
Bisphenol a Induces Autophagy Defects and AIF-Dependent Apoptosis via HO-1 and AMPK to Degenerate N2a Neurons


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
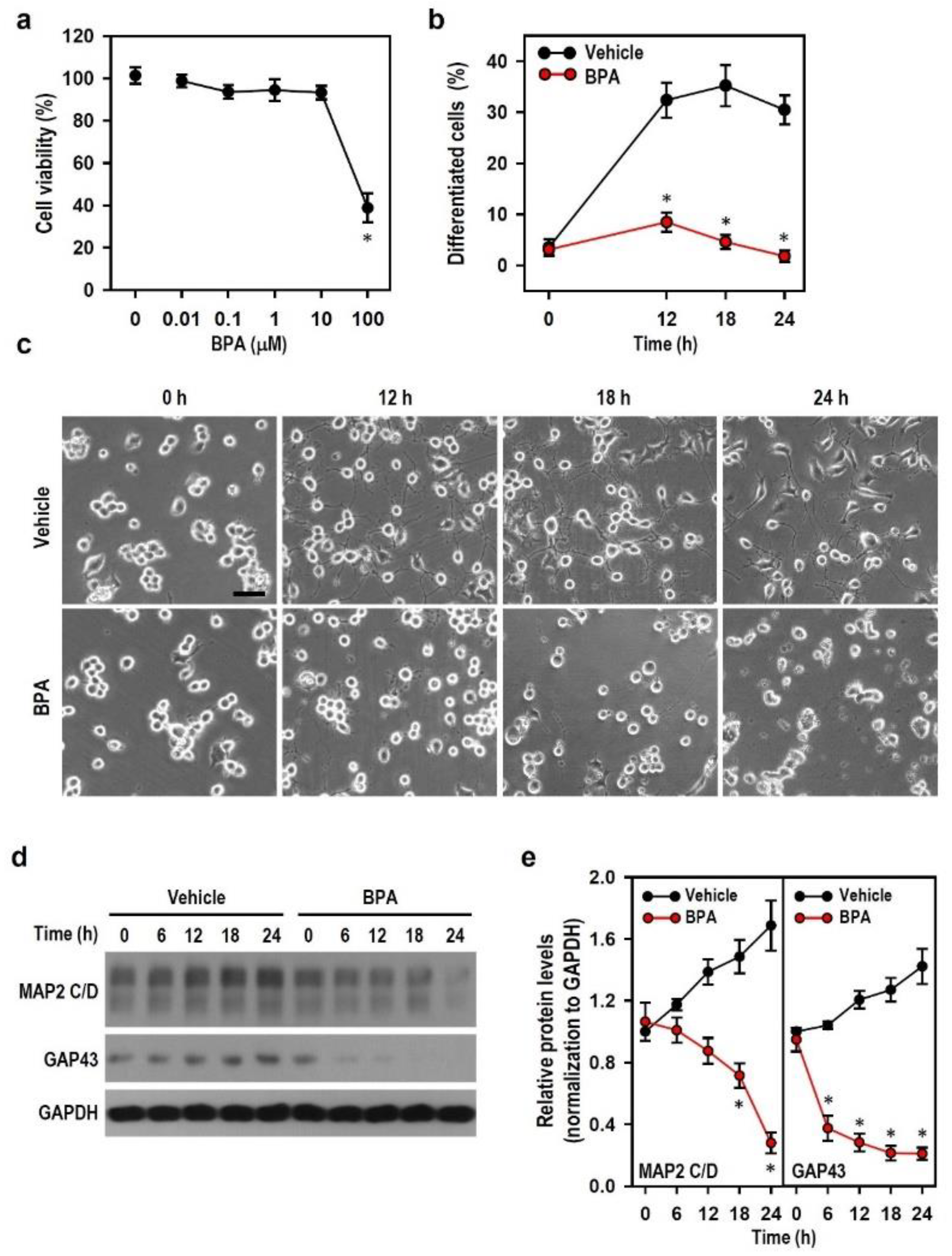
2.1. BPA Exerts Neurotoxic Effect to Impair the Development of N2a Neurons
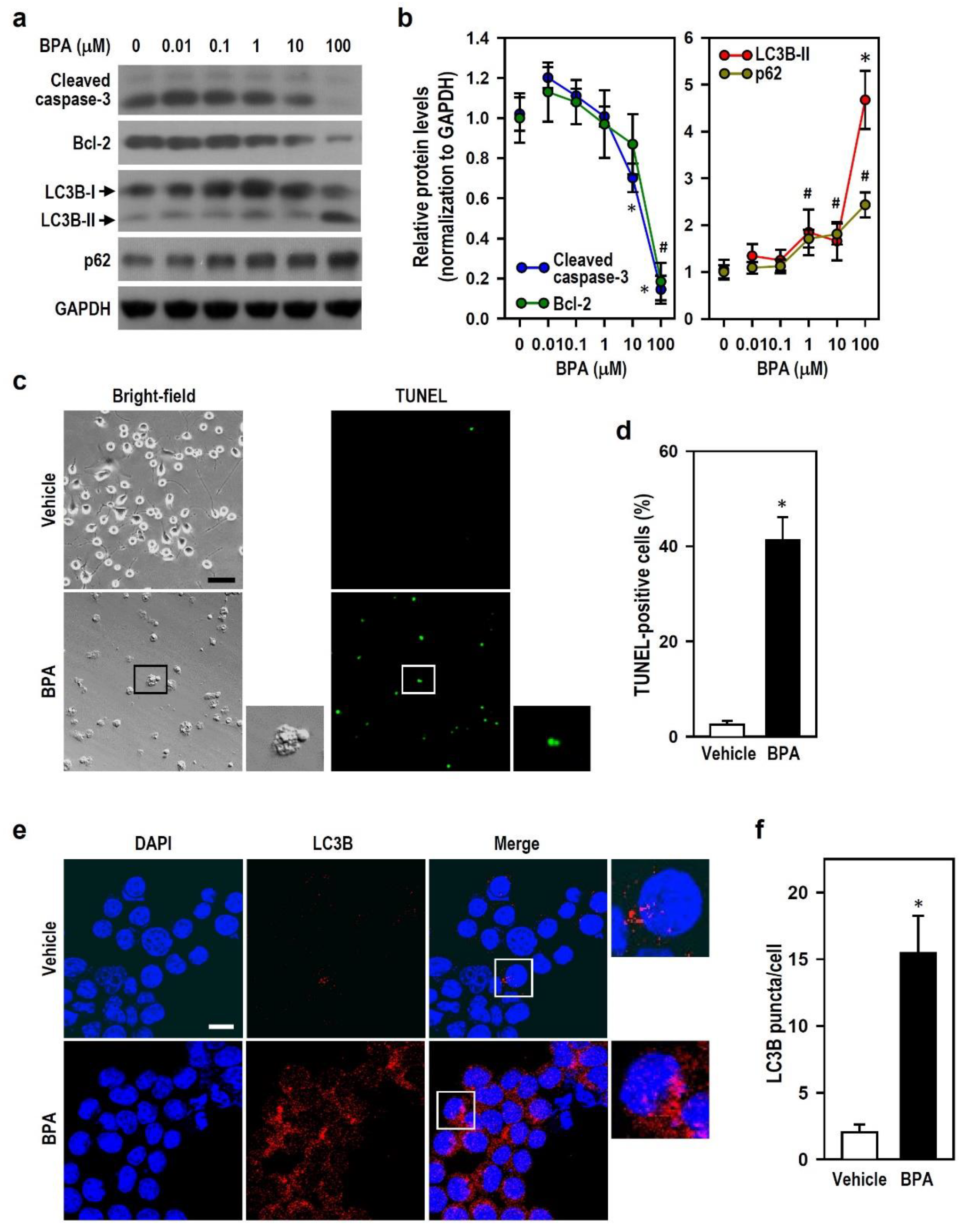
2.2. BPA Blocks Autophagy Flux and Induces Caspase-3-Independent Apoptosis in N2a Neurons
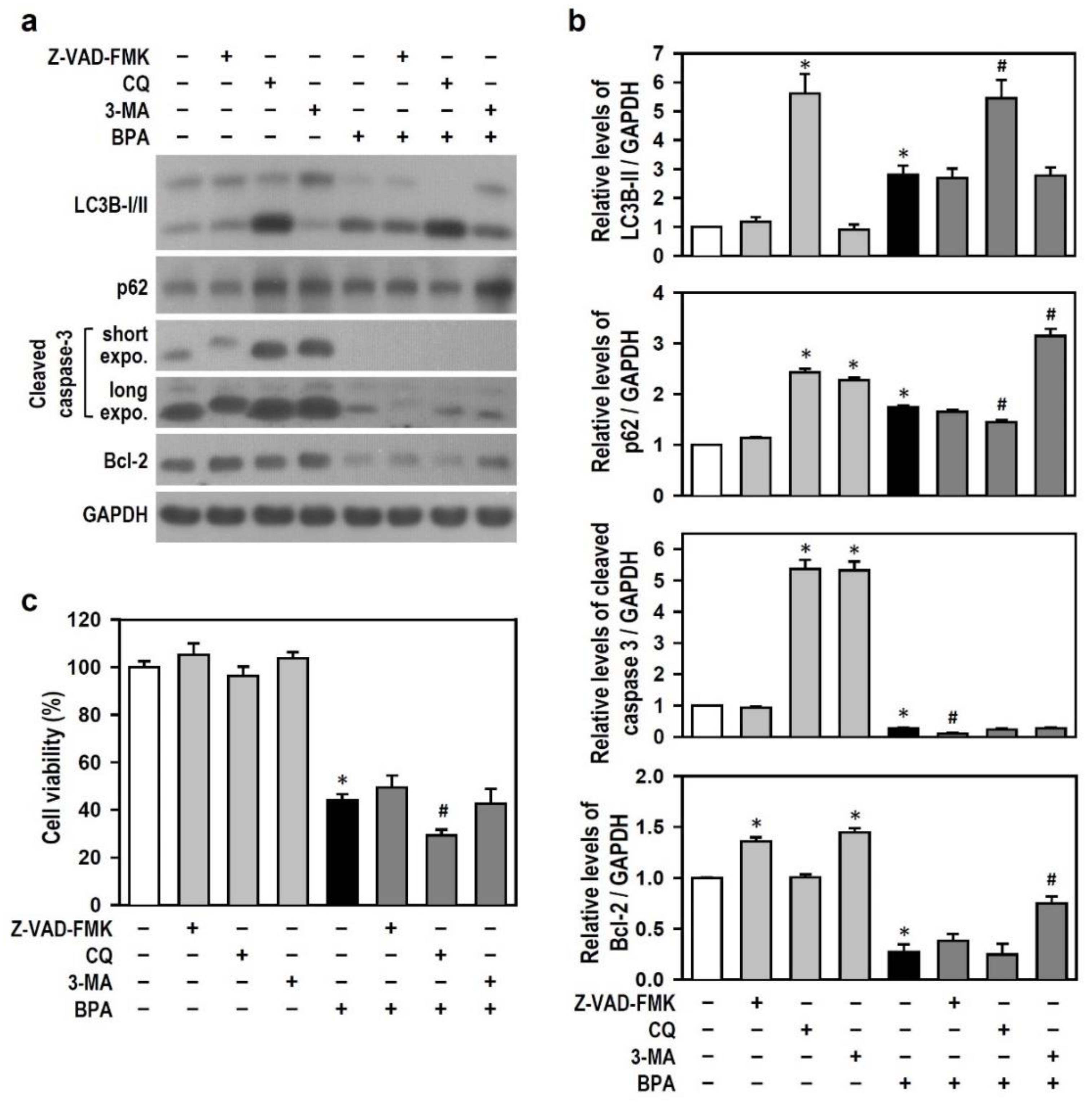
2.3. BPA Inhibits the Generation of Active Caspase-3, and BPA-Induced Autophagy Flux Inhibition Involves the Increases in LC3B-II and p62 Production
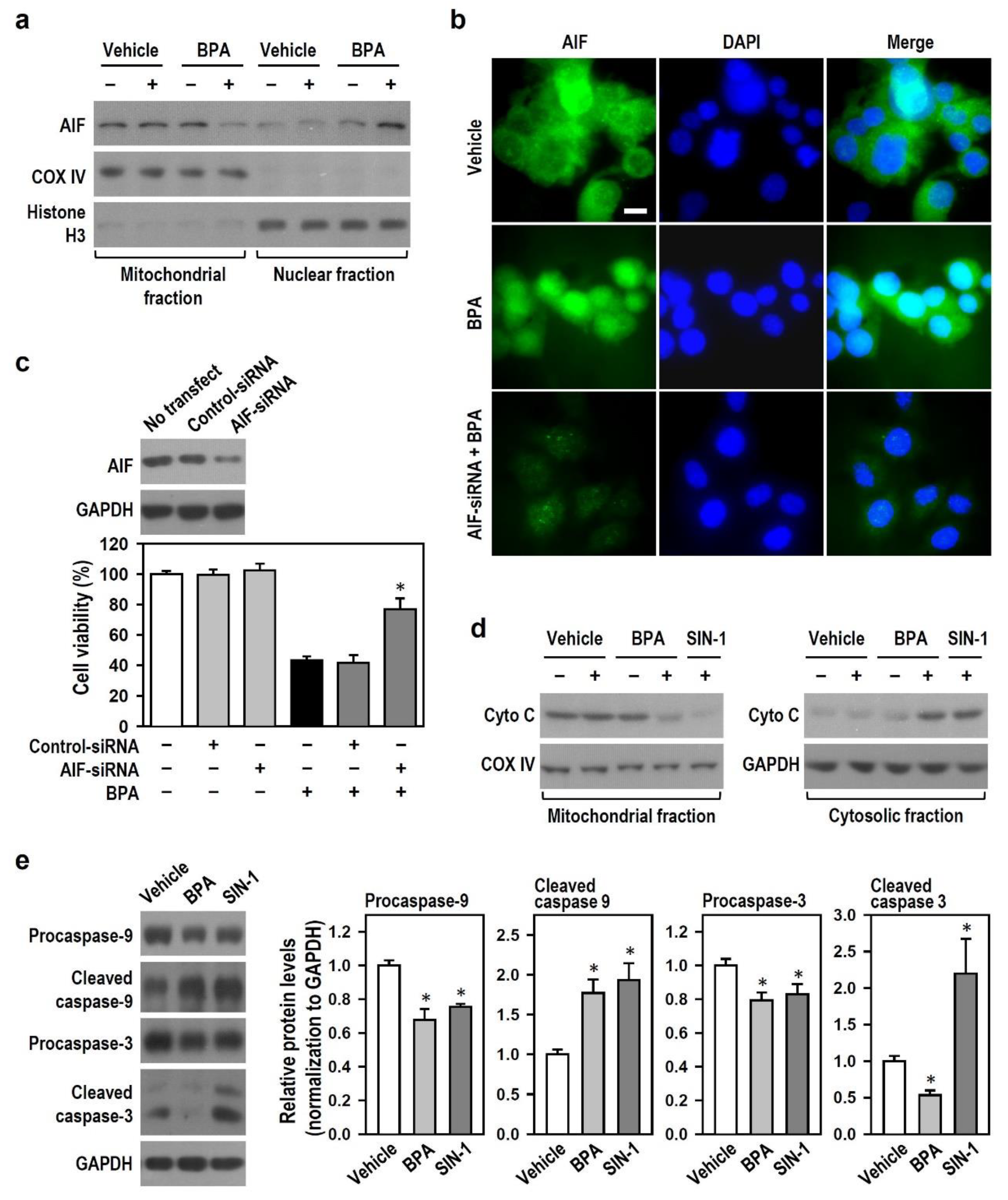
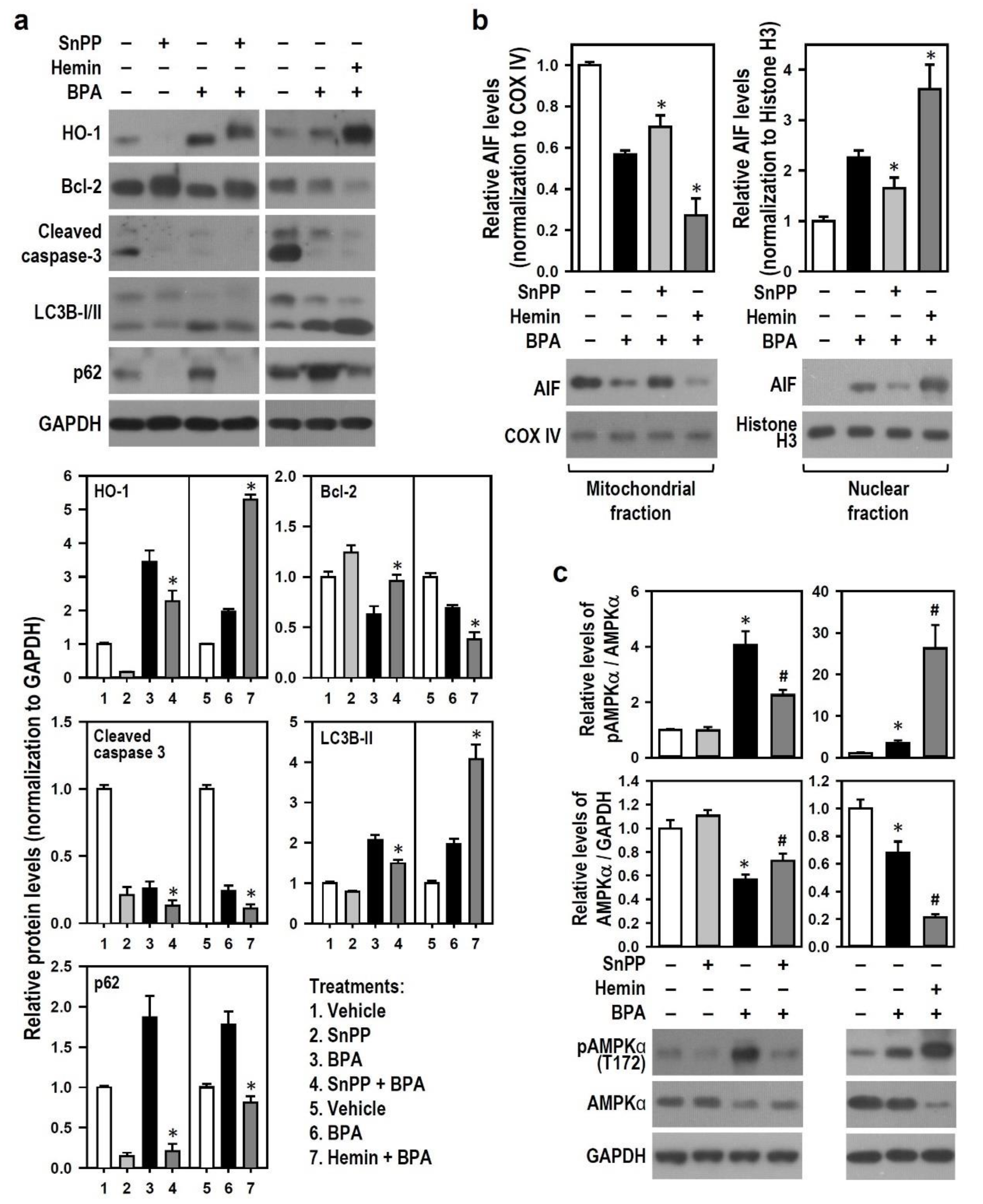
2.4. BPA Shuts Down Caspase-3-Driven Apoptosis and Activates AIF-Related Apoptosis in N2a Neurons
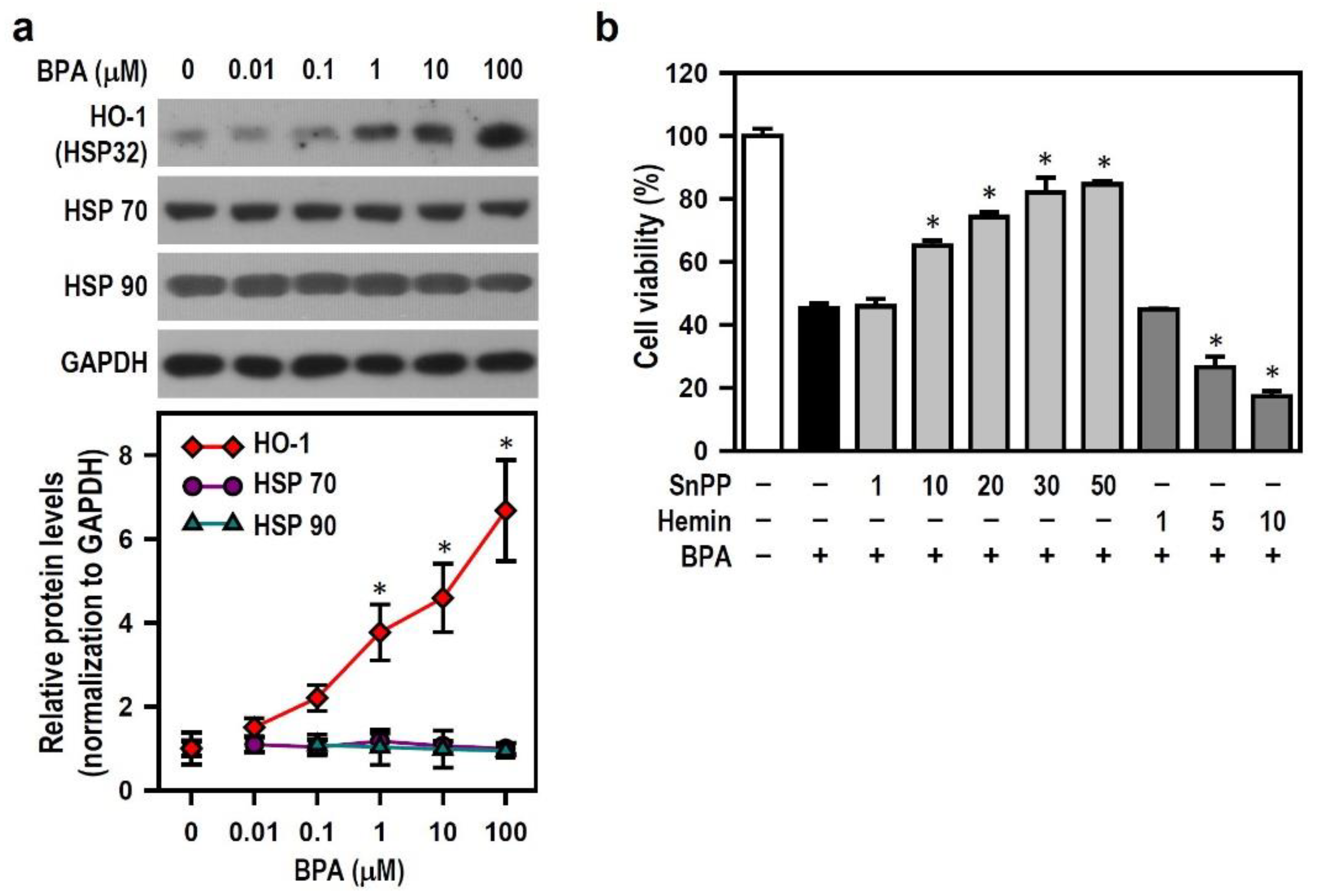
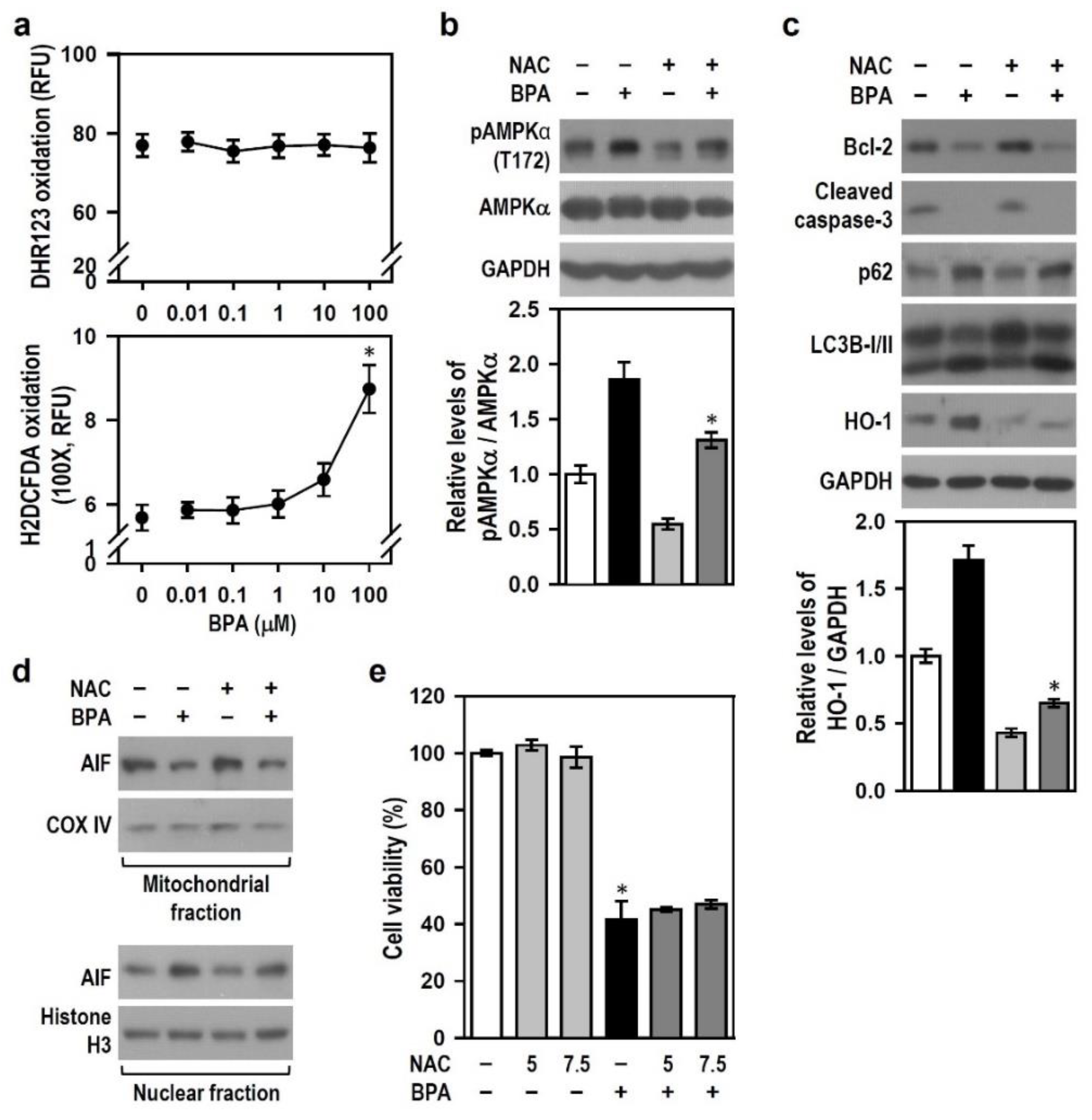
2.5. BPA-Induced HO-1 Mediates N2a Neurotoxicity via AMPK Activation, Autophagy Initiation, AIF-Dependent Apoptosis and Bcl-2 Down-Regulation
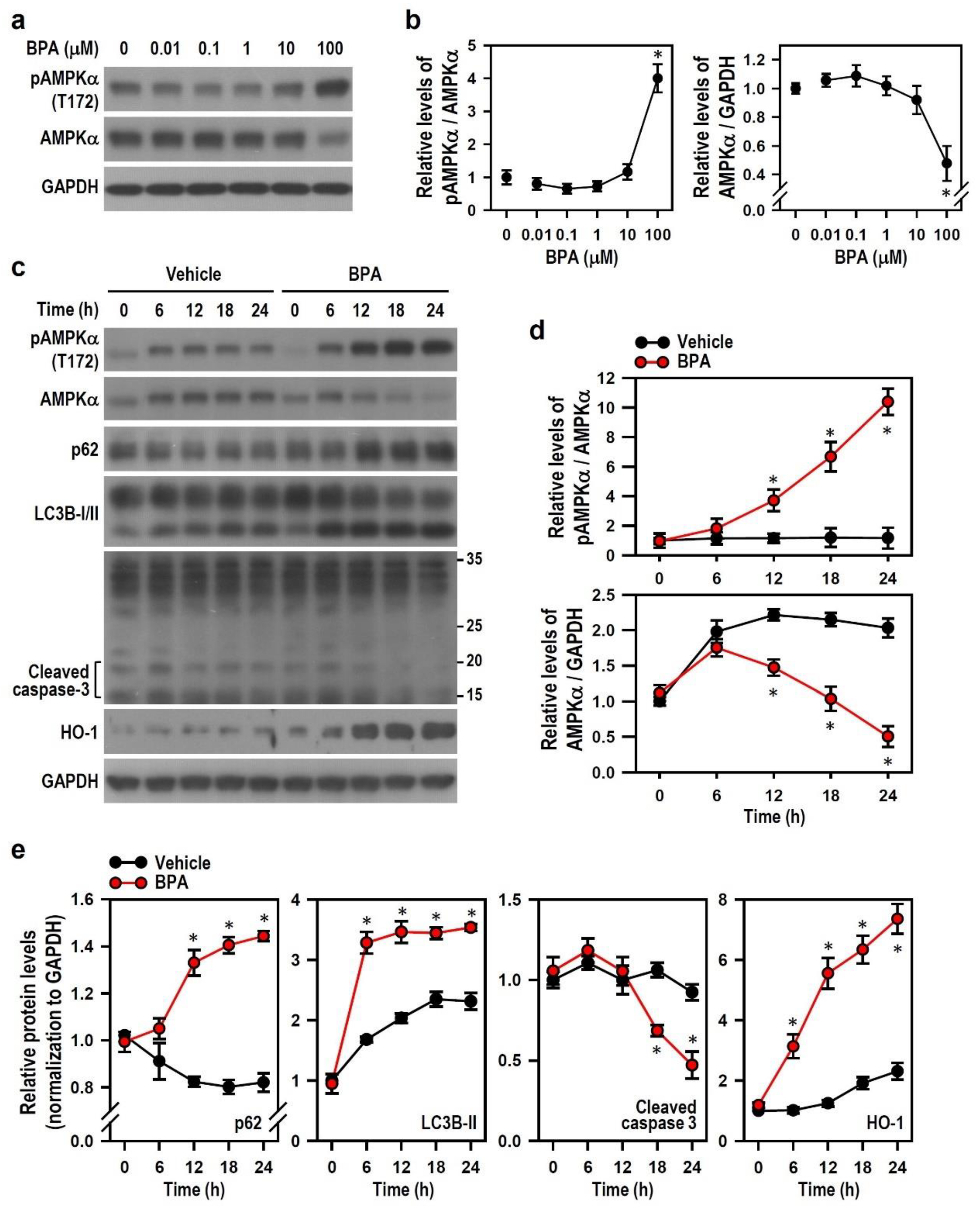
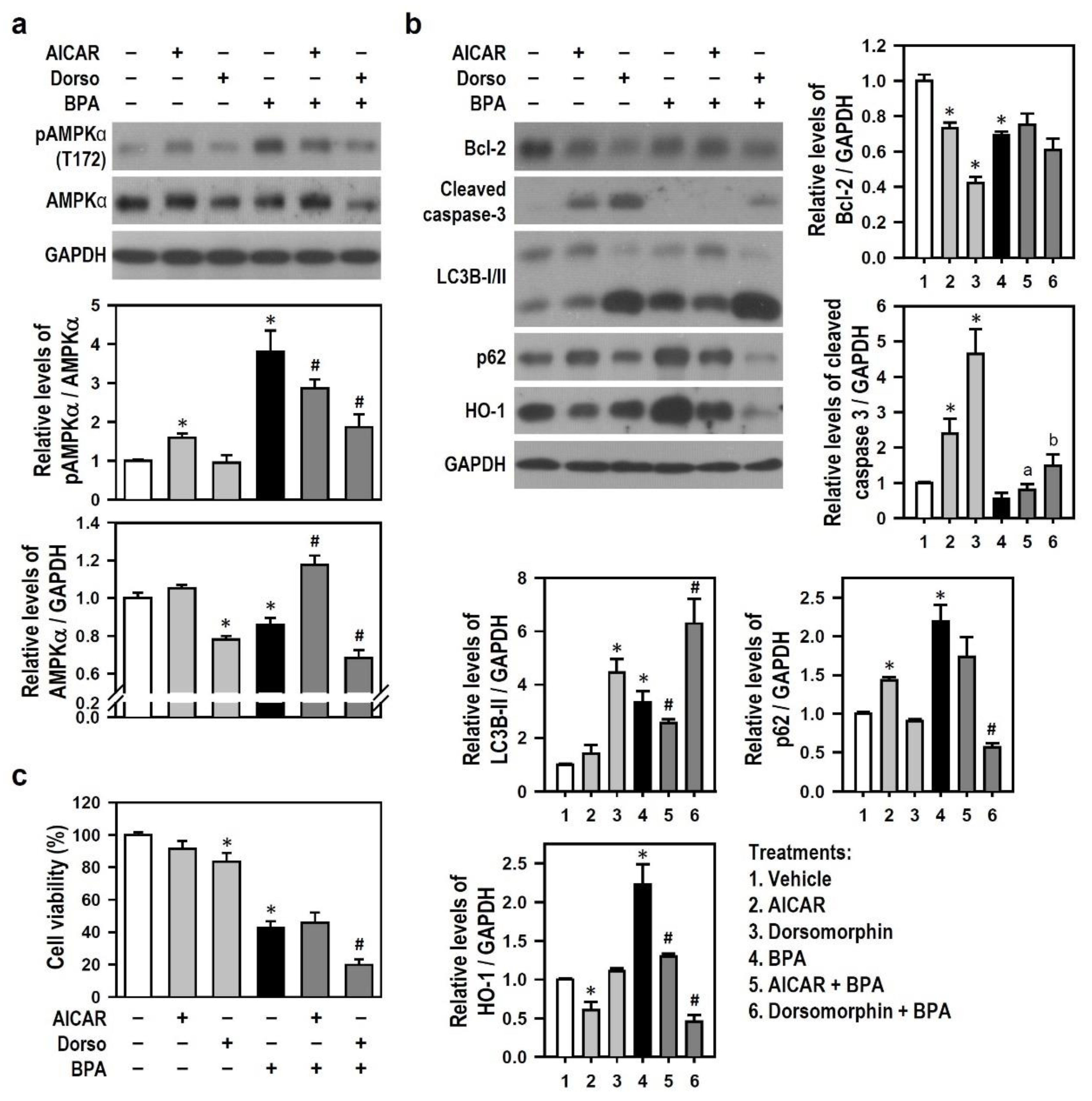
2.6. BPA-Activated AMPK Reduces HO-1 Induction and Autophagy Initiation
2.7. Reactive Oxygen Species (ROS) Induced by BPA Involve AMPK Activation and HO-1 Induction but Does Not Mediate Apoptosis and Autophagy Flux Inhibition
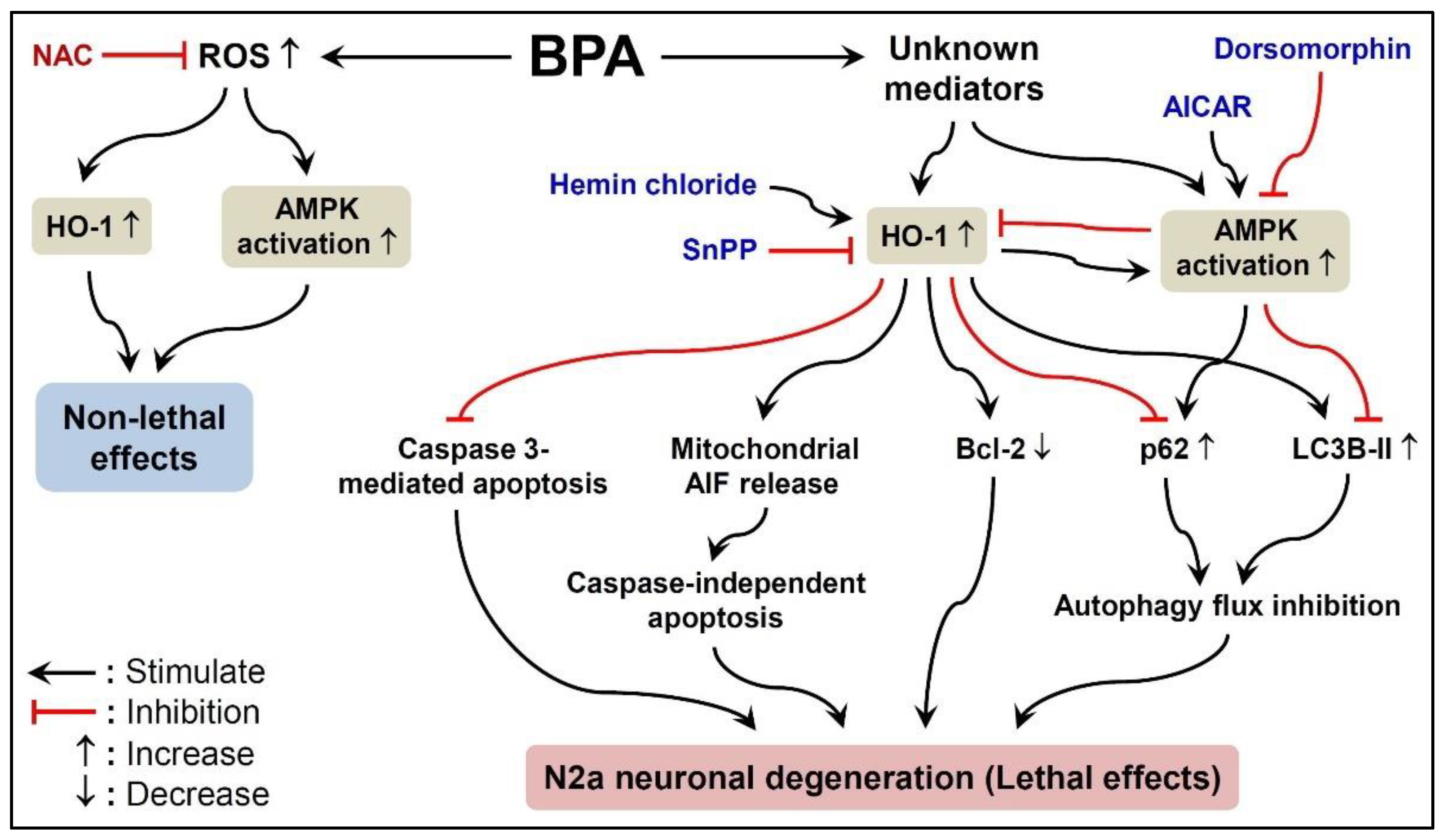
3. Discussion
4. Materials and Methods
4.1. Chemicals, Antibodies, Kits and Reagents
4.2. Cell Culture and Induction of Neuronal Differentiation
4.3. Quantification of Axon Outgrowth
4.4. 3-(4,5-Dimethylthianol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) Reduction Assay
4.5. Determination of Intracellular Oxygen Free Radicals
4.6. TUNEL Assay
4.7. Immunofluorescence Staining
4.8. RNAi and Transient Transfection
4.9. Subcellular Fractionation
4.10. Western Blots
