
A Novel Nanobody-Horseradish Peroxidase Fusion Based-Competitive ELISA to Rapidly Detect Avian Corona-Virus-Infectious Bronchitis Virus Antibody in Chicken Serum

 , and
, and
Abstract
:1. Introduction
2. Results
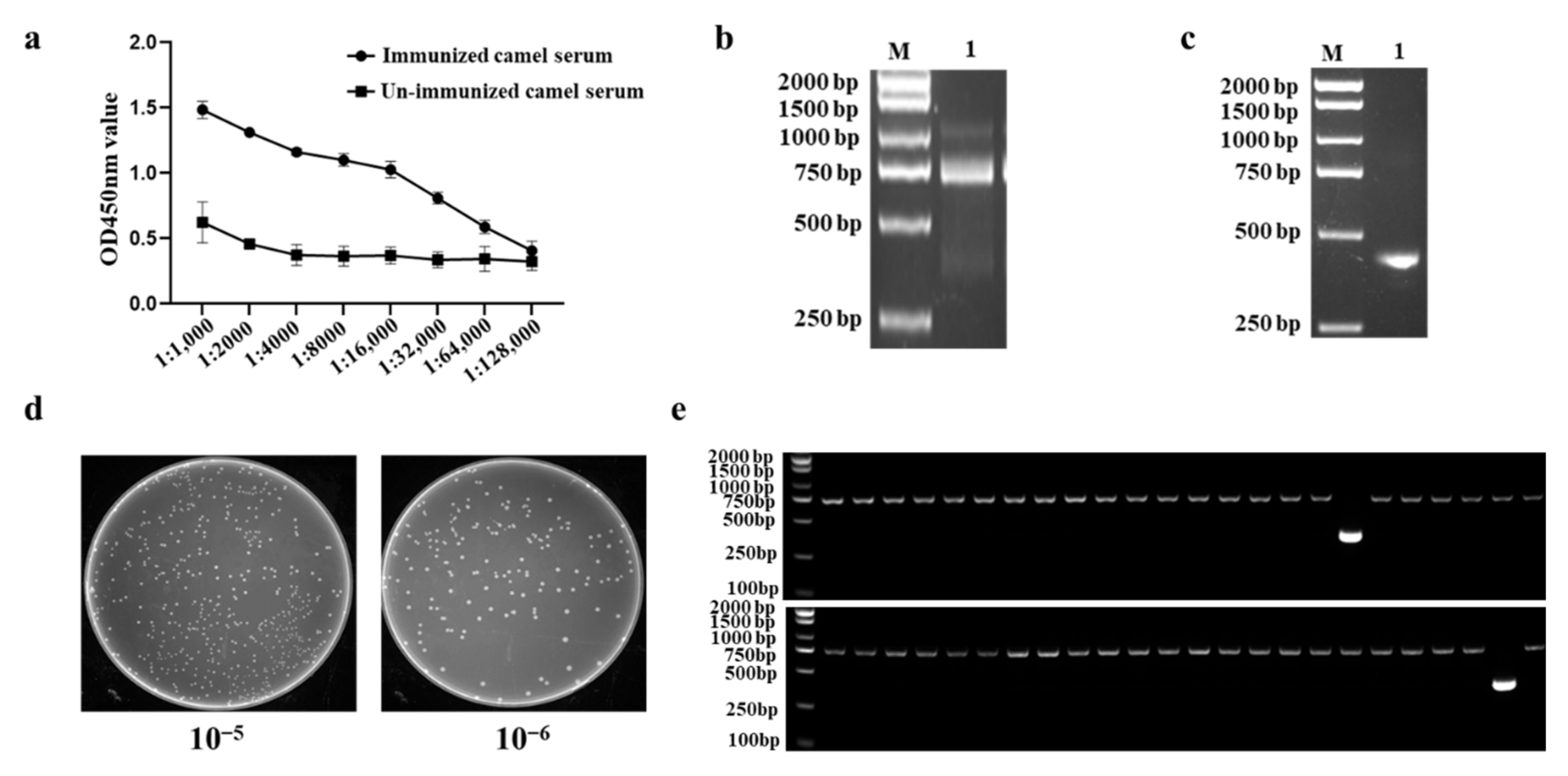
2.1. Expression, Purification and Identification of IBV-N Protein
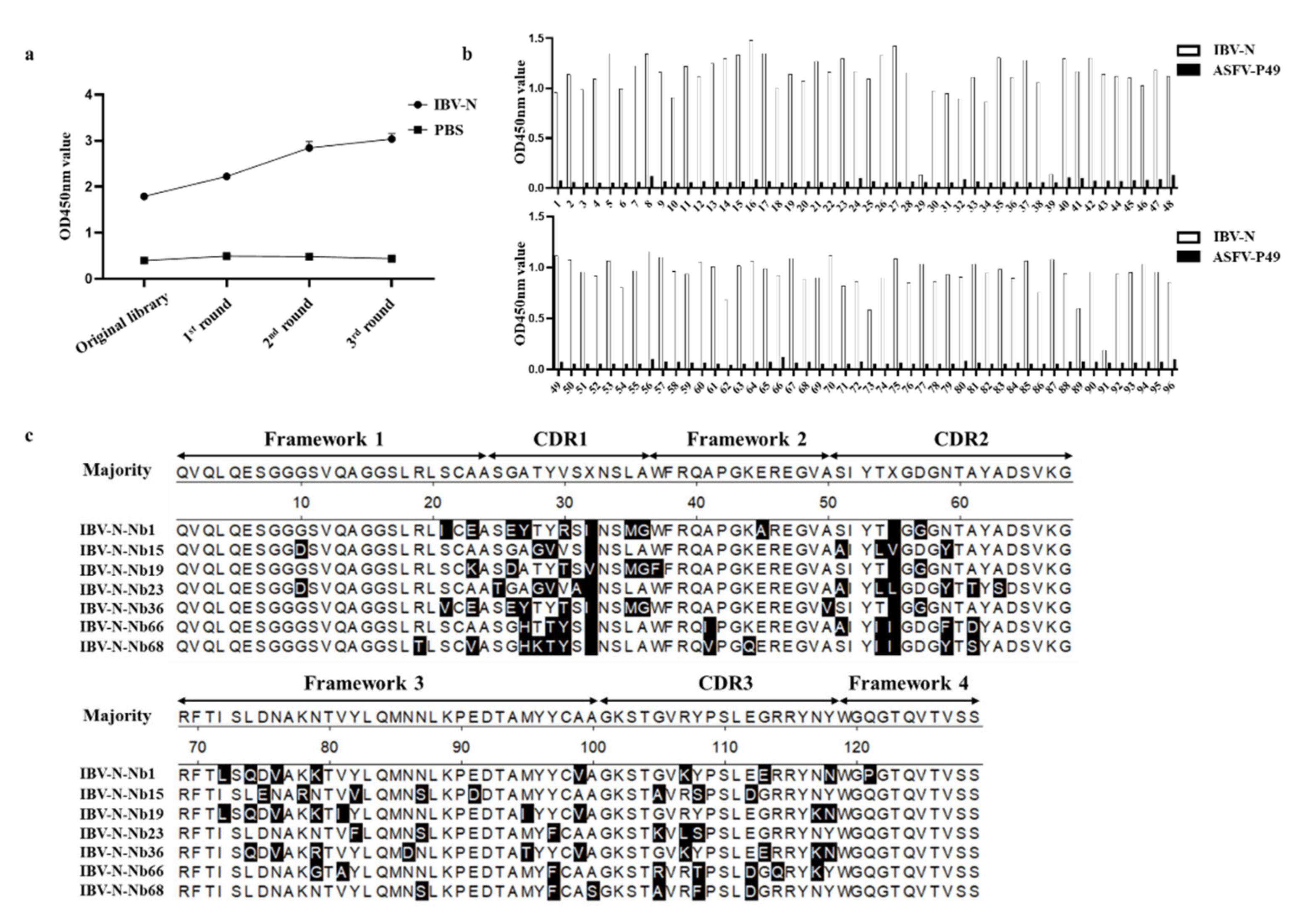
2.2. Construction of a Phage Display VHH Library
2.3. Screening and Sequencing of Nanobodies against IBV-N Protein
2.4. Expression of RANbodies against IBV-N Protein
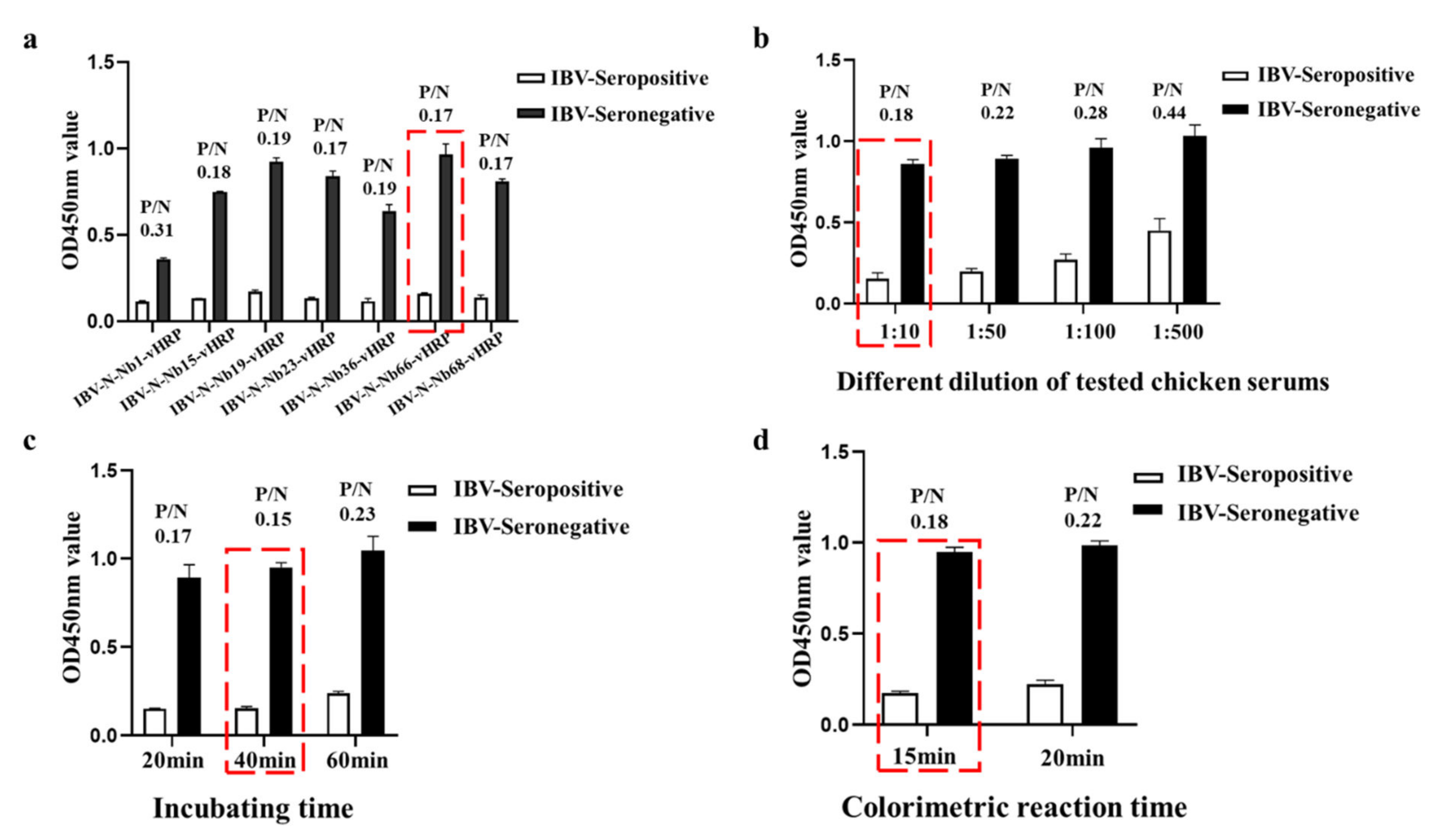
2.5. Development the cELISA for Detecting IBV Antibody
2.6. Cut-Off Values for the cELISA
2.7. Specificity, Sensitivity and Reproducibility of the cELISA
2.8. Agreements of the cELISA with a Commercial ELISA Kit
3. Discussion
4. Materials and Methods
4.1. Serum Samples, Cells, and Vectors
4.2. Experiment Animals
4.3. Expression, Purification and Identification of IBV-N Protein
4.4. Immunization of Camel
4.5. Generation of VHH Library
4.6. Screening of Specific VHH against IBV-N Protein
4.7. Production of Nanobody-Horseradish Peroxidase Fusion against IBV-N Protein
4.8. Development of the Competitive ELISA using RANbodies
4.9. Cut-off Value
4.10. Sensitivity, Specificity and Repeatability of cELISA
4.11. Comparisons of the cELISA with the Commercial ELISA Kit
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, P.; Gu, K.; Li, H.; Zhao, Y.; Li, C.; Wen, R.; Zhou, C.; Lei, C.; Yang, X.; Wang, H. Infectious Bronchitis Virus Nsp14 Degrades JAK1 to Inhibit the JAK-STAT Signaling Pathway in HD11 Cells. Viruses 2022, 14, 1045. [Google Scholar] [CrossRef] [PubMed]
