agabitini, G.; González-Prieto, R. TULIP2: An Improved Method for the Identification of Ubiquitin E3-Specific Targets. Front. Chem. 2019, 7, 802. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]



| Organism | Effector Name | Main Characterized Binding Partners | |
|---|---|---|---|
| No Ubiquitination Shown | Ubiquitination Shown | ||
| S. enterica | SlrP | ERdj3 | Thioredoxin |
| SspH1 | PKN1 | ||
| SspH2 | Profilins, filamin, SGT1 | Nod1 | |
| S. flexneri | IpaH1.4 | HOIL-1L | HOIP |
| IpaH2.5 | HOIL-1L | HOIP | |
| IpaH4.5 | Rab31 | p65, TBK1, RPN13, NLRP3 | |
| IpaH7.8 | Glomulin, GSDMB, GSDMD | ||
| IpaH9.8 | Ste7, U2AF35, NEMO, GBP1, GBP2, GBP4 | ||
| IpaHa | TRAF2 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bullones-Bolaños, A.; Bernal-Bayard, J.; Ramos-Morales, F. The NEL Family of Bacterial E3 Ubiquitin Ligases. Int. J. Mol. Sci. 2022, 23, 7725. https://doi.org/10.3390/ijms23147725
Bullones-Bolaños A, Bernal-Bayard J, Ramos-Morales F. The NEL Family of Bacterial E3 Ubiquitin Ligases. International Journal of Molecular Sciences. 2022; 23(14):7725. https://doi.org/10.3390/ijms23147725
Chicago/Turabian StyleBullones-Bolaños, Andrea, Joaquín Bernal-Bayard, and Francisco Ramos-Morales. 2022. "The NEL Family of Bacterial E3 Ubiquitin Ligases" International Journal of Molecular Sciences 23, no. 14: 7725. https://doi.org/10.3390/ijms23147725