Links between Infections, Lung Cancer, and the Immune System


 ,
,  , ,
, ,  , and
, and
Abstract
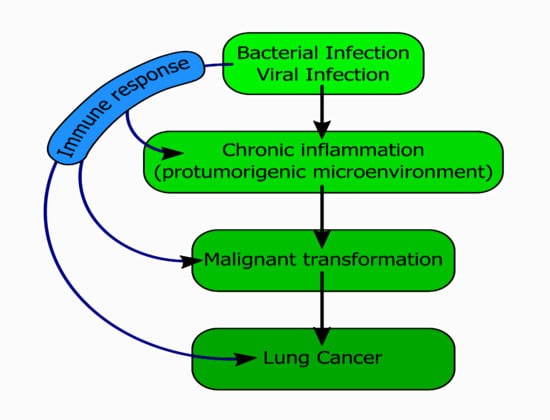
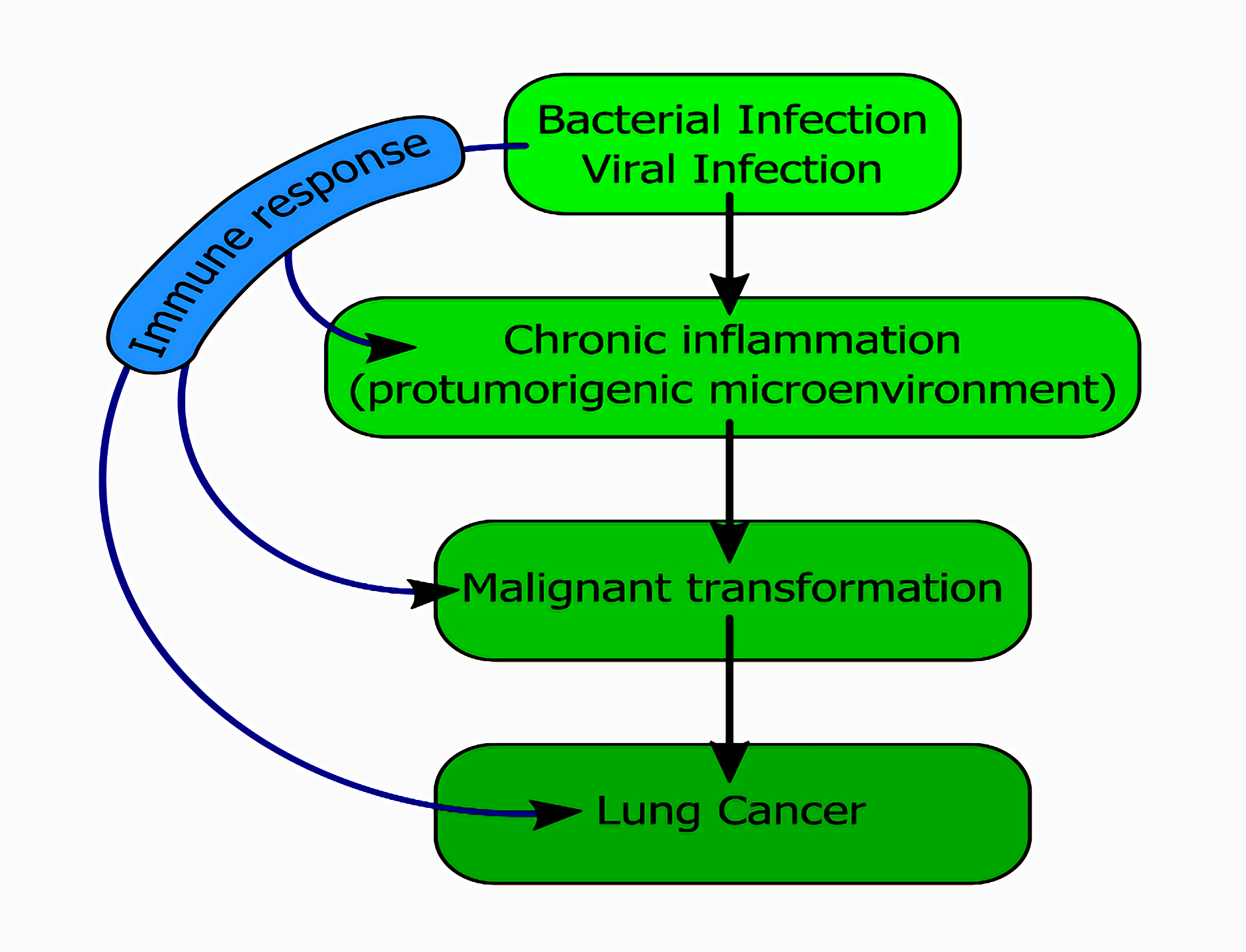
:
1. Introduction
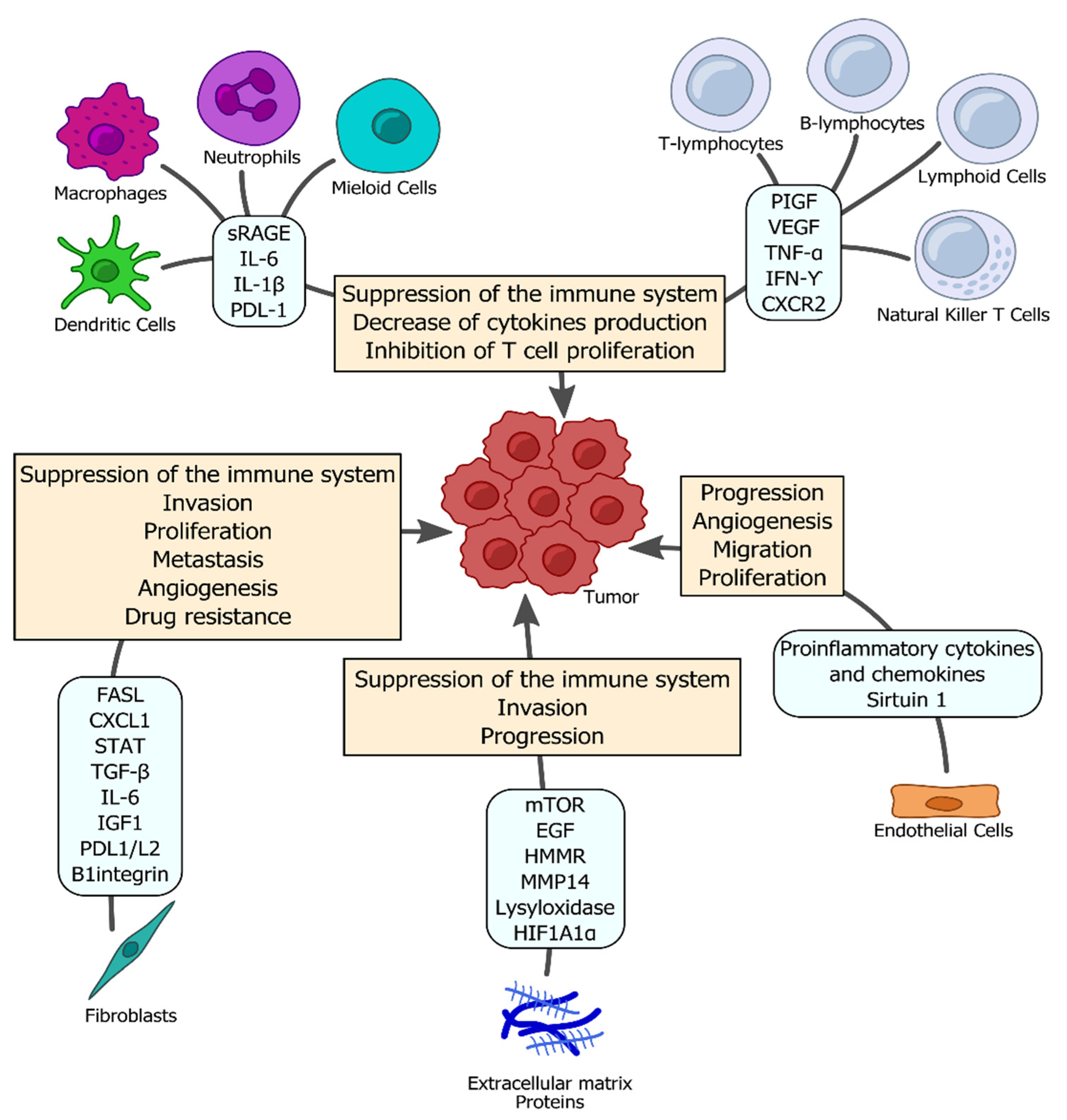
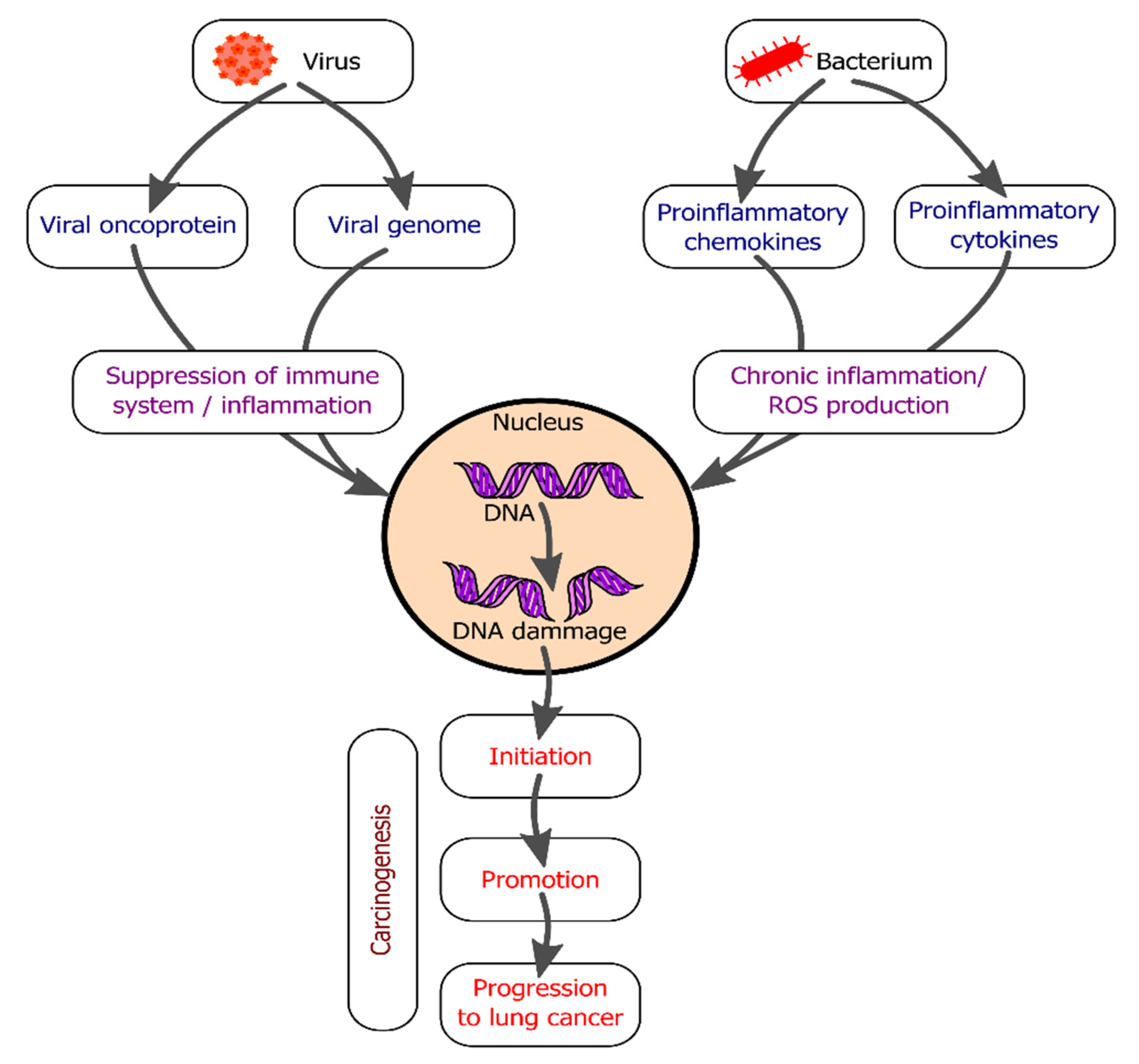
2. Immune System Responses to Infectious Factors Leading to Tumorigenesis
2.1. Bacterial Infections and Lung Cancer Development
2.1.1. Chlamydophila pneumonia
2.1.2. Mycobacterium tuberculosis
2.1.3. Cryptococcus sp.
2.1.4. Helicobacter pylori
2.2. Viral Infections and Lung Cancer Development
2.2.1. Human Immunodeficiency Virus (HIV)
2.2.2. Human Papilloma Virus (HPV)
2.2.3. Epstein–Barr Virus
2.2.4. Cytomegalovirus
2.2.5. Influenza Virus
2.2.6. Measles Virus (MV)
2.2.7. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
3. Infectious Complications of Lung Cancer
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.M.; Xu, Q.P.; Li, X.; Xiao, R.D.; Cai, L.; He, F. The association between human papillomavirus infection and lung cancer: A system review and meta-analysis. Oncotarget 2017, 8, 96419–96432. [Google Scholar] [CrossRef] [Green Version]
- Akinosoglou, K.S.; Karkoulias, K.; Marangos, M. Infectious complications in patients with lung cancer. Eur. Rev. Med. Pharm. Sci. 2013, 17, 8–18. [Google Scholar]
- Qiao, D.; Wang, Z.; Lu, Y.; Wen, X.; Li, H.; Zhao, H. A retrospective study of risk and prognostic factors in relation to lower respiratory tract infection in elderly lung cancer patients. Am. J. Cancer Res. 2015, 5, 423–432. [Google Scholar] [PubMed]
- Palucka, A.K.; Coussens, L.M. The basis of oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [Green Version]
- Maddi, A.; Sabharwal, A.; Violante, T.; Manuballa, S.; Genco, R.; Patnaik, S.; Yendamuri, S. The microbiome and lung cancer. J. Thorac. Dis. 2019, 11, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern recognition receptors and the host cell death molecular machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.D.; Malachowa, N.; DeLeo, F.R. Neutrophils and bacterial immune evasion. J. Innate Immun. 2018, 10, 432–441. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut microbiota and immune system interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Chen, Y.; Tan, W.; Wang, C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial-mesenchymal transition. Oncotargets Therapy 2018, 11, 3817–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipanyula, M.J.; Seke Etet, P.F.; Vecchio, L.; Farahna, M.; Nukenine, E.N.; Nwabo Kamdje, A.H. Signaling pathways bridging microbial-triggered inflammation and cancer. Cell Signal. 2013, 25, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lawrence, T.; Nizet, V. Innate immunity gone awry: Linking microbial infections to chronic inflammation and cancer. Cell 2006, 124, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- zur Hausen, H.; de Villiers, E.M. Cancer “causation” by infections--individual contributions and synergistic networks. Seminars Oncol. 2014, 41, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.A.; Cousins, R.J. Kinetics of zinc metabolism in the rat: Effect of dibutyryl cAMP. Am. J. Physiol. 1989, 256, E420–E430. [Google Scholar] [CrossRef]
