
Focus on the Small GTPase Rab1: A Key Player in the Pathogenesis of Parkinson’s Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to PD Pathogenesis
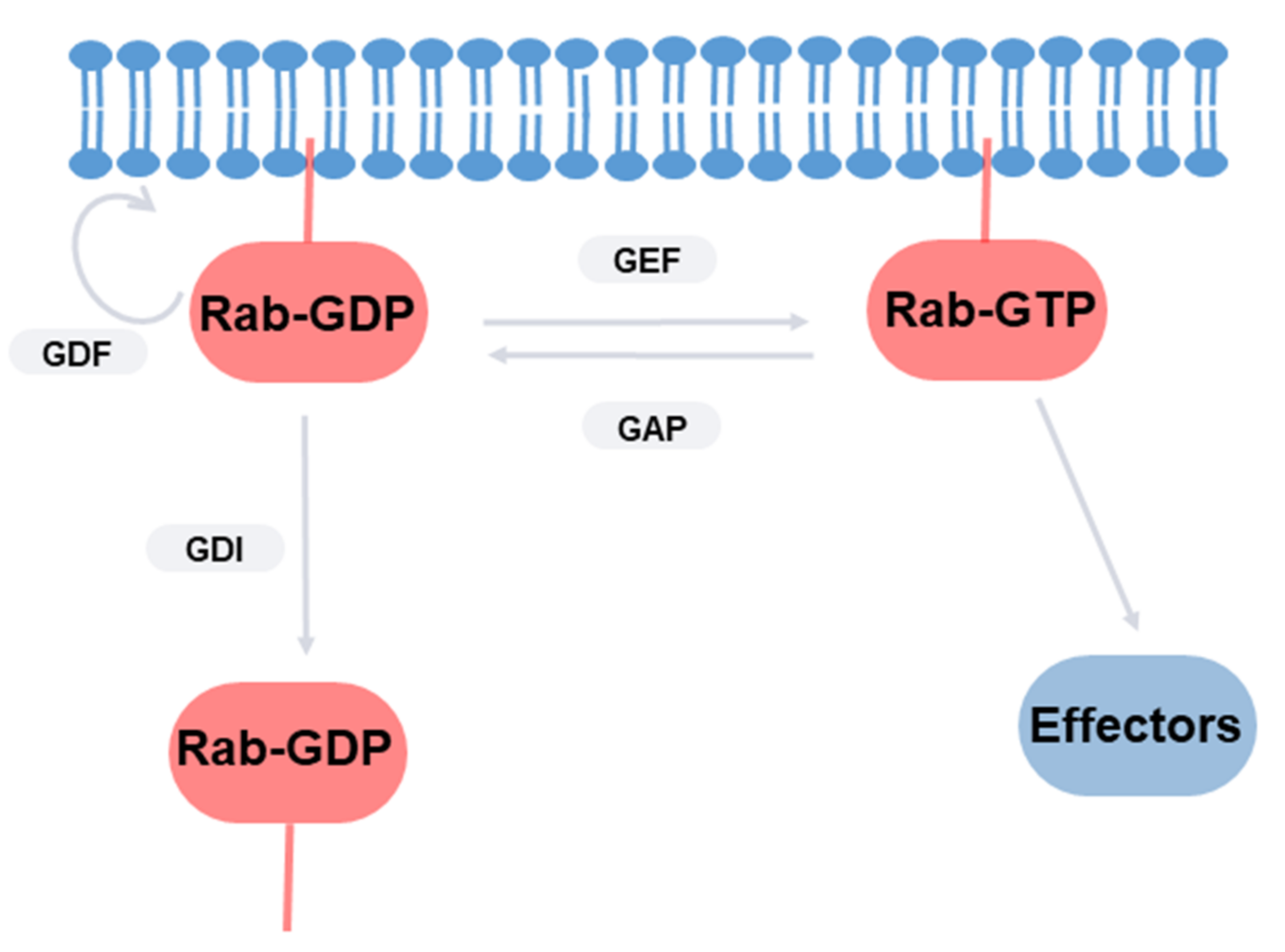
2. Rab GTPases
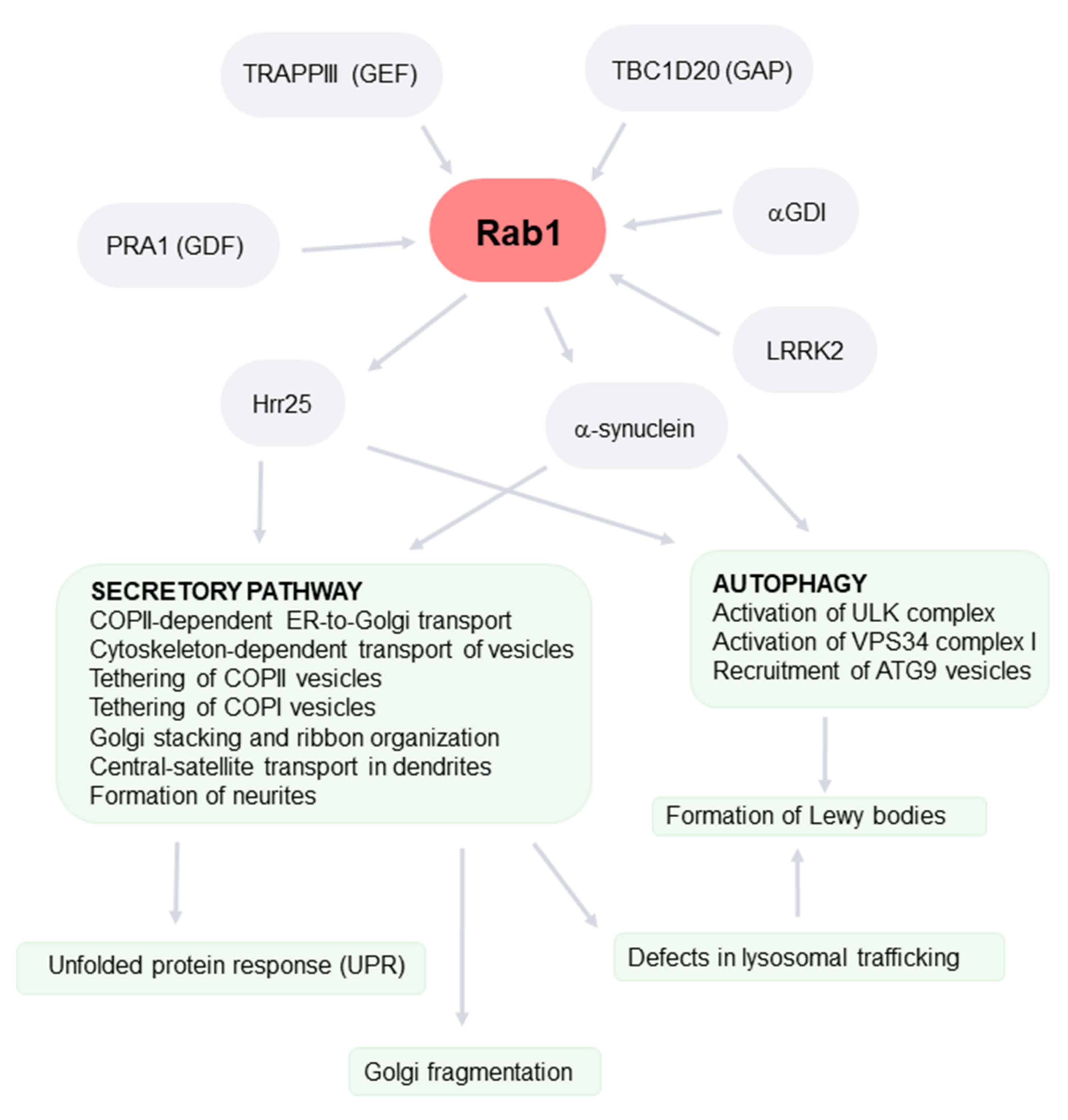
3. Rab1
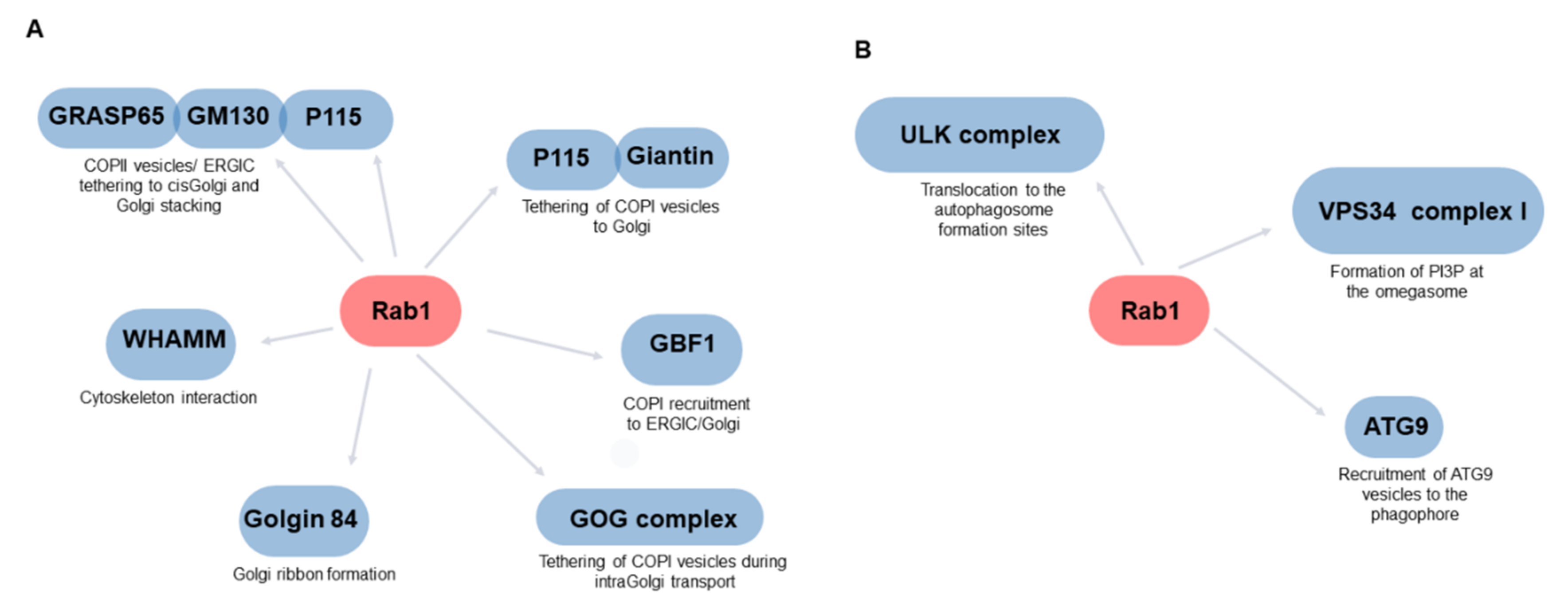
4. The Role of Rab1 in the Secretory Pathway
5. Rab1 as a Regulator of Autophagy
6. Rab1, Secretory Pathway, and PD
7. Relationship between PD-Associated Golgi Fragmentation and Rab1
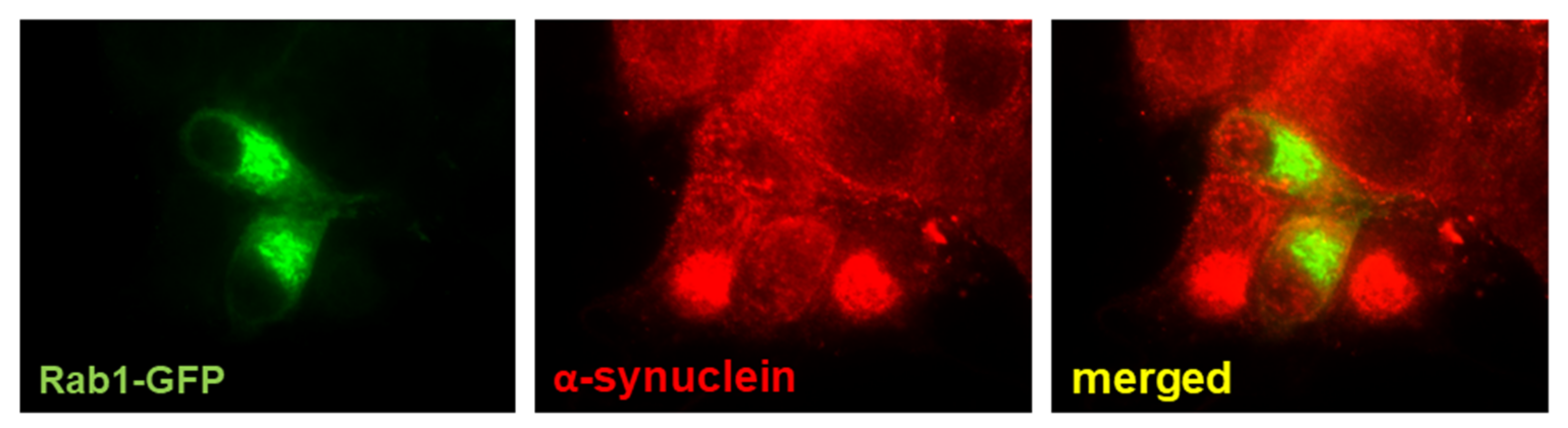
8. Rab1, Autophagy, and PD
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balestrino, R.; Schapira, A. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A. Evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 2016, 12, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Mollenhauer, B.; Beach, T.G.; Adler, C.H.; Coffey, C.S.; Kopil, C.M.; Dave, K.D.; Foroud, T.; Chahine, L.; Jennings, D. The Systemic Synuclein Sampling Study: Toward a biomarker for Parkinson’s disease. Biomark. Med. 2017, 11, 359–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordelon, Y.; Keener, A. Parkinsonism. Semin. Neurol. 2016, 36, 330–334. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef]
- Galvan, A.; Wichmann, T. Pathophysiology of parkinsonism. Clin. Neurophysiol. 2008, 119, 1459–1474. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, T.; Adachi, E.; Orimo, S.; Nakamura, A.; Mizusawa, H.; Uchihara, T. Pale neurites, premature α-synuclein aggregates with centripetal extension from axon collaterals. Brain Pathol. 2012, 22, 67–78. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Mehra, S.; Sahay, S.; Maji, S.K. Alpha-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys Acta Proteins Proteom 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Stefanis, L. Alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 4, a009399. [Google Scholar]
- Oliveira, L.M.A.; Gasser, T.; Edwards, R.; Zweckstetter, M.; Melki, R.; Stefanis, L.; Lashuel, H.A.; Sulzer, D.; Vekrellis, K.; Halliday, G.M.; et al. Alpha-synuclein research: Defining strategic moves in the battle against Parkinson’s disease. NPJ Parkison’s Dis. 2021, 7, 1–23. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, Y.; Xi, H.; Jiang, J.; Yu, Y.; Dong, W. The Membrane Interaction of Alpha-Synuclein. Front. Cell. Neurosci. 2021, 15. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Hay, J.C. Alpha-synuclein Toxicity in the Early Secretory Pathway: How It Drives Neurodegeneration in Parkinsons Disease. Front. Neurosci. 2015, 12, 9–433. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. [Google Scholar] [CrossRef]
- Berge, N.V.D.; Ferreira, N.; Mikkelsen, T.W.; Alstrup, A.K.O.; Tamgüney, G.; Karlsson, P.; Terkelsen, A.J.; Nyengaard, J.R.; Jensen, P.H.; Borghammer, P. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain 2021, 144, 1853–1868. [Google Scholar] [CrossRef]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzón-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Bu, M.; Farrer, M.J.; Khoshbouei, H. Parkinsons Dynamic control of the dopamine transporter in neurotransmission and homeostasis. NPJ Parkinson’s Dis. 2021, 7, 22. [Google Scholar]
- Hunn, B.H.M.; Vingill, S.; Threlfell, S.; Alegre-Abarrategui, J.; Magdelyns, M.; Deltheil, T.; Bengoa-Vergniory, N.; Oliver, P.L.; Cioroch, M.; Doig, N.M.; et al. Impairment of Macroautophagy in Dopamine Neurons Has Opposing Effects on Parkinsonian Pathology and Behavior. Cell Rep. 2019, 29, 920–931.e7. [Google Scholar] [CrossRef] [Green Version]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef]
- Singh, P.K.; Muqit, M.M.K. Parkinson’s: A Disease of Aberrant Vesicle Trafficking. Annu. Rev. Cell Dev. Biol. 2020, 36, 237–264. [Google Scholar] [CrossRef]
- Matsuda, W.; Furuta, T.; Nakamura, K.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single Nigrostriatal Dopaminergic Neurons Form Widely Spread and Highly Dense Axonal Arborizations in the Neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Lewis, P.; Revesz, T.; Lees, A.; Paisan-Ruiz, C. The genetics of Parkinson’s syndromes: A critical review. Curr. Opin. Genet. Dev. 2009, 19, 254–265. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; Van Der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Gialluisi, A.; Reccia, M.G.; Modugno, N.; Nutile, T.; Lombardi, A.; Di Giovannantonio, L.G.; Pietracupa, S.; Ruggiero, D.; Scala, S.; Gambardella, S.; et al. Identification of sixteen novel candidate genes for late onset Parkinson’s disease. Mol. Neurodegener. 2021, 16, 1–18. [Google Scholar] [CrossRef]
- Lee, V.M.Y.; Trojanowski, J.Q. Mechanisms of Parkinson’s disease linked to pathological α-synuclein: New targets for drug discovery. Neuron 2006, 51, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Ray, B.; Mahalakshmi, A.M.; Tuladhar, S.; Bhat, A.; Srinivasan, A.; Pellegrino, C.; Kannan, A.; Bolla, S.R.; Chidambaram, S.B.; Sakharkar, M.K. “Janus-Faced” α-Synuclein: Role in Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 673395. [Google Scholar] [CrossRef]
- Alessi, D.R.; Sammler, E. LRRK2 kinase in Parkinson’s disease. Science 2018, 360, 36–37. [Google Scholar] [CrossRef] [Green Version]