- Lugovskaya, N.N.; Scherbakov, A.V.; Yakovleva, A.S.; Tsyvanyuk, M.A.; Mudrak, N.S.; Drygin, V.V.; Borisov, A.V. Detection of antibodies to avian infectious bronchitis virus by a recombinant nucleocapsid protein-based enzyme-linked immunosorbent assay. J. Virol. Methods 2006, 135, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.; Saenz, L.; Hidalgo, H. Molecular and Antigenic Characterization of GI-13 and GI-16 Avian Infectious Bronchitis Virus Isolated in Chile from 2009 to 2017 Regarding 4/91 Vaccine Introduction. Animals 2019, 9, 656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, H.; Faburay, B.; Turan, N.; Cotton-Caballero, M.; Cetinkaya, B.; Gurel, A.; Yilmaz, A.; Cizmecigil, U.Y.; Aydin, O.; Tarakci, E.A.; et al. Production of Recombinant N Protein of Infectious Bronchitis Virus Using the Baculovirus Expression System and Its Assessment as a Diagnostic Antigen. Appl. Biochem. Biotechnol. 2019, 187, 506–517. [Google Scholar] [CrossRef]
- Thor, S.W.; Hilt, D.A.; Kissinger, J.C.; Paterson, A.H.; Jackwood, M.W. Recombination in avian gamma-coronavirus infectious bronchitis virus. Viruses 2011, 3, 1777–1799. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Weng, X.; Neethirajan, S. Immunosensor Based on Antibody-Functionalized MoS2 for Rapid Detection of Avian Coronavirus on Cotton Thread. IEEE Sens. J. 2018, 18, 4358–4363. [Google Scholar] [CrossRef]
- Chen, H.; Coote, B.; Attree, S.; Hiscox, J.A. Evaluation of a nucleoprotein-based enzyme-linked immunosorbent assay for the detection of antibodies against infectious bronchitis virus. Avian. Pathol. 2003, 32, 519–526. [Google Scholar] [CrossRef]
- Ding, M.D.; Yang, X.; Wang, H.N.; Zhang, A.Y.; Zhang, Z.K.; Fan, W.Q.; Cao, H.P. Development of an ELISA based on a multi-fragment antigen of infectious bronchitis virus for antibodies detection. Biotechnol. Lett. 2015, 37, 2453–2459. [Google Scholar] [CrossRef]
- Boots, A.M.; Van Lierop, M.J.; Kusters, J.G.; Van Kooten, P.J.; Van der Zeijst, B.A.; Hensen, E.J. MHC class II-restricted T-cell hybridomas recognizing the nucleocapsid protein of avian coronavirus IBV. Immunology 1991, 72, 10–14. [Google Scholar]