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abraham, A.; Chakraborty, P. A review on sources and health impacts of bisphenol A. Rev. Environ. Health 2020, 35, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Sonavane, M.; Gassman, N.R. Bisphenol A co-exposure effects: A key factor in understanding BPA’s complex mechanism and health outcomes. Crit. Rev. Toxicol. 2019, 49, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.S. Bisphenol A: An endocrine disruptor with widespread exposure and multiple effects. J. Steroid Biochem. Mol. Biol. 2011, 127, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Chahoud, I.; Heindel, J.J.; Padmanabhan, V.; Paumgartten, F.J.; Schoenfelder, G. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Environ. Health Perspect. 2010, 118, 1055–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, A.; Chianese, R.; Troisi, J.; Richards, S.; Nori, S.L.; Fasano, S.; Guida, M.; Plunk, E.; Viggiano, A.; Pierantoni, R.; et al. Neuro-toxic and reproductive effects of BPA. Curr. Neuropharmacol. 2019, 17, 1109–1132. [Google Scholar] [CrossRef] [PubMed]
- Zeliger, H.I. Exposure to lipophilic chemicals as a cause of neurological impairments, neurodevelopmental disorders and neurodegenerative diseases. Interdiscip. Toxicol. 2013, 6, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Matsuki, N.; Ohno, Y.; Nakazawa, K. Effects of 17beta-estradiol and xenoestrogens on the neuronal survival in an organotypic hippocampal culture. Neuroendocrinology 2002, 76, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.-H.; Baek, J.-H.; Lee, S.-Y.; Jang, C.-G. Prenatal and postnatal exposure to bisphenol a induces anxiolytic behaviors and cognitive deficits in mice. Synapse 2010, 64, 432–439. [Google Scholar] [CrossRef]
- Zhang, H.; Kuang, H.; Luo, Y.; Liu, S.; Meng, L.; Pang, Q.; Fan, R. Low-dose bisphenol A exposure impairs learning and memory ability with alterations of neuromorphology and neurotransmitters in rats. Sci. Total Environ. 2019, 697, 134036. [Google Scholar] [CrossRef]
- Wang, T.; Xie, C.; Yu, P.; Fang, F.; Zhu, J.; Cheng, J.; Gu, A.; Wang, J.; Xiao, H. Involvement of insulin signaling disturbances in bisphenol a-induced alzheimer’s disease-like neurotoxicity. Sci. Rep. 2017, 7, 7497. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Fang, F.; Chen, D.; Gao, Y.; Liu, J.; Gao, R.; Wang, J.; Xiao, H. Bisphenol A disrupts glucose transport and neurophysiological role of IR/IRS/AKT/GSK3beta axis in the brain of male mice. Environ. Toxicol. Pharmacol. 2016, 43, 7–12. [Google Scholar] [CrossRef]