- Bonet-Ponce, L.; Cookson, M.R. LRRK2 recruitment, activity, and function in organelles. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Berwick, D.C.; Heaton, G.R.; Azeggagh, S.; Harvey, K. LRRK2 Biology from structure to dysfunction: Research progresses, but the themes remain the same. Mol. Neurodegener. 2019, 14, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Madureira, M.; Connor-Robson, N.; Wade-Martins, R. “LRRK2: Autophagy and Lysosomal Activity”. Front. Neurosci. 2020, 14, 498. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Alessi, D.R. Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr. Opin. Cell Biol. 2020, 63, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Follett, J.; Farrer, M.J. LRRK2; a dynamic regulator of cellular trafficking. Brain Res. 2021, 1761, 147394. [Google Scholar] [CrossRef]
- Rassu, M.; Del Giudice, M.G.; Sanna, S.; Taymans, J.M.; Morari, M.; Brugnoli, A.; Frassineti, M.; Masala, A.; Esposito, S.; Galioto, M.; et al. Role of LRRK2 in the regulation of dopamine receptor trafficking. PLoS ONE 2017, 12, e0179082. [Google Scholar] [CrossRef]
- Cho, H.J.; Yu, J.; Xie, C.; Rudrabhatla, P.; Chen, X.; Wu, J.; Parisiadou, L.; Liu, G.; Sun, L.; Ma, B.; et al. Leucine-rich repeat kinase 2 regulates Sec16A at ER exit sites to allow ER-Golgi export. EMBO J. 2014, 33, 2314–2331. [Google Scholar] [CrossRef] [Green Version]
- Mir, R.; Tonelli, F.; Lis, P.; Macartney, T.; Polinski, N.K.; Martinez, T.N.; Chou, M.Y.; Howden, A.J.M.; König, T.; Hotzy, C.; et al. The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem. J. 2018, 475, 1861–1883. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Fu, Y.; Halliday, G.M.; Sue, C.M. PARK Genes Link Mitochondrial Dysfunction and Alpha-Synuclein Pathology in Sporadic Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef]
- Yang, X.Z.; Li, X.X.; Zhang, Y.J.; Rodriguez-Rodriguez, L.; Xiang, M.Q.; Wang, H.Y.; Zheng, X.F. Rab1 in cell signaling, cancer and other diseases. Oncogene 2016, 35, 5699–56704. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, X.; Yuan, Z.; Radford, S.J.; Liu, C.; Libutti, S.K.; Zheng, X.F.S. Amino acids-Rab1A-mTORC1 signaling controls whole-body glucose homeostasis. Cell Rep. 2021, 34, 108830. [Google Scholar] [CrossRef]
- Du, J.; von Wrisberg, M.-K.; Gulen, B.; Stahl, M.; Pett, C.; Hedberg, C.; Lang, K.; Schneider, S.; Itzen, A. Rab1-AMPylation by Legionella DrrA is allosterically activated by Rab1. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Kawabata, M.; Matsuo, H.; Koito, T.; Murata, M.; Kubori, T.; Nagai, H.; Tagaya, M.; Arasaki, K. Legionella hijacks the host Golgi-to-ER retrograde pathway for the association of Legionella-containing vacuole with the ER. PLoS Pathog. 2021, 17, e1009437. [Google Scholar] [CrossRef]
- Rendón, W.O.; Martínez-Alonso, E.; Tomás, M.; Martínez-Martínez, N.; Martínez-Menárguez, J.A. Golgi fragmentation is Rab and SNARE dependent in cellular models of Parkinson’s disease. Histochem. Cell Biol. 2012, 139, 671–684. [Google Scholar] [CrossRef]
- Tomás, M.; Martínez-Alonso, E.; Martínez-Martínez, N.; Cara-Esteban, M.; Martínez-Menárguez, J.A. Fragmentation of the Golgi complex of dopaminergic neurons in human substantia nigra: New cytopathological findings in Parkinson’s disease. Histol. Histopathol. 2021, 36, 47–60. [Google Scholar]
- Soo, K.Y.; Halloran, M.; Sundaramoorthy, V.; Parakh, S.; Toth, R.P.; Southam, K.A.; McLean, C.A.; Lock, P.; King, A.; Farg, M.A.; et al. Rab1-dependent ER-Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropathol. 2015, 130, 679–697. [Google Scholar] [CrossRef]
- Soo, K.Y.; Sultana, J.M.; King, A.; Atkinson, R.; Warraich, S.T.; Sundaramoorthy, V.; Blair, I.; Farg, M.A.; Atkin, J.D. ALS-associated mutant FUS inhibits macroautophagy which is restored by overexpression of Rab1. Cell Death Discov. 2015, 1, 15030. [Google Scholar] [CrossRef]
- Dugan, J.M.; deWit, C.; McConlogue, L.; Maltese, W.A. The Ras-related GTP-binding protein, Rab1B, regulates early steps in exocytic transport and processing of beta-amyloid precursor protein. J. Biol. Chem. 1995, 270, 10982–10989. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.L. Rabs, Membrane Dynamics, and Parkinson’s Disease. J. Cell. Physiol. 2017, 232, 1626–1633. [Google Scholar] [CrossRef]
- Kiral, F.R.; Kohrs, F.E.; Jin, E.J.; Hiesinger, P.R. Rab GTPases and Membrane Trafficking in Neurodegeneration. Curr. Biol. 2018, 28, R471–R486. [Google Scholar] [CrossRef] [Green Version]
- Lipatova, Z.; Hain, A.U.; Nazarko, V.Y.; Segev, N. Ypt/Rab GTPases: Principles learned from yeast. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Wilson, G.R.; Stephenson, S.; Bozaoglu, K.; Farrer, M.J.; Lockhart, P. The emerging role of Rab GTPases in the pathogenesis of Parkinson’s disease. Mov. Disord. 2018, 33, 196–207. [Google Scholar] [CrossRef]
- Aloisi, A.L.; Bucci, C. Rab GTPases-cargo direct interactions: Fine modulators of intracellular trafficking. Histol. Histopathol. 2013, 28, 839–849. [Google Scholar] [PubMed]
- Voss, S.; Li, F.; Rätz, A.; Röger, M.; Wu, Y.W. Spatial Cycling of Rab GTPase, Driven by the GTPase Cycle, Controls Rab’s Subcellular Distribution. Biochemistry 2019, 58, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Kirsten, M.L.; Baron, R.A.; Seabra, M.C.; Ces, O. Rab1a and Rab5a preferentially bind to binary lipid compositions with higher stored curvature elastic energy. Mol. Membr. Biol. 2013, 30, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Waschbüsch, D.; Khan, A.R. Phosphorylation of Rab GTPases in the regulation of membrane trafficking. Traffic 2020, 21, 712–719. [Google Scholar] [CrossRef]