- de Martel, C.; Franceschi, S. Infections and cancer: Established associations and new hypotheses. Crit Rev. Oncolhematol. 2009, 70, 183–194. [Google Scholar] [CrossRef]
- Cohen, J.R.; Tenenbaum, N.; Citron, M. Greenfield filter as primary therapy for deep venous thrombosis and/or pulmonary embolism in patients with cancer. Surgery 1991, 109, 12–15. [Google Scholar]
- de Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- MacMahon, B. A code of ethical conduct for epidemiologists? J. Clin. Epidemiol. 1991, 44 (Suppl 1), 147s–149s. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Bui, J.D.; Schreiber, R.D. Cancer immunosurveillance, immunoediting and inflammation: Independent or interdependent processes? Curr. Opin. Immunol. 2007, 19, 203–208. [Google Scholar] [CrossRef]
- Ingels, A.; Sanchez Salas, R.E.; Ravery, V.; Fromont-Hankard, G.; Validire, P.; Patard, J.J.; Pignot, G.; Prapotnich, D.; Olivier, F.; Galiano, M.; et al. T-helper 1 immunoreaction influences survival in muscle-invasive bladder cancer: Proof of concept. Ecancermedicalscience 2014, 8, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haabeth, O.A.; Lorvik, K.B.; Hammarström, C.; Donaldson, I.M.; Haraldsen, G.; Bogen, B.; Corthay, A. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat. Commun. 2011, 2, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, G.C.; Accardi, G.; Monastero, R.; Nicoletti, F.; Libra, M. Ageing: From inflammation to cancer. Immun. Ageing 2018, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, G.; Grange, J.M.; Fadda, E.; Fedeli, U.; Buja, A.; Lange, J.H. Lung cancer risk: Effect of dairy farming and the consequence of removing that occupational exposure. Am. J. Epidemiol. 2005, 161, 1037–1046. [Google Scholar] [CrossRef]
- Rylander, R. Endotoxin in the environment--exposure and effects. J. Endotoxin Res. 2002, 8, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Iheagwara, U.K.; Beatty, P.L.; Van, P.T.; Ross, T.M.; Minden, J.S.; Finn, O.J. Influenza virus infection elicits protective antibodies and T cells specific for host cell antigens also expressed as tumor-associated antigens: A new view of cancer immunosurveillance. Cancer Immunol. Res. 2014, 2, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Anderson, L.A.; Landgren, O.; Engels, E.A. Common community acquired infections and subsequent risk of chronic lymphocytic leukaemia. Brit. J. Haematol. 2009, 147, 444–449. [Google Scholar] [CrossRef] [Green Version]