- Gu, K.; Song, Z.; Zhou, C.; Ma, P.; Li, C.; Lu, Q.; Liao, Z.; Huang, Z.; Tang, Y.; Li, H.; et al. Development of nanobody-horseradish peroxidase-based sandwich ELISA to detect Salmonella Enteritidis in milk and in vivo colonization in chicken. J. Nanobiotechnology 2022, 20, 167. [Google Scholar] [CrossRef]
- Salvador, J.P.; Vilaplana, L.; Marco, M.P. Nanobody: Outstanding features for diagnostic and therapeutic applications. Anal. Bioanal. Chem. 2019, 411, 1703–1713. [Google Scholar] [CrossRef]
- Yamagata, M.; Sanes, J.R. Reporter-nanobody fusions (RANbodies) as versatile, small, sensitive immunohistochemical reagents. Proc. Natl. Acad. Sci. USA 2018, 115, 2126–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.; Jiang, B.; Guan, Z.; He, J.; Yang, D.; Xie, N.; Nie, G.; Xie, C.; Yan, X. Fenobody: A Ferritin-Displayed Nanobody with High Apparent Affinity and Half-Life Extension. Anal. Chem. 2018, 90, 5671–5677. [Google Scholar] [CrossRef]
- Jordan, B. Vaccination against infectious bronchitis virus: A continuous challenge. Vet. Microbiol. 2017, 206, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Legnardi, M.; Tucciarone, C.M.; Franzo, G.; Cecchinato, M. Infectious Bronchitis Virus Evolution, Diagnosis and Control. Vet. Sci. 2020, 7, 79. [Google Scholar] [CrossRef]
- Laconi, A.; Berends, A.J.; de Laat, E.C.H.; Urselmann, T.; Verheije, H.M. Infectious bronchitis virus Mass-type (GI-1) and QX-like (GI-19) genotyping and vaccine differentiation using SYBR green RT-qPCR paired with melting curve analysis. J. Virol. Methods 2020, 275, 113771. [Google Scholar] [CrossRef] [PubMed]
- Gibertoni, A.M.; Montassier Mde, F.; Sena, J.A.; Givisiez, P.E.; Furuyama, C.R.; Montassier, H.J. Development and application of a Saccharomyces cerevisiae-expressed nucleocapsid protein-based enzyme-linked immunosorbent assay for detection of antibodies against infectious bronchitis virus. J. Clin. Microbiol. 2005, 43, 1982–1984. [Google Scholar] [CrossRef] [Green Version]