- Ng, E.L.; Tang, B.L. Rab GTPases and their roles in brain neurons and glia. Brain Res. Rev. 2008, 58, 236–246. [Google Scholar] [CrossRef]
- Villarroel-Campos, D.; Bronfman, F.C.; Gonzalez-Billault, C. Rab GTPase signaling in neurite outgrowth and axon specification. Cytoskeleton 2016, 73, 498–507. [Google Scholar] [CrossRef]
- Zheng, L.-Q.; Chi, S.-M.; Li, C.-X. Rab23’s genetic structure, function and related diseases: A review. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- D’Adamo, P.; Masetti, M.; Bianchi, V.; Morè, L.; Mignogna, M.L.; Giannandrea, M.; Gatti, S. RAB GTPases and RAB-interacting proteins and their role in the control of cognitive functions. Neurosci. Biobehav. Rev. 2014, 46, 302–314. [Google Scholar] [CrossRef]
- Sastre, A.A.; Montoro, M.L.; Lacerda, H.; Llavero, F.; Zugaza, J. Small GTPases of the Rab and Arf Families: Key Regulators of Intracellular Trafficking in Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 4425. [Google Scholar] [CrossRef]
- Gonçalves, S.A.; Macedo, D.; Raquel, H.; Simões, P.D.; Giorgini, F.; Ramalho, J.S.; Barral, D.C.; Ferreira-Moita, L.; Outeiro, T.F. shRNA-Based Screen Identifies Endocytic Recycling Pathway Components That Act as Genetic Modifiers of Alpha-Synuclein Aggregation, Secretion and Toxicity. PLoS Genet. 2016, 12, e1005995. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.-J.; Kim, D.-K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Dinter, E.; Saridaki, T.; Nippold, M.; Plum, S.; Diederichs, L.; Komnig, D.; Fensky, L.; May, C.; Marcus, K.; Voigt, A.; et al. Rab7 induces clearance of α-synuclein aggregates. J. Neurochem. 2016, 138, 758–774. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Yi, S.; Sahni, N.; Loh, K.H.; Auluck, P.K.; Baru, V.; Udeshi, N.D.; Freyzon, Y.; Carr, S.A.; et al. In Situ Peroxidase Labeling and Mass-Spectrometry Connects Alpha-Synuclein Directly to Endocytic Trafficking and mRNA Metabolism in Neurons. Cell Syst. 2017, 4, 242–250.e4. [Google Scholar] [CrossRef] [Green Version]
- Dalfo, E.; Gómez-Isla, T.; Rosa, J.L.; Bodelón, M.N.; Cuadrado-Tejedor, M.; Barrachina, M.; Ambrosio, S.; Ferrer, I. Abnormal α-Synuclein Interactions with Rab Proteins in α-Synuclein A30P Transgenic Mice. J. Neuropathol. Exp. Neurol. 2004, 63, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Koss, D.J.; Campesan, S.; Giorgini, F.; Outeiro, T.F. Dysfunction of RAB39B- Mediated Vesicular Trafficking in Lewy Body Diseases. Mov. Disord. 2021, 36, 1744–1758. [Google Scholar] [CrossRef]
- Touchot, N.; Zahraoui, A.; Vielh, E.; Tavitian, A. Biochemical properties of the YPT-related rab 1B protein. FEBS Lett. 1989, 256, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Zahraoui, A.; Touchot, N.; Chardin, P.; Tavitian, A. The human Rab genes encode a family of GTP-binding proteins related to yeast YPT1 and SEC4 products involved in secretion. J. Biol. Chem. 1989, 264, 12394–12401. [Google Scholar] [CrossRef]
- Gurkan, C.; Lapp, H.; Alory, C.; Su, A.I.; HogenEsch, J.B.; Balch, W.E. Large-Scale Profiling of Rab GTPase Trafficking Networks: The Membrome. Mol. Biol. Cell 2005, 16, 3847–3864. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhou, X.; Zhang, R.; Sun, H.; You, F.; Jiang, Z. Differences in IFNβ secretion upon Rab1 inactivation in cells exposed to distinct innate immune stimuli. Cell. Mol. Immunol. 2021, 18, 1590–1592. [Google Scholar] [CrossRef]
- Allan, B.B.; Moyer, B.D.; Balch, W.E. Rab1 Recruitment of p115 into a cis-SNARE Complex: Programming Budding COPII Vesicles for Fusion. Science 2000, 289, 444–448. [Google Scholar] [CrossRef]
- Moyer, B.D.; Allan, B.B.; Balch, W.E. Rab1 interaction with a GM130 effector complex regulates COPII vesicle cis—Golgi tethering. Traffic 2001, 2, 268–276. [Google Scholar] [CrossRef]
- Weide, T.; Bayer, M.; Köster, M.; Siebrasse, J.P.; Peters, R.; Barnekow, A. The Golgi matrix protein GM130: A specific interacting partner of the small GTPase rab1b. EMBO Rep. 2001, 2, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Satoh, A.; Wang, Y.; Malsam, J.; Beard, M.B.; Warren, G. Golgin-84 is a rab1 Binding Partner Involved in Golgi Structure. Traffic 2003, 4, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Beard, M.; Satoh, A.; Shorter, J.; Warren, G. A Cryptic Rab1-binding Site in the p115 Tethering Protein. J. Biol. Chem. 2005, 280, 25840–25848. [Google Scholar] [CrossRef] [Green Version]
- Bayer, M.; Fischer, J.; Kremerskothen, J.; Ossendorf, E.; Matanis, T.; Konczal, M.; Weide, T.; Barnekow, A. Identification and characterization of Iporin as a novel interaction partner for rab1. BMC Cell Biol. 2005, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.; Weide, T.; Barnekow, A. The MICAL proteins and rab1: A possible link to the cytoskeleton? Biochem. Biophys. Res. Commun. 2005, 328, 415–423. [Google Scholar] [CrossRef]
- Cavieres, V.A.; Cerda-Troncoso, C.; Rivera-Dictter, A.; Castro, R.I.; Luchsinger, C.; Santibañez, N.; Burgos, P.V.; Mar-dones, G.A. Human Golgi phosphoprotein 3 is an effector of RAB1A and RAB1B. PLoS ONE 2020, 15, e0237514. [Google Scholar] [CrossRef] [PubMed]
- Galindo, A.; Planelles-Herrero, V.J.; Degliesposti, G.; Munro, S. Cryo-EM structure of metazoan TRAPPIII, the multi-subunit complex that activates the GTPase Rab1. EMBO J. 2021, 40, e107608. [Google Scholar] [CrossRef] [PubMed]