- Fan, J.; Dawson, T.M.; Dawson, V.L. Cell death mechanisms of neurodegeneration. Adv. Neurobiol. 2017, 15, 403–425. [Google Scholar]
- Ryu, J.R.; Hong, C.J.; Kim, J.Y.; Kim, E.-K.; Sun, W.; Yu, S.-W. Control of adult neurogenesis by programmed cell death in the mammalian brain. Mol. Brain 2016, 9, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Yosef, T.; Damri, O.; Agam, G. Dual Role of autophagy in diseases of the central nervous system. Front. Cell. Neurosci. 2019, 13, 196. [Google Scholar] [CrossRef] [PubMed]
- Napoletano, F.; Baron, O.; Vandenabeele, P.; Mollereau, B.; Fanto, M. Intersections between Regulated cell death and autophagy. Trends Cell Biol. 2019, 29, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Jazwa, A.; Cuadrado, A. Targeting heme oxygenase-1 for neuroprotection and neuroinflammation in neurodegenerative diseases. Curr. Drug Targets 2010, 11, 1517–1531. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the nervous system: Does it favor neuronal cell survival or induce neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Peng, Y.; Hui, Y.; Zhang, S.; Zhou, Y.; Li, D.; Li, J.; Si, Z.; Li, J.; Wang, D.; et al. Overexpression of heme oxygenase 1 impairs cognitive ability and changes the plasticity of the synapse. J. Alzheimer’s Dis. 2015, 47, 595–608. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Rau, C.-S.; Hsieh, M.-W.; Chen, Y.-C.; Jeng, S.-F.; Lu, T.-H.; Chen, S.-S. Simvastatin-induced heme oxygenase-1 increases apoptosis of neuro 2A cells in response to glucose deprivation. Toxicol. Sci. 2007, 101, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Lacoste, B.; Pistell, P.J.; Ingram, D.K.; Hamel, E.; Alaoui-Jamali, M.A.; Szarek, W.A.; Vlahakis, J.Z.; Jie, S.; Song, W.; et al. Neurotherapeutic effects of novel HO-1 inhibitors in vitro and in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2014, 131, 778–790. [Google Scholar] [CrossRef]