- Bronzoni, R.V.; Fatima, M.; Montassier, S.; Pereira, G.T.; Gama, N.M.; Sakai, V.; Montassier, H.J. Detection of infectious bronchitis virus and specific anti-viral antibodies using a Concanavalin A-Sandwich-ELISA. Viral. Immunol. 2005, 18, 569–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.W.; Wang, C.H.; Cheng, I.C. A type-specific blocking ELISA for the detection of infectious bronchitis virus antibody. J. Virol. Methods 2011, 173, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Li, X.; Zhao, J.; Zhu, J.; Luo, Y.; Duan, H.; Ji, P.; Wang, K.; Liu, B.; Wang, X.; et al. Nanobodyhorseradish peroxidase and -EGFP fusions as reagents to detect porcine parvovirus in the immunoassays. J. Nanobiotechnol. 2020, 18, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojarad, A.E.; Gargaria, S.L.M. Aptamer-nanobody based ELASA for detection of Vibrio cholerae O1. Iran. J. Microbiol. 2020, 12, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Rasoulinejad, S.; Gargari, S.L.M. Aptamer-nanobody based ELASA for specific detection of Acinetobacter baumannii isolates. J. Biotechnol. 2016, 231, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Pagneux, Q.; Roussel, A.; Saada, H.; Cambillau, C.; Amigues, B.; Delauzun, V.; Engelmann, I.; Alidjinou, E.K.; Ogiez, J.; Rolland, A.S.; et al. SARS-CoV-2 detection using a nanobody-functionalized voltammetric device. Commun Med. 2022, 2, 56. [Google Scholar] [CrossRef]
- Delfin-Riela, T.; Rossotti, M.; Alvez-Rosado, R.; Leizagoyen, C.; Gonzalez-Sapienza, G. Highly Sensitive Detection of Zika Virus Nonstructural Protein 1 in Serum Samples by a Two-Site Nanobody ELISA. Biomolecules 2020, 10, 1652. [Google Scholar] [CrossRef]