- Joiner, A.M.; Phillips, B.P.; Yugandhar, K.; Sanford, E.J.; Smolka, M.B.; Yu, H.; Miller, E.A.; Fromme, J.C. Structural basis of TRAPPIII-mediated Rab1 activation. EMBO J. 2021, 40, e107607. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Chin, H.F.; Lazarova, D.; Menon, S.; Fu, C.; Cai, H.; Sclafani, A.; Rodgers, D.W.; De La Cruz, E.M.; Ferro-Novick, S.; et al. The Structural Basis for Activation of the Rab Ypt1p by the TRAPP Membrane-Tethering Complexes. Cell 2008, 133, 1202–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Yu, S.; Menon, S.; Cai, Y.; Lazarova, D.L.; Fu, C.; Reinisch, K.M.; Hay, J.C.; Ferro-Novick, S. TRAPPI tethers COPII vesicles by binding the coat subunit Sec23. Nature 2007, 445, 941–944. [Google Scholar] [CrossRef]
- Tan, D.; Cai, Y.; Wang, J.; Zhang, J.; Menon, S.; Chou, H.-T.; Ferro-Novick, S.; Reinisch, K.M.; Walz, T. The EM structure of the TRAPPIII complex leads to the identification of a requirement for COPII vesicles on the macroautophagy pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 19432–19437. [Google Scholar] [CrossRef] [Green Version]
- Lynch-Day, M.A.; Bhandari, D.; Menon, S.; Huang, J.; Cai, H.; Bartholomew, C.R.; Brumell, J.H.; Ferro-Novick, S.; Klionsky, D.J. Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 7811–7816. [Google Scholar] [CrossRef] [Green Version]
- Van Bergen, N.J.; Guo, Y.; Al-Deri, N.; Lipatova, Z.; Stanga, D.; Zhao, S.; Murtazina, R.; Gyurkovska, V.; Pehlivan, D.; Mitani, T.; et al. Deficiencies in vesicular transport mediated by TRAPPC4 are associated with severe syndromic intellectual disability. Brain 2019, 143, 112–130. [Google Scholar] [CrossRef]
- Barr, F.; Lambright, D.G. Rab GEFs and GAPs. Curr. Opin. Cell Biol. 2010, 22, 461–470. [Google Scholar] [CrossRef]
- Du, L.-L.; Novick, P. Yeast Rab GTPase-activating Protein Gyp1p Localizes to the Golgi Apparatus and Is a Negative Regulator of Ypt1p. Mol. Biol. Cell 2001, 12, 1215–1226. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.L.; Highland, C.M.; Fromme, J.C. Arf1 orchestrates Rab GTPase conversion at the trans-Golgi network. Mol. Biol. Cell 2021, 32, 1104–1120. [Google Scholar] [CrossRef]
- Kim, J.J.; Lipatova, Z.; Majumdar, U.; Segev, N. Regulation of Golgi Cisternal Progression by Ypt/Rab GTPases. Dev. Cell 2016, 36, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Novick, P. Regulation of membrane traffic by Rab GEF and GAP cascades. Small GTPases 2016, 7, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Mitter, A.L.; Schlotterhose, P.; Krick, R. Gyp1 has a dual function as Ypt1 GAP and interaction partner of Atg8 in selective autophagy. Autophagy 2019, 15, 1031–1050. [Google Scholar] [CrossRef]
- Haas, A.K.; Yoshimura, S.-I.; Stephens, D.; Preisinger, C.; Fuchs, E.; Barr, F. Analysis of GTPase-activating proteins: Rab1 and Rab43 are key Rabs required to maintain a functional Golgi complex in human cells. J. Cell Sci. 2007, 120, 2997–3010. [Google Scholar] [CrossRef] [Green Version]
- Sidjanin, D.J.; Park, A.K.; Ronchetti, A.; Martins, J.; Jackson, W.T. TBC1D20 mediates autophagy as a key regulator of autophagosome maturation. Autophagy 2016, 12, 1759–1775. [Google Scholar] [CrossRef] [Green Version]
- Calero, M.; Collins, R.N. Saccharomyces cerevisiae Pra1p/Yip3p interacts with Yip1p and Rab proteins. Biochem. Biophys. Res. Commun. 2002, 290, 676–681. [Google Scholar] [CrossRef]
- Lee, H.J.; Kang, S.J.; Lee, K.; Im, H. Human α-synuclein modulates vesicle trafficking through its interaction with prenylated Rab acceptor protein 1. Biochem. Biophys. Res. Commun. 2011, 412, 526–531. [Google Scholar] [CrossRef]
- Wu, S.K.; Zeng, K.; Wilson, I.A.; Balch, W.E. Structural insights into the function of the Rab GDI superfamily. Trends Biochem. Sci. 1996, 21, 472–476. [Google Scholar] [CrossRef]
- Saraste, J.; Lahtinen, U.; Goud, B. Localization of the small GTP-binding protein rab1p to early compartments of the secretory pathway. J. Cell Sci. 1995, 108, 1541–1552. [Google Scholar] [CrossRef]
- Saraste, J. Spatial and Functional Aspects of ER-Golgi Rabs and Tethers. Front. Cell Dev. Biol. 2016, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Sannerud, R.; Marie, M.; Nizak, C.; Dale, H.A.; Pernet-Gallay, K.; Perez, F.; Goud, B.; Saraste, J. Rab1 Defines a Novel Pathway Connecting the Pre-Golgi Intermediate Compartment with the Cell Periphery. Mol. Biol. Cell 2006, 17, 1514–1526. [Google Scholar] [CrossRef] [Green Version]
- Monetta, P.; Slavin, I.; Romero, N.; Alvarez, C. Rab1b Interacts with GBF1 and Modulates both ARF1 Dynamics and COPI Association. Mol. Biol. Cell 2007, 18, 2400–2410. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Ballew, N.; Barlowe, C. Initial docking of ER-derived vesicles requires Uso1p and Ypt1p but is independent of SNARE proteins. EMBO J. 1998, 17, 2156–2165. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Barlowe, C. Asymmetric Requirements for a Rab Gtpase and Snare Proteins in Fusion of Copii Vesicles with Acceptor Membranes. J. Cell Biol. 2000, 149, 55–66. [Google Scholar] [CrossRef]