- Dong, C.; Zheng, H.; Huang, S.; You, N.; Xu, J.; Ye, X.; Zhu, Q.; Feng, Y.; You, Q.; Miao, H.; et al. Heme oxygenase-1 enhances autophagy in podocytes as a protective mechanism against high glucose-induced apoptosis. Exp. Cell Res. 2015, 337, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, L.; Qiao, Y.; Zhou, X.; Wu, G.; Wang, L.; Peng, Y.; Dong, X.; Huang, H.; Si, L.; et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS ONE 2013, 8, e75927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domise, M.; Vingtdeux, V. AMPK in neurodegenerative diseases. Exp. Suppl. 2016, 107, 153–177. [Google Scholar] [CrossRef]
- Villanueva-Paz, M.; Cotán, D.; Garrido-Maraver, J.; Oropesa-Ávila, M.; de la Mata, M.; Delgado-Pavón, A.; de Lavera, I.; Alcocer-Gómez, E.; Álvarez-Córdoba, M.; Sánchez-Alcázar, J.A. AMPK Regulation of cell growth, apoptosis, autophagy, and bioenergetics. Exp. Suppl. 2016, 107, 45–71. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Saito, M.; Das, B.C. Involvement of AMP-activated protein kinase in neuroinflammation and neurodegeneration in the adult and developing brain. Int. J. Dev. Neurosci. 2019, 77, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Poels, J.; Spasić, M.R.; Callaerts, P.; Norga, K.K. Expanding roles for AMP-activated protein kinase in neuronal survival and autophagy. BioEssays 2009, 31, 944–952. [Google Scholar] [CrossRef]
- Shah, S.Z.A.; Zhao, D.; Hussain, T.; Yang, L. Role of the AMPK pathway in promoting autophagic flux via modulating mitochondrial dynamics in neurodegenerative diseases: Insight into prion diseases. Ageing Res. Rev. 2017, 40, 51–63. [Google Scholar] [CrossRef]
- Tavakol, S.; Ashrafizadeh, M.; Deng, S.; Azarian, M.; Abdoli, A.; Motavaf, M.; Poormoghadam, D.; Khanbabaei, H.; Afshar, E.G.; Mandegary, A.; et al. Autophagy modulators: Mechanistic aspects and drug delivery systems. Biomolecules 2019, 9, 530. [Google Scholar] [CrossRef] [Green Version]
- Rusten, T.E.; Stenmark, H. p62, an autophagy hero or culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef]
- Gil, R.S.; Ooi, L.; Yerbury, J.J.; Ecroyd, H. The heat shock response in neurons and astroglia and its role in neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 65. [Google Scholar] [CrossRef]