- Gelkop, S.; Sobarzo, A.; Brangel, P.; Vincke, C.; Romao, E.; Fedida-Metula, S.; Strom, N.; Ataliba, I.; Mwiine, F.N.; Ochwo, S.; et al. The Development and Validation of a Novel Nanobody-Based Competitive ELISA for the Detection of Foot and Mouth Disease 3ABC Antibodies in Cattle. Front. Vet. Sci. 2018, 5, 250. [Google Scholar] [CrossRef]
- Morales-Yanez, F.; Trashin, S.; Hermy, M.; Sariego, I.; Polman, K.; Muyldermans, S.; De Wael, K. Fast One-Step Ultrasensitive Detection of Toxocara canis Antigens by a Nanobody-Based Electrochemical Magnetosensor. Anal. Chem. 2019, 91, 11582–11588. [Google Scholar] [CrossRef] [Green Version]
- Zanganeh, S.; Rouhani Nejad, H.; Mehrabadi, J.F.; Hosseini, R.; Shahi, B.; Tavassoli, Z.; Aramvash, A. Rapid and Sensitive Detection of Staphylococcal Enterotoxin B by Recombinant Nanobody Using Phage Display Technology. Appl. Biochem. Biotechnol. 2019, 187, 493–505. [Google Scholar] [CrossRef]
- Xu, B.; Wang, K.; Vasylieva, N.; Zhou, H.; Xue, X.; Wang, B.; Li, Q.X.; Hammock, B.D.; Xu, T. Development of a nanobody-based ELISA for the detection of the insecticides cyantraniliprole and chlorantraniliprole in soil and the vegetable bok choy. Anal. Bioanal. Chem. 2021, 413, 2503–2511. [Google Scholar] [CrossRef] [PubMed]
- Trashin, S.; Morales-Yanez, F.; Thiruvottriyur Shanmugam, S.; Paredis, L.; Carrion, E.N.; Sariego, I.; Muyldermans, S.; Polman, K.; Gorun, S.M.; De Wael, K. Nanobody-Based Immunosensor Detection Enhanced by Photocatalytic-Electrochemical Redox Cycling. Anal. Chem. 2021, 93, 13606–13614. [Google Scholar] [CrossRef]
- Folorunsho, O.G.; Oloketuyi, S.F.; Mazzega, E.; Budasheva, H.; Beran, A.; Cabrini, M.; Korte, D.; Franko, M.; de Marco, A. Nanobody-Dependent Detection of Microcystis aeruginosa by ELISA and Thermal Lens Spectrometry. Appl. Biochem. Biotechnol. 2021, 193, 2729–2741. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Ohmuro-Matsuyama, Y.; Kitaguchi, T.; Ueda, H. Creation of a Nanobody-Based Fluorescent Immunosensor Mini Q-body for Rapid Signal-On Detection of Small Hapten Methotrexate. ACS Sens. 2020, 5, 3457–3464. [Google Scholar] [CrossRef] [PubMed]