- Morsomme, P.; Riezman, H. The Rab GTPase Ypt1p and tethering factors couple protein sorting at the ER to vesicle targeting to the Golgi apparatus. Dev. Cell 2002, 2, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Tisdale, E.J.; Bourne, J.R.; Khosravi-Far, R.; Der, C.J.; Balch, W.E. GTP-binding mutants of rab1 and rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. J. Cell Biol. 1992, 119, 749–761. [Google Scholar] [CrossRef]
- Slavin, I.; García, I.A.; Monetta, P.; Martinez, H.; Romero, N.; Alvarez, C. Role of Rab1b in COPII dynamics and function. Eur. J. Cell Biol. 2011, 90, 301–311. [Google Scholar] [CrossRef]
- Westrate, L.M.; Hoyer, M.J.; Nash, M.J.; Voeltz, G.K. Vesicular and uncoated Rab1-dependent cargo carriers facilitate ER to Golgi transport. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Witkos, T.; Lowe, M. The Golgin Family of Coiled-Coil Tethering Proteins. Front. Cell Dev. Biol. 2016, 3, 86. [Google Scholar] [CrossRef] [Green Version]
- Diao, A.; Rahman, D.; Pappin, D.; Lucocq, J.; Lowe, M. The coiled-coil membrane protein golgin-84 is a novel rab effector required for Golgi ribbon formation. J. Cell Biol. 2003, 160, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, Z.; Yang, Y.; Li, Q.; Zeng, R.; Kang, J.; Wu, J. Rab1A mediates proinsulin to insulin conversion in beta-cells by maintaining Golgi stability through interactions with golgin-84. Protein Cell 2016, 7, 692–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamena, F.; Diefenbacher, M.; Kilchert, C.; Schwarz, H.; Spang, A. Ypt1p is essential for retrograde Golgi-ER transport and for Golgi maintenance in S. cerevisiae. J. Cell Sci. 2008, 121, 1293–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvorova, E.S.; Duden, R.; Lupashin, V. The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J. Cell Biol. 2002, 157, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willett, R.; Ungar, D.; Lupashin, V. The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem. Cell Biol. 2013, 140, 271–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, S.D.; Lee, A.R.; Selegue, J.E.; Cavanagh, A.T.; Bashirullah, A. A novel function for Rab1 and Rab11 during secretory granule maturation. J. Cell Sci. 2021, 134, 259037. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.J.; Mathiowetz, A.J.; Hong, S.; Welch, M.D.; Campellone, K.G. Rab1 recruits WHAMM during membrane remodeling but limits actin nucleation. Mol. Biol. Cell 2016, 27, 967–978. [Google Scholar] [CrossRef]
- Campellone, K.G.; Webb, N.J.; Znameroski, E.A.; Welch, M.D. WHAMM is an Arp2/3 complex activator that binds microtubules and functions in ER to Golgi transport. Cell 2008, 134, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, A.; Quiroz, J.A.; Wolkoff, A.W. Rab1a regulates sorting of early endocytic vesicles. Am. J. Physiol. Liver Physiol. 2014, 306, G412–G424. [Google Scholar] [CrossRef] [Green Version]
- Rivero, S.; Cardenas, J.; Bornens, M.; Ríos, R.M. Microtubule nucleation at the cis-side of the Golgi apparatus requires AKAP450 and GM130. EMBO J. 2009, 28, 1016–1028. [Google Scholar] [CrossRef]
- Mikhaylova, M.; Bera, S.; Kobler, O.; Frischknecht, R.; Kreutz, M.R. A Dendritic Golgi Satellite between ERGIC and Retromer. Cell Rep. 2015, 14, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Saraste, J.; Marie, M. Intermediate compartment (IC): From pre-Golgi vacuoles to a semi-autonomous membrane system. Histochem. Cell Biol. 2018, 150, 407–430. [Google Scholar] [CrossRef] [Green Version]
- Solis, G.P.; Bilousov, O.; Koval, A.; Lüchtenborg, A.M.; Lin, C.; Katanaev, V.L. Golgi-Resident Galphao Promotes Protrusive Membrane Dynamics. Cell 2017, 170, 939–955. [Google Scholar] [CrossRef] [Green Version]
- de Lima, N.C.R.; Melo, T.Q.; Sakugawa, A.Y.; Melo, K.P.; Ferrari, M.F. Restoration of Rab1 Levels Prevents Endoplasmic Reticulum Stress in Hippocampal Cells during Protein Aggregation Triggered by Rotenone. Neuroscience 2019, 419, 5–13. [Google Scholar] [CrossRef]
- Tsvetanova, N.G. The secretory pathway in control of endoplasmic reticulum homeostasis. Small GTPases 2013, 4, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.H.; Park, J.H.; Lee, E.S.; Paeng, S.K.; Chae, H.B.; Chi, Y.H.; Lee, S.Y. Exploring Novel Functions of the Small GTPase Ypt1p under Heat-Shock by Characterizing a Temperature-Sensitive Mutant Yeast Strain, ypt1-G80D. Int. J. Mol. Sci. 2019, 20, 132. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, R.; Zhu, L. Chaperone-Mediated Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 435–452. [Google Scholar]
- Schuck, S. Microautophagy—Distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Lamark, T.; Johansen, T. Mechanisms of Selective Autophagy. Annu. Rev. Cell Dev. Biol. 2021, 37, 143–169. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, M.R.; Ruiz-Garcia, D.; Martin-Solana, E.; Chichon, F.J.; Carrascosa, J.L.; Fernandez, J.-J. 3D electron tomography of brain tissue unveils distinct Golgi structures that sequester cytoplasmic contents in neurons. J. Cell Sci. 2016, 130, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Rana, T.; Behl, T.; Sehgal, A.; Mehta, V.; Singh, S.; Bhatia, S.; Al-Harrasi, A.; Bungau, S. Exploring the Role of Autophagy Dysfunction in Neurodegenerative Disorders. Mol. Neurobiol. 2021, 58, 4886–4905. [Google Scholar] [CrossRef]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.; Ktistakis, N.T. Omegasomes: PI3P platforms that manufacture autophagosomes. Essays Biochem. 2013, 55, 17–27. [Google Scholar] [CrossRef]
- Nascimbeni, A.C.; Codogno, P.; Morel, E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017, 284, 1267–1278. [Google Scholar] [CrossRef]
- Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.-L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [Green Version]
- Uemura, T.; Yamamoto, M.; Kametaka, A.; Sou, Y.-S.; Yabashi, A.; Yamada, A.; Annoh, H.; Kametaka, S.; Komatsu, M.; Waguri, S. A Cluster of Thin Tubular Structures Mediates Transformation of the Endoplasmic Reticulum to Autophagic Isolation Membrane. Mol. Cell. Biol. 2014, 34, 1695–1706. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.-M.; Mizushima, N. At the end of the autophagic road: An emerging understanding of lysosomal functions in autophagy. Trends Biochem. Sci. 2014, 39, 61–71. [Google Scholar] [CrossRef]
- Ganesan, D.; Cai, Q. Understanding amphisomes. Biochem. J. 2021, 478, 1959–1976. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.J.; Gubas, A.; Tooze, S.A. A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 2018, 293, 5386–5395. [Google Scholar] [CrossRef] [Green Version]
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 21, 439–458. [Google Scholar] [CrossRef]
- Matoba, K.; Noda, N.N. Structural catalog of core Atg proteins opens new era of autophagy research. J. Biochem. 2021, 169, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Tamura, N.; Kono, N.; Shimanaka, Y.; Arai, H.; Yamamoto, H.; Mizushima, N. Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. EMBO J. 2017, 36, 1719–1735. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ma, K.; Gao, R.; Mu, C.; Chen, L.; Liu, Q.; Luo, Q.; Feng, D.; Zhu, Y.; Chen, Q. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2016, 27, 184–201. [Google Scholar] [CrossRef] [Green Version]
- De Tito, S.; Hervás, J.H.; van Vliet, A.R.; Tooze, S.A. The Golgi as an Assembly Line to the Autophagosome. Trends Biochem. Sci. 2020, 45, 484–496. [Google Scholar] [CrossRef]
- Matoba, K.; Kotani, T.; Tsutsumi, A.; Tsuji, T.; Mori, T.; Noshiro, D.; Sugita, Y.; Nomura, N.; Iwata, S.; Ohsumi, Y.; et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 2020, 27, 1185–1193. [Google Scholar] [CrossRef]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbé, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [Green Version]
- Mizushimam, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 con-jugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef] [Green Version]
- Slobodkin, M.R.; Elazar, Z. The Atg8 family: Multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 2013, 55, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Noda, T. Autophagy in the context of the cellular membrane-trafficking system: The enigma of Atg9 vesicles. Biochem. Soc. Trans. 2017, 45, 1323–1331. [Google Scholar] [CrossRef] [Green Version]
- Jedd, G.; Richardson, C.; Litt, R.; Segev, N. The Ypt1 GTPase is essential for the first two steps of the yeast secretory pathway. J. Cell Biol. 1995, 131, 583–590. [Google Scholar] [CrossRef]
- Zoppino, F.C.M.; Militello, R.D.; Slavin, I.; Álvarez, C.; Colombo, M.I. Autophagosome Formation Depends on the Small GTPase Rab1 and Functional ER Exit Sites. Traffic 2010, 11, 1246–1261. [Google Scholar] [CrossRef]
- Huang, J.; Birmingham, C.L.; Shahnazari, S.; Shiu, J.; Zheng, Y.T.; Smith, A.C.; Campellone, K.G.; Heo, W.D.; Gruenheid, S.; Meyer, T.; et al. Antibacterial autophagy occurs at PI(3)P-enriched domains of the endoplasmic reticulum and requires Rab1 GTPase. Autophagy 2011, 7, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Lipatova, Z.; Belogortseva, N.; Zhang, X.Q.; Kim, J.; Taussig, D.; Segev, N. Regulation of selective autophagy onset by a Ypt/Rab GTPase module. Proc. Natl. Acad. Sci. USA 2012, 109, 6981–6986. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, Y.; Ohashi, R.; Kawamura, T.; Iwanari, H.; Kodama, T.; Naito, M.; Hamakubo, T. Phosphatidylinositol 3-Phosphatase Myotubularin-related Protein 6 (MTMR6) Is Regulated by Small GTPase Rab1B in the Early Secretory and Autophagic Pathways. J. Biol. Chem. 2013, 288, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.; Ferro-Novick, S. Ypt1 and COPII vesicles act in autophagosome biogenesis and the early secretory pathway. Biochem. Soc. Trans. 2015, 43, 92–96. [Google Scholar] [CrossRef]
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Barz, S.; Kriegenburg, F.; Sánchez-Martín, P.; Kraft, C. Small but mighty: Atg8s and Rabs in membrane dynamics during autophagy. Biochim. Biophys. Acta Bioenerg. 2021, 1868, 119064. [Google Scholar] [CrossRef]
- Wang, J.; Menon, S.; Yamasaki, A.; Chou, H.-T.; Walz, T.; Jiang, Y.; Ferro-Novick, S. Ypt1 recruits the Atg1 kinase to the preautophagosomal structure. Proc. Natl. Acad. Sci. USA 2013, 110, 9800–9805. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK 1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Tremel, S.; Ohashi, Y.; Morado, D.R.; Bertram, J.; Perisic, O.; Brandt, L.T.L.; von Wrisberg, M.K.; Chen, Z.A.; Maslen, S.L.; Kovtun, O.; et al. Structural basis for VPS34 kinase activation by Rab1 and Rab5 on membranes. Nat. Commun. 2021, 12, 1564. [Google Scholar] [CrossRef] [PubMed]
- Backues, S.K.; Klionsky, D.J. Atg11: A Rab-dependent, coiled-coil membrane protein that acts as a tether for autophagy. Autophagy 2012, 8, 1275–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakuta, S.; Yamaguchi, J.; Suzuki, C.; Sasaki, M.; Kazuno, S.; Uchiyama, Y. Small GTPase Rab1B is associated with ATG9A vesicles and regulates autophagosome formation. FASEB J. 2017, 31, 3757–3773. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Nühlen, S.; Judith, D.; Frith, D.; Snijders, B.; Behrends, C.; Tooze, S.A. TBC 1D14 regulates autophagy via the TRAPP complex and ATG 9 traffic. EMBO J. 2015, 35, 281–301. [Google Scholar] [CrossRef]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [Green Version]
- Graef, M.; Friedman, J.; Graham, C.; Babu, M.; Nunnari, J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell 2013, 24, 2918–2931. [Google Scholar] [CrossRef]
- Shima, T.; Kirisako, H.; Nakatogawa, H. COPII vesicles contribute to autophagosomal membranes. J. Cell Biol. 2019, 218, 1503–1510. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Huang, W.; Wang, W. Multifaceted roles of COPII subunits in autophagy. Biochim. Biophys. Acta Bioenerg. 2019, 1867, 118627. [Google Scholar] [CrossRef]
- Ge, L.; Melville, D.; Zhang, M.; Schekman, R. The ER–Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2013, 2, e00947. [Google Scholar] [CrossRef]
- Ge, L.; Zhang, M.; Schekman, R. Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. eLife 2014, 3, e04135. [Google Scholar] [CrossRef]
- Ge, L.; Zhang, M.; Kenny, S.J.; Liu, D.; Maeda, M.; Saito, K.; Mathur, A.; Xu, K.; Schekman, R. Remodeling of ER -exit sites initiates a membrane supply pathway for autophagosome biogenesis. EMBO Rep. 2017, 18, 1586–1603. [Google Scholar] [CrossRef]
- Jeong, Y.-T.; Simoneschi, D.; Keegan, S.; Melville, D.; Adler, N.S.; Saraf, A.; Florens, L.; Washburn, M.P.; Cavasotto, C.N.; Fenyö, D.; et al. The ULK1-FBXW5-SEC23B nexus controls autophagy. eLife 2018, 7, e42253. [Google Scholar] [CrossRef]
- Gan, W.; Zhang, C.; Siu, K.Y.; Satoh, A.; Tanner, J.A.; Yu, S. ULK1 phosphorylates Sec23A and mediates autophagy-induced inhibition of ER-to-Golgi traffic. BMC Cell Biol. 2017, 18, 22. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Davis, S.; Menon, S.; Zhang, J.; Ding, J.; Cervantes, S.; Miller, E.; Jiang, Y.; Ferro-Novick, S. Ypt1/Rab1 regulates Hrr25/CK1delta kinase activity in ER-Golgi traffic and macroautophagy. J. Cell Biol. 2015, 210, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chen, X.; Xiong, Q.; Chen, Y.; Zhao, H.; Tahir, M.; Song, J.; Zhou, B.; Wang, J. Casein Kinase 1 Family Member CK1delta/Hrr25 Is Required for Autophagosome Completion. Front. Cell Dev. Biol. 2020, 8, 460. [Google Scholar]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeo-stasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Thayanidhi, N.; Helm, J.R.; Nycz, D.C.; Bentley, M.; Liang, Y.; Hay, J.C. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 2010, 21, 1850–1863. [Google Scholar] [CrossRef] [Green Version]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-Synuclein–induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef] [Green Version]
- Glick, B.S.; Nakano, A. Membrane Traffic within the Golgi Apparatus. Annu. Rev. Cell Dev. Biol. 2009, 25, 113–132. [Google Scholar] [CrossRef] [Green Version]
- Klumperman, J. Architecture of the Mammalian Golgi. Cold Spring Harb. Perspect. Biol. 2011, 3, a005181. [Google Scholar] [CrossRef]
- Li, J.; Ahat, E.; Wang, Y. Golgi Structure and Function in Health; Stress, and Diseases. Results Probl. Cell Differ. 2019, 67, 441–485. [Google Scholar]
- Coune, P.G.; Bensadoun, J.C.; Aebischer, P.; Schneider, B.L. Rab1A over-expression prevents Golgi apparatus frag-men-tation and partially corrects motor deficits in an alpha-synuclein based rat model of Parkinson’s disease. J. Parkinsons Dis. 2011, 1, 373–387. [Google Scholar] [CrossRef]
- Gosavi, P.; Gleeson, P.A. The Function of the Golgi Ribbon Structure—An Enduring Mystery Unfolds! Bioessays 2017, 39, 1700063. [Google Scholar] [CrossRef]
- Martínez-Menárguez, J.A.; Tomás, M.; Martínez-Martínez, N.; Martínez-Alonso, E. Golgi Fragmentation in Neuro-degenerative Diseases: Is There a Common Cause? Cells 2019, 8, 748. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Hirling, H. Rescuing Defective Vesicular Trafficking Protects against α-Synuclein Toxicity in Cellular and Animal Models of Parkinson’s Disease. ACS Chem. Biol. 2006, 1, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Sevlever, D.; Jiang, P.; Yen, S.-H.C. Cathepsin D Is the Main Lysosomal Enzyme Involved in the Degradation of α-Synuclein and Generation of Its Carboxy-Terminally Truncated Species. Biochemistry 2008, 47, 9678–9687. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.S.; Nuoffer, C.; Meinkoth, J.L.; McCaffery, M.; Feramisco, J.R.; Balch, W.E.; Farquhar, M.G. A Rab1 mutant affecting guanine nucleotide exchange promotes disassembly of the Golgi apparatus. J. Cell Biol. 1994, 125, 557–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizawa, M.; Fukuda, M. Small GTPase Rab2B and Its Specific Binding Protein Golgi-associated Rab2B Interactor-like 4 (GARI-L4) Regulate Golgi Morphology. J. Biol. Chem. 2015, 290, 22250–22261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galea, G.; Bexiga, M.G.; Panarella, A.; O’Neill, E.D.; Simpson, J.C. A high-content screening microscopy approach to dissect the role of Rab proteins in Golgi-to-ER retrograde trafficking. J. Cell Sci. 2015, 128, 2339–2349. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [Green Version]
- Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol. Dis. 2011, 43, 690–697. [Google Scholar] [CrossRef]
- Friedman, L.G.; Lachenmayer, M.L.; Wang, J.; He, L.; Poulose, S.M.; Komatsu, M.; Holstein, G.R.; Yue, Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J. Neurosci. 2012, 32, 7585–7593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Pantazopoulou, M.; Brembati, V.; Kanellidi, A.; Bousset, L.; Melki, R.; Stefanis, L. Distinct alpha-Synuclein species induced by seeding are selectively cleared by the Lysosome or the Proteasome in neuronally differentiated SH-SY5Y cells. J. Neurochem. 2020, 156, 880–896. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Huang, Y.; Chegini, F.; Chua, G.; Murphy, K.; Gai, W.; Halliday, G.M. Macroautophagy in sporadic and the genetic form of Parkinson’s disease with the A53T alpha-synuclein mutation. Transl. Neurodegener. 2012, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.-F.; Zhang, Y.-J.; Zhou, H.-Y.; Wang, H.-M.; Tian, L.-P.; Liu, J.; Ding, J.-Q.; Chen, S.-D. Curcumin Ameliorates the Neurodegenerative Pathology in A53T α-synuclein Cell Model of Parkinson’s Disease Through the Downregulation of mTOR/p70S6K Signaling and the Recovery of Macroautophagy. J. Neuroimmune Pharmacol. 2013, 8, 356–369. [Google Scholar] [CrossRef]
- Sarkar, S.; Olsen, A.L.; Sygnecka, K.; Lohr, K.M.; Feany, M.B. α-synuclein impairs autophagosome maturation through abnormal actin stabilization. PLoS Genet. 2021, 17, e1009359. [Google Scholar] [CrossRef]
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Carasa, I.F.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [Green Version]
- Alegre-Abarrategui, J.; Christian, H.; Lufino, M.M.; Mutihac, R.; Venda, L.L.; Ansorge, O.; Wade-Martins, R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009, 18, 4022–4034. [Google Scholar] [CrossRef] [Green Version]
- Plowey, E.D.; Cherra, S.J., 3rd; Liu, Y.J.; Chu, C.T. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 2008, 105, 1048–1056. [Google Scholar] [CrossRef] [Green Version]
- Ramonet, D.; Daher, J.P.L.; Lin, B.M.; Stafa, K.; Kim, J.; Banerjee, R.; Westerlund, M.; Pletnikova, O.; Glauser, L.; Yang, L.; et al. Dopaminergic Neuronal Loss, Reduced Neurite Complexity and Autophagic Abnormalities in Transgenic Mice Expressing G2019S Mutant LRRK2. PLoS ONE 2011, 6, e18568. [Google Scholar] [CrossRef]
- Boecker, C.A.; Goldsmith, J.; Dou, D.; Cajka, G.G.; Holzbaur, E.L.F. Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr. Biol. 2021, 31, 2140–2154. [Google Scholar] [CrossRef] [PubMed]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonet-Ponce, L.; Cookson, M.R. The role of Rab GTPases in the pathobiology of Parkinson’ disease. Curr. Opin. Cell Biol. 2019, 59, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Nirujogi, R.S.; Tonelli, F.; Taylor, M.; Lis, P.; Zimprich, A.; Sammler, E.; Alessi, D.R. Development of a multiplexed targeted mass spectrometry assay for LRRK2-phosphorylated Rabs and Ser910/Ser935 biomarker sites. Biochem. J. 2021, 478, 299–326. [Google Scholar] [CrossRef]
- Liu, K.; Shi, N.; Sun, Y.; Zhang, T.; Sun, X. Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem. Res. 2013, 38, 201–207. [Google Scholar] [CrossRef]
- Lizama, B.N.; Chu, C.T. Neuronal autophagy and mitophagy in Parkinson’s disease. Mol. Asp. Med. 2021, 100972, in press. [Google Scholar] [CrossRef]
- Savitt, D.; Jankovic, J. Targeting α-Synuclein in Parkinson’s Disease: Progress Towards the Development of Disease-Modifying Therapeutics. Drugs 2019, 79, 797–810. [Google Scholar] [CrossRef]
- Lukman, S.; Nguyen, M.N.; Sim, K.; Teo, J.C. Discovery of Rab1 binding sites using an ensemble of clustering methods. Proteins Struct. Funct. Bioinform. 2017, 85, 859–871. [Google Scholar] [CrossRef]
- Fleming, J.; Outeiro, T.F.; Slack, M.; Lindquist, S.L.; Bulawa, C.E. Detection of Compounds That Rescue Rab1-Synuclein. Toxicity 2008, 439, 339–351. [Google Scholar] [CrossRef]
- Gray, J.L.; Von Delft, F.; Brennan, P.E. Targeting the Small GTPase Superfamily through Their Regulatory Proteins. Angew. Chem. Int. Ed. 2019, 59, 6342–6366. [Google Scholar] [CrossRef] [Green Version]
- Gendaszewska-Darmach, E.; Garstka, M.A.; Błażewska, K.M. Targeting Small GTPases and Their Prenylation in Diabetes Mellitus. J. Med. Chem. 2021, 64, 9677–9710. [Google Scholar] [CrossRef]
- Carroll, C.B.; Wyse, R.K.H. Simvastatin as a Potential Disease-Modifying Therapy for Patients with Parkinson’s Disease: Rationale for Clinical Trial, and Current Progress. J. Parkinsons Dis. 2017, 7, 545–568. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Menárguez, J.Á.; Martínez-Alonso, E.; Cara-Esteban, M.; Tomás, M. Focus on the Small GTPase Rab1: A Key Player in the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 12087. https://doi.org/10.3390/ijms222112087
Martínez-Menárguez JÁ, Martínez-Alonso E, Cara-Esteban M, Tomás M. Focus on the Small GTPase Rab1: A Key Player in the Pathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(21):12087. https://doi.org/10.3390/ijms222112087
Chicago/Turabian StyleMartínez-Menárguez, José Ángel, Emma Martínez-Alonso, Mireia Cara-Esteban, and Mónica Tomás. 2021. "Focus on the Small GTPase Rab1: A Key Player in the Pathogenesis of Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 21: 12087. https://doi.org/10.3390/ijms222112087