- Bosteels, C.; Neyt, K.; Vanheerswynghels, M.; van Helden, M.J.; Sichien, D.; Debeuf, N.; De Prijck, S.; Bosteels, V.; Vandamme, N.; Martens, L.; et al. Inflammatory type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity 2020, 52, 1039–1056.e9. [Google Scholar] [CrossRef]
- Newman, J.H.; Chesson, C.B.; Herzog, N.L.; Bommareddy, P.K.; Aspromonte, S.M.; Pepe, R.; Estupinian, R.; Aboelatta, M.M.; Buddhadev, S.; Tarabichi, S.; et al. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, A.C.; Newton, D.J.; Carding, S.R.; Kaye, P.M. Evidence for the involvement of lung-specific gammadelta T cell subsets in local responses to Streptococcus pneumoniae infection. Eur. J. Immunol. 2007, 37, 3404–3413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.; Liu, T.; Zhou, W.Y.; Zhuang, Y.; Peng, L.S.; Zhang, J.Y.; Yin, Z.N.; Mao, X.H.; Guo, G.; Shi, Y.; et al. Role of gamma-delta T cells in host response against Staphylococcus aureus-induced pneumonia. BMC Immunol. 2012, 13, 38. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Hu, S. Lung-resident γδ T cells and their roles in lung diseases. Immunology 2017, 151, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.J.; Sethi, S.; Murphy, T.; Nariya, S.; Boushey, H.A.; Lynch, S.V. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J. Clin. Microbiol. 2014, 52, 2813–2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammen, M.J.; Sethi, S. COPD and the microbiome. Respirology 2016, 21, 590–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Q.; Jiang, F.; Yin, R.; Wang, J.; Xia, W.; Dong, G.; Ma, W.; Yang, Y.; Xu, L.; Hu, J. Interplay between the lung microbiome and lung cancer. Cancer Lett. 2018, 415, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Tsay, J.J.; Wu, B.G.; Badri, M.H.; Clemente, J.C.; Shen, N.; Meyn, P.; Li, Y.; Yie, T.A.; Lhakhang, T.; Olsen, E.; et al. Airway microbiota is associated with upregulation of the PI3K pathway in lung cancer. Am. J. Resp. Crit. Care Med. 2018, 198, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Redecke, V.; Dalhoff, K.; Bohnet, S.; Braun, J.; Maass, M. Interaction of Chlamydia pneumoniae and human alveolar macrophages: Infection and inflammatory response. Am. J. Res. Cell Mol. Biol. 1998, 19, 721–727. [Google Scholar] [CrossRef] [Green Version]
- Gaydos, C.A. Growth in vascular cells and cytokine production by Chlamydia pneumoniae. J. Infect. Dis 2000, 181 (Suppl 3), S473–S478. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Shane, R.B.; Berencsi, K.; Gonczol, E.; Zaki, M.H.; Margolis, D.J.; Trinchieri, G.; Rook, A.H. Chlamydia pneumoniae inhibits apoptosis in human peripheral blood mononuclear cells through induction of IL-10. J. Immunol. 2000, 164, 5522–5529. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.; Lu, H.; Hu, H.; Shi, L.; McClarty, G.A.; Nance, D.M.; Greenberg, A.H.; Zhong, G. Inhibition of apoptosis in chlamydia-infected cells: Blockade of mitochondrial cytochrome c release and caspase activation. J. Exp. Med. 1998, 187, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Biasucci, L.M.; Liuzzo, G.; Ciervo, A.; Petrucca, A.; Piro, M.; Angiolillo, D.J.; Crea, F.; Cassone, A.; Maseri, A. Antibody response to chlamydial heat shock protein 60 is strongly associated with acute coronary syndromes. Circulation 2003, 107, 3015–3017. [Google Scholar] [CrossRef]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Rosin, M.P.; Anwar, W.A.; Ward, A.J. Inflammation, chromosomal instability, and cancer: The schistosomiasis model. Cancer Res. 1994, 54, 1929s–1933s. [Google Scholar] [PubMed]
- Dheda, K.; Booth, H.; Huggett, J.F.; Johnson, M.A.; Zumla, A.; Rook, G.A. Lung remodeling in pulmonary tuberculosis. J. Infect. Dis. 2005, 192, 1201–1209. [Google Scholar] [CrossRef]
- Cooper, A.M.; Khader, S.A. The role of cytokines in the initiation, expansion, and control of cellular immunity to tuberculosis. Immunol. Rev. 2008, 226, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Fried, L.E.; Arbiser, J.L. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid Redox Signal. 2009, 11, 1139–1148. [Google Scholar] [CrossRef]
- Liuzzo, G.; Trotta, F.; Pedicino, D. Interleukin-17 in atherosclerosis and cardiovascular disease: The good, the bad, and the unknown. Eur. Heart J. 2013, 34, 556–559. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.T.; Linderman, J.J.; Kirschner, D.E. Multiple mechanisms allow Mycobacterium tuberculosis to continuously inhibit MHC class II-mediated antigen presentation by macrophages. Proc. Natl. Acad. Sci. USA 2005, 102, 4530–4535. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.H.; McDonald, R.A.; Wells, J.C.; Huffnagle, G.B.; Lukacs, N.W.; Toews, G.B. The gamma interferon receptor is required for the protective pulmonary inflammatory response to Cryptococcus neoformans. Infect. Immun. 2005, 73, 1788–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, S.; Kakugawa, T.; Yura, H.; Tomonaga, M.; Harada, T.; Hara, A.; Hara, S.; Nakano, M.; Yamasaki, E.; Sakamoto, N.; et al. Identification of Helicobacter pylori VacA in human lung and its effects on lung cells. Biochem Biophys. Res. Commun. 2015, 460, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Samareh-Fekri, M.; Hashemi Bajgani, S.M.; Shafahi, A.; Asadi-Zarandi, M.; Mollaie, H.; Jamali Paghalhe, A. Detection of Helicobacter pylori in the bronchoalveolar lavage of patients with lung cancer using real-time PCR. Jundishapur J. Microbiol. 2016, 9, e32144. [Google Scholar] [CrossRef] [Green Version]
- GonzÁlez, I.; Araya, P.; Rojas, A. Helicobacter pylori infection and lung cancer: New insights and future challenges. Zhongguo Fei Ai Za Zhi 2018, 21, 658–662. [Google Scholar] [PubMed]
- Kang, J.H.; Hwang, S.M.; Chung, I.Y. S100A8, S100A9 and S100A12 activate airway epithelial cells to produce MUC5AC via extracellular signal-regulated kinase and nuclear factor-κB pathways. Immunology 2015, 144, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurila, A.L.; Anttila, T.; Läärä, E.; Bloigu, A.; Virtamo, J.; Albanes, D.; Leinonen, M.; Saikku, P. Serological evidence of an association between Chlamydia pneumoniae infection and lung cancer. Int. J. Cancer 1997, 74, 31–34. [Google Scholar] [CrossRef]
- Jackson, L.A.; Wang, S.P.; Nazar-Stewart, V.; Grayston, J.T.; Vaughan, T.L. Association of Chlamydia pneumoniae immunoglobulin A seropositivity and risk of lung cancer. Cancer Epidemiol. Biomark. Prev. 2000, 9, 1263–1266. [Google Scholar]
- Eickhoff, M.; Thalmann, J.; Hess, S.; Martin, M.; Laue, T.; Kruppa, J.; Brandes, G.; Klos, A. Host cell responses to Chlamydia pneumoniae in gamma interferon-induced persistence overlap those of productive infection and are linked to genes involved in apoptosis, cell cycle, and metabolism. Infect. Immun. 2007, 75, 2853–2863. [Google Scholar] [CrossRef] [Green Version]
- Hess, S.; Peters, J.; Bartling, G.; Rheinheimer, C.; Hegde, P.; Magid-Slav, M.; Tal-Singer, R.; Klos, A. More than just innate immunity: Comparative analysis of Chlamydophila pneumoniae and Chlamydia trachomatis effects on host-cell gene regulation. Cell Microbiol. 2003, 5, 785–795. [Google Scholar] [CrossRef]
- von Hertzen, L.C. Chlamydia pneumoniae and its role in chronic obstructive pulmonary disease. Ann. Med. 1998, 30, 27–37. [Google Scholar] [CrossRef]
- Khan, S.; Imran, A.; Khan, A.A.; Abul Kalam, M.; Alshamsan, A. Systems biology approaches for the prediction of possible role of Chlamydia pneumoniae oroteins in the etiology of lung cancer. PLoS ONE 2016, 11, e0148530. [Google Scholar]
- Mayer, J.; Woods, M.L.; Vavrin, Z.; Hibbs, J.B., Jr. Gamma interferon-induced nitric oxide production reduces Chlamydia trachomatis infectivity in McCoy cells. Infect. Immun. 1993, 61, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Keikha, M.; Esfahani, B.N. The relationship between tuberculosis and lung cancer. Adv. Biomed. Res. 2018, 7, 58. [Google Scholar]
- Bhatt, M.; Kant, S.; Bhaskar, R. Pulmonary tuberculosis as differential diagnosis of lung cancer. South. Asian J. Cancer 2012, 1, 36–42. [Google Scholar] [CrossRef]
- Engels, E.A. Inflammation in the development of lung cancer: Epidemiological evidence. Expert Rev. Anticancer Therap. 2008, 8, 605–615. [Google Scholar] [CrossRef]
- Engels, E.A.; Shen, M.; Chapman, R.S.; Pfeiffer, R.M.; Yu, Y.Y.; He, X.; Lan, Q. Tuberculosis and subsequent risk of lung cancer in Xuanwei, China. Int. J. Cancer 2009, 124, 1183–1187. [Google Scholar] [CrossRef]
- Nalbandian, A.; Yan, B.S.; Pichugin, A.; Bronson, R.T.; Kramnik, I. Lung carcinogenesis induced by chronic tuberculosis infection: The experimental model and genetic control. Oncogene 2009, 28, 1928–1938. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.Y.; Li, X.L.; Yu, X.S.; Guan, P.; Yin, Z.H.; He, Q.C.; Zhou, B.S. Facts and fiction of the relationship between preexisting tuberculosis and lung cancer risk: A systematic review. Int. J. Cancer 2009, 125, 2936–2944. [Google Scholar] [CrossRef]
- Park, S.K.; Cho, L.Y.; Yang, J.J.; Park, B.; Chang, S.H.; Lee, K.S.; Kim, H.; Yoo, K.Y.; Lee, C.T. Lung cancer risk and cigarette smoking, lung tuberculosis according to histologic type and gender in a population based case-control study. Lung Cancer 2010, 68, 20–26. [Google Scholar] [CrossRef]
- Wang, X.R.; Yu, I.T.; Chiu, Y.L.; Qiu, H.; Fu, Z.; Goggins, W.; Au, J.S.; Tse, L.A.; Wong, T.W. Previous pulmonary disease and family cancer history increase the risk of lung cancer among Hong Kong women. Cancer Causes Control. 2009, 20, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Li, J.; Lu, J.; Zhong, R.; Zhong, H. Mycobacterium tuberculosis antigens repress Th1 immune response suppression and promotes lung cancer metastasis through PD-1/PDl-1 signaling pathway. Cell Death Dis. 2019, 10, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicenas, S.; Vencevicius, V. Lung cancer in patients with tuberculosis. World J. Surg. Oncol. 2007, 5, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.H.; Wu, C.H.; Wu, W.S.; Huang, C.Y.; Su, W.J.; Tsai, C.M.; Lee, Y.C.; Perng, R.P.; Chen, Y.M. Association between tumor epidermal growth factor receptor mutation and pulmonary tuberculosis in patients with adenocarcinoma of the lungs. J. Thoracic Oncol. 2012, 7, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Rihawi, A.; Huang, G.; Al-Hajj, A.; Bootwala, Z. A case of tuberculosis and adenocarcinoma coexisting in the same lung lobe. Int. J. Mycobacteriol. 2016, 5, 80–82. [Google Scholar] [CrossRef]
- Huang, J.; Lan, C.; Li, H.; Chen, S.; Lin, Q.; Weng, H. Concomitant lung adenocarcinoma and pulmonary cryptococcosis confirmed by pathologic examinations. Medicine 2019, 98, e18316. [Google Scholar] [CrossRef]
- Harada, T.; Hakuma, N.; Kamimura, A.; Ito, K.; Okamoto, K. Pulmonary cryptococcosis within a pulmonary carcinoma-review of reported cases. Intern. Med. 2006, 45, 369–372. [Google Scholar] [CrossRef] [Green Version]
- Robinson, T.D.; Barnes, D.J.; Watson, G.F. Coexistent cryptococcosis and carcinoma within a solitary pulmonary nodule. Aust. N. Z. J. Med. 1999, 29, 561–562. [Google Scholar] [CrossRef] [PubMed]
- Fa, Z.; Xie, Q.; Fang, W.; Zhang, H.; Zhang, H.; Xu, J.; Pan, W.; Xu, J.; Olszewski, M.A.; Deng, X.; et al. RIPK3/Fas-associated death domain axis regulates pulmonary immunopathology to cryptococcal infection independent of necroptosis. Front. Immunol. 2017, 8, 1055. [Google Scholar] [CrossRef] [Green Version]
- Jenny-Avital, E.R.; Abadi, M. Immune reconstitution cryptococcosis after initiation of successful highly active antiretroviral therapy. Clin. Infect. Dis. 2002, 35, e128–e133. [Google Scholar] [CrossRef]
- Cavaleiro-Pinto, M.; Peleteiro, B.; Lunet, N.; Barros, H. Helicobacter pylori infection and gastric cardia cancer: Systematic review and meta-analysis. Cancer Causes Control. 2011, 22, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Salama, N.R.; Hartung, M.L.; Müller, A. Life in the human stomach: Persistence strategies of the bacterial pathogen Helicobacter pylori. Rev. Microbiol. 2013, 11, 385–399. [Google Scholar] [CrossRef]
- Corrales, L.; Rosell, R.; Cardona, A.F.; Martín, C.; Zatarain-Barrón, Z.L.; Arrieta, O. Lung cancer in never smokers: The role of different risk factors other than tobacco smoking. Crit. Rev. Oncol. Hematol. 2020, 148, 102895. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Collard, H.R.; Raghu, G.; Sweet, M.P.; Hays, S.R.; Campos, G.M.; Golden, J.A.; King, T.E., Jr. Does chronic microaspiration cause idiopathic pulmonary fibrosis? Am. J. Med. 2010, 123, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Starosta, V.; Kitz, R.; Hartl, D.; Marcos, V.; Reinhardt, D.; Griese, M. Bronchoalveolar pepsin, bile acids, oxidation, and inflammation in children with gastroesophageal reflux disease. Chest 2007, 132, 1557–1564. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Rojas, A.; Pérez-Castro, R.; González, I.; Delgado, F.; Romero, J.; Rojas, I. The emerging role of the receptor for advanced glycation end products on innate immunity. Int. Rev. Immunol. 2014, 33, 67–80. [Google Scholar] [CrossRef]
- Grulich, A.E.; van Leeuwen, M.T.; Falster, M.O.; Vajdic, C.M. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: A meta-analysis. Lancet 2007, 370, 59–67. [Google Scholar] [CrossRef]
- Prabhu, P.R