- Kijanka, M.; van Donselaar, E.G.; Muller, W.H.; Dorresteijn, B.; Popov-Celeketic, D.; El Khattabi, M.; Verrips, C.T.; van Bergen En Henegouwen, P.M.P.; Post, J.A. A novel immuno-gold labeling protocol for nanobody-based detection of HER2 in breast cancer cells using immuno-electron microscopy. J. Struct. Biol. 2017, 199, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Aghamollaei, H.; Ghanei, M.; Rasaee, M.J.; Latifi, A.M.; Bakherad, H.; Fasihi-Ramandi, M.; Taheri, R.A.; Gargari, S.L.M. Isolation and characterization of a novel nanobody for detection of GRP78 expressing cancer cells. Biotechnol. Appl. Biochem. 2021, 68, 239–246. [Google Scholar] [CrossRef]
- Bernardinelli, G.; Oloketuyi, S.; Werner, S.F.; Mazzega, E.; Hogberg, B.; de Marco, A. A compact nanobody-DNAzyme conjugate enables antigen detection and signal amplification. New Biotechnol. 2020, 56, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hulstein, J.J.; de Groot, P.G.; Silence, K.; Veyradier, A.; Fijnheer, R.; Lenting, P.J. A novel nanobody that detects the gain-of-function phenotype of von Willebrand factor in ADAMTS13 deficiency and von Willebrand disease type 2B. Blood 2005, 106, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Vincke, C.; Gutierrez, C.; Wernery, U.; Devoogdt, N.; Hassanzadeh-Ghassabeh, G.; Muyldermans, S. Generation of single domain antibody fragments derived from camelids and generation of manifold constructs. Methods Mol. Biol. 2012, 907, 145–176. [Google Scholar] [CrossRef]
- Pinto Torres, J.E.; Goossens, J.; Ding, J.; Li, Z.; Lu, S.; Vertommen, D.; Naniima, P.; Chen, R.; Muyldermans, S.; Sterckx, Y.G.; et al. Development of a Nanobody-based lateral flow assay to detect active Trypanosoma congolense infections. Sci. Rep. 2018, 8, 9019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Y.; Jia, C.; Zheng, X.; Zhu, H.; Zhang, X.; Xu, H.; Liu, B.; Zhao, Q.; Zhou, E.M. A nanobody-horseradish peroxidase fusion protein-based competitive ELISA for rapid detection of antibodies against porcine circovirus type 2. J. Nanobiotechnol. 2021, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Wang, K.; Lu, Q.; Ji, P.; Liu, B.; Zhu, J.; Liu, Q.; Sun, Y.; Zhang, J.; Zhou, E.M.; et al. Nanobody-horseradish peroxidase fusion protein as an ultrasensitive probe to detect antibodies against Newcastle disease virus in the immunoassay. J. Nanobiotechnol. 2019, 17, 35. [Google Scholar] [CrossRef] [Green Version]
- Peng, P.; Gao, Y.; Zhou, Q.; Jiang, T.; Zheng, S.; Huang, M.; Xue, C.; Cao, Y.; Xu, Z. Development of an indirect ELISA for detecting swine acute diarrhoea syndrome coronavirus IgG antibodies based on a recombinant spike protein. Transbound Emerg. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Round of Screening | Input (Pfu/Well) | P Output (Pfu/Well) | N Output (Pfu/Well) | Recovery (P/Input) | P/N |
|---|---|---|---|---|---|
| 1st Round | 5.0 × 1010 | 3.0 × 104 | 3.06 × 103 | 6.0 × 10−5 | 9.8 |
| 2nd Round | 5.0 × 1010 | 6.0 × 105 | 2.7 × 104 | 1.2 × 10−5 | 22.2 |
| 3rd Round | 5.0 × 1010 | 3.6 × 106 | 1.5 × 105 | 1.8 × 10−4 | 2.4 × 103 |
| Coated IBV-N Protein | Different Dilution of the IBV-N-Nb66-vHRP | |||||||
|---|---|---|---|---|---|---|---|---|
| 1:2 | 1:4 | 1:8 | 1:16 | 1:32 | 1:64 | 1:128 | 1:256 | |
| 1600 ng/well | 1.544 ± 0.020 | 1.330 ± 0.008 | 1.207 ± 0.070 | 1.157 ± 0.106 | 0.833 ± 0.103 | 0.678 ± 0.002 | 0.421 ± 0.020 | 0.287 ± 0.002 |
| 800 ng/well | 1.513 ± 0.020 | 1.313 ± 0.014 | 1.136 ± 0.069 | 1.086 ± 0.053 | 0.742 ± 0.027 | 0.653 ± 0.005 | 0.313 ± 0.115 | 0.291 ± 0.001 |
| 400 ng/well | 0.952 ± 0.005 | 0.834 ± 0.001 | 0.831 ± 0.010 | 0.779 ± 0.020 | 0.674 ± 0.050 | 0.479 ± 0.012 | 0.308 ± 0.030 | 0.210 ± 0.020 |
| 200 ng/well | 0.543 ± 0.143 | 0.437 ± 0.061 | 0.387 ± 0.018 | 0.366 ± 0.001 | 0.321 ± 0.005 | 0.223 ± 0.020 | 0.141 ± 0.010 | 0.130 ± 0.017 |
| Item | Range | Mid-Value |
|---|---|---|
| Coefficient of variation in intra-plates | 0.55–1.65% | 1.10% |
| Coefficient of variation between inter-plates | 2.58–6.03% | 4.17% |
| Samples | Number | The Developed cELISA | IDEXX ELISA Kit | Agreement (%) | Kappa Value | |
|---|---|---|---|---|---|---|
| + | - | |||||
| Chicken serum samples with unknown exposure to the IBV virus or vaccine immunization | 83 | + | 82 | 1 | 98.0% (343/350) | 0.94 |
| 67 | - | 2 | 65 | |||
| Chicken serum samples with a live attenuated vaccine strain of H120 strain | 195 | + | 191 | 4 | ||
| 5 | - | 0 | 5 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, K.; Song, Z.; Ma, P.; Liao, Z.; Yang, M.; Zhou, C.; Li, C.; Zhao, Y.; Li, H.; Yang, X.; et al. A Novel Nanobody-Horseradish Peroxidase Fusion Based-Competitive ELISA to Rapidly Detect Avian Corona-Virus-Infectious Bronchitis Virus Antibody in Chicken Serum. Int. J. Mol. Sci. 2022, 23, 7589. https://doi.org/10.3390/ijms23147589
Gu K, Song Z, Ma P, Liao Z, Yang M, Zhou C, Li C, Zhao Y, Li H, Yang X, et al. A Novel Nanobody-Horseradish Peroxidase Fusion Based-Competitive ELISA to Rapidly Detect Avian Corona-Virus-Infectious Bronchitis Virus Antibody in Chicken Serum. International Journal of Molecular Sciences. 2022; 23(14):7589. https://doi.org/10.3390/ijms23147589
Chicago/Turabian StyleGu, Kui, Zengxu Song, Peng Ma, Ziwei Liao, Ming Yang, Changyu Zhou, Chao Li, Yu Zhao, Hao Li, Xin Yang, and et al. 2022. "A Novel Nanobody-Horseradish Peroxidase Fusion Based-Competitive ELISA to Rapidly Detect Avian Corona-Virus-Infectious Bronchitis Virus Antibody in Chicken Serum" International Journal of Molecular Sciences 23, no. 14: 7589. https://doi.org/10.3390/ijms23147589