- Agarwal, S.; Tiwari, S.K.; Seth, B.; Yadav, A.; Singh, A.; Mudawal, A.; Chauhan, L.K.; Gupta, S.K.; Choubey, V.; Tripathi, A.; et al. Activation of Autophagic flux against xenoestrogen bisphenol-a-induced hippocampal neurodegeneration via AMP kinase (AMPK)/mammalian target of rapamycin (mTOR) pathways. J. Biol. Chem. 2015, 290, 21163–21184. [Google Scholar] [CrossRef] [Green Version]
- Goto, A.; Egawa, T.; Sakon, I.; Oshima, R.; Ito, K.; Serizawa, Y.; Sekine, K.; Tsuda, S.; Goto, K.; Hayashi, T. Heat stress acutely activates insulin-independent glucose transport and 5′-AMP-activated protein kinase prior to an increase in HSP72 protein in rat skeletal muscle. Physiol. Rep. 2015, 3, e12601. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.M.; Gillespie, J.G.; Hardie, G. Role of the AMP-activated protein kinase in the cellular stress response. Curr. Biol. 1994, 4, 315–324. [Google Scholar] [CrossRef]
- Huang, F.-M.; Chang, Y.-C.; Lee, S.-S.; Ho, Y.-C.; Yang, M.-L.; Lin, H.-W.; Kuan, Y.-H. Bisphenol A exhibits cytotoxic or genotoxic potential via oxidative stress-associated mitochondrial apoptotic pathway in murine macrophages. Food Chem. Toxicol. 2018, 122, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Kumar, C.M.; Suranagi, U.D.; Mediratta, P.K. Protective effect of N-acetylcysteine on bisphenol A-induced cognitive dysfunction and oxidative stress in rats. Food Chem. Toxicol. 2011, 49, 1404–1409. [Google Scholar] [CrossRef]
- Agarwal, S.; Yadav, A.; Tiwari, S.K.; Seth, B.; Chauhan, L.K.; Khare, P.; Ray, R.S.; Chaturvedi, R.K. Dynamin-related protein 1 inhibition mitigates bisphenol A-mediated alterations in mitochondrial dynamics and neural stem cell proliferation and differentiation. J. Biol. Chem. 2016, 291, 15923–15939. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.K.; Agarwal, S.; Seth, B.; Yadav, A.; Ray, R.S.; Mishra, V.N.; Chaturvedi, R.K. Inhibitory effects of bisphenol-a on neural stem cells proliferation and differentiation in the rat brain are dependent on wnt/beta-catenin pathway. Mol. Neurobiol. 2015, 52, 1735–1757. [Google Scholar] [CrossRef]
- Tiwari, S.K.; Agarwal, S.; Tripathi, A.; Chaturvedi, R.K. Bisphenol—A mediated inhibition of hippocampal neurogenesis attenuated by curcumin via canonical Wnt pathway. Mol. Neurobiol. 2016, 53, 3010–3029. [Google Scholar] [CrossRef]
- Sevrioukova, I.F. Apoptosis-Inducing factor: Structure, function, and redox regulation. Antioxid. Redox Signal. 2011, 14, 2545–2579. [Google Scholar] [CrossRef] [Green Version]
- Terasaka, H.; Kadoma, Y.; Sakagami, H.; Fujisawa, S. Cytotoxicity and apoptosis-inducing activity of bisphenol A and hydroquinone in HL-60 cells. Anticancer. Res. 2005, 25, 2241–2247. [Google Scholar] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Higdon, A.N.; Benavides, G.A.; Chacko, B.K.; Ouyang, X.; Johnson, M.S.; Landar, A.; Zhang, J.; Darley-Usmar, V.M. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: The protective role of autophagy. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1394–H1409. [Google Scholar] [CrossRef] [Green Version]
- Gyongyosi, A.; Szoke, K.; Fenyvesi, F.; Fejes, Z.; Debreceni, I.B.; Nagy, B.; Tosaki, A.; Lekli, I. Inhibited autophagy may contribute to heme toxicity in cardiomyoblast cells. Biochem. Biophys. Res. Commun. 2019, 511, 732–738. [Google Scholar] [CrossRef]
- Schipper, H.M.; Cissé, S.; Stopa, E.G. Expression of heme oxygenase-1 in the senescent and Alzheimer’s-diseased brain. Ann. Neurol. 1995, 37, 758–768. [Google Scholar] [CrossRef]
- Schipper, H.M.; Bennett, D.A.; Liberman, A.; Bienias, J.L.; Schneider, J.A.; Kelly, J.; Arvanitakis, Z. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 252–261. [Google Scholar] [CrossRef]
- Schipper, H.M.; Liberman, A.; Stopa, E.G. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem. 2009, 110, 469–485. [Google Scholar] [CrossRef]
- Song, W.; Zukor, H.; Lin, S.-H.; Liberman, A.; Tavitian, A.; Mui, J.; Vali, H.; Fillebeen, C.; Pantopoulos, K.; Wu, T.-D.; et al. Unregulated brain iron deposition in transgenic mice over-expressingHMOX1 in the astrocytic compartment. J. Neurochem. 2012, 123, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Su, H.; Song, S.; Paudel, H.K.; Schipper, H.M. Over-expression of heme oxygenase-1 promotes oxidative mitochondrial damage in rat astroglia. J. Cell. Physiol. 2006, 206, 655–663. [Google Scholar] [CrossRef]
- Suttner, D.M.; Dennery, P.A. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999, 13, 1800–1809. [Google Scholar] [CrossRef]
- Tsou, Y.-H.; Shih, C.-T.; Ching, C.-H.; Huang, J.-Y.; Jen, C.J.; Yu, L.; Kuo, Y.-M.; Wu, F.-S.; Chuang, J.-I. Treadmill exercise activates Nrf2 antioxidant system to protect the nigrostriatal dopaminergic neurons from MPP+ toxicity. Exp. Neurol. 2015, 263, 50–62. [Google Scholar] [CrossRef]
- Yang, C.-M.; Lin, C.-C.; Hsieh, H.-L. High-glucose-derived oxidative stress-dependent heme oxygenase-1 expression from astrocytes contributes to the neuronal apoptosis. Mol. Neurobiol. 2017, 54, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.G.; Ikram, M.; Jo, M.H.; Yoo, L.; Chung, K.C.; Nah, S.-Y.; Hwang, H.; Rhim, H.; Kim, M.O. Gintonin mitigates MPTP-induced loss of nigrostriatal dopaminergic neurons and accumulation of α-Synuclein via the Nrf2/HO-1 pathway. Mol. Neurobiol. 2018, 56, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Hu, Y.; Liu, L.; Cai, J.; Peng, C.; Li, Q. Gastrodin protects against MPP+-induced oxidative stress by up regulates heme oxygenase-1 expression through p38 MAPK/Nrf2 pathway in human dopaminergic cells. Neurochem. Int. 2014, 75, 79–88. [Google Scholar] [CrossRef]
- Vucicevic, L.; Misirkic, M.; Kristina, J.; Vilimanovich, U.; Sudar, E.; Isenovic, E.; Prica, M.; Harhaji-Trajkovic, L.; Stevovic, T.K.; Vladimir, B.; et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy 2011, 7, 40–50. [Google Scholar] [CrossRef]
- Liu, X.; Chhipa, R.R.; Nakano, I.; Dasgupta, B. The AMPK inhibitor compound C Is a Potent AMPK-independent antiglioma agent. Mol. Cancer Ther. 2014, 13, 596–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.-L.; Bs, W.P.; Liou, D.; Marambaud, P.; Wang, P. AMPK inhibitor compound C suppresses cell proliferation by induction of apoptosis and autophagy in human colorectal cancer cells. J. Surg. Oncol. 2012, 106, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-M.; Peyton, K.J.; Shebib, A.R.; Wang, H.; Korthuis, R.J.; Durante, W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H84–H93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, E.; Dopheide, J.; Schuhmacher, S.; Thomas, S.R.; Chen, K.; Daiber, A.; Wenzel, P.; Munzel, T.; Keaney, J.F. Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation 2008, 118, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Tsogbadrakh, B.; Ryu, H.; Ju, K.D.; Lee, J.; Yun, S.; Yu, K.-S.; Kim, H.J.; Ahn, C.; Oh, K.-H. AICAR, an AMPK activator, protects against cisplatin-induced acute kidney injury through the JAK/STAT/SOCS pathway. Biochem. Biophys. Res. Commun. 2019, 509, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Xin, F.; Jiang, L.; Liu, X.; Geng, C.; Wang, W.; Zhong, L.; Yang, G.; Chen, M. Bisphenol A induces oxidative stress-associated DNA damage in INS-1 cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 769, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Asahi, J.; Kamo, H.; Baba, R.; Doi, Y.; Yamashita, A.; Murakami, D.; Hanada, A.; Hirano, T. Bisphenol A induces endoplasmic reticulum stress-associated apoptosis in mouse non-parenchymal hepatocytes. Life Sci. 2010, 87, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.K.; Sharma, A.; Singh, N.; Kakkar, P. Activation of autophagic flux via LKB1/AMPK/mTOR axis against xenoestrogen Bisphenol-A exposure in primary rat hepatocytes. Food Chem. Toxicol. 2020, 141, 111314. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-T.; Hsieh, C.-F.; Wang, J.-Y. Bisphenol a Induces Autophagy Defects and AIF-Dependent Apoptosis via HO-1 and AMPK to Degenerate N2a Neurons. Int. J. Mol. Sci. 2021, 22, 10948. https://doi.org/10.3390/ijms222010948
Lee C-T, Hsieh C-F, Wang J-Y. Bisphenol a Induces Autophagy Defects and AIF-Dependent Apoptosis via HO-1 and AMPK to Degenerate N2a Neurons. International Journal of Molecular Sciences. 2021; 22(20):10948. https://doi.org/10.3390/ijms222010948
Chicago/Turabian StyleLee, Ching-Tien, Cheng-Fang Hsieh, and Jiz-Yuh Wang. 2021. "Bisphenol a Induces Autophagy Defects and AIF-Dependent Apoptosis via HO-1 and AMPK to Degenerate N2a Neurons" International Journal of Molecular Sciences 22, no. 20: 10948. https://doi.org/10.3390/ijms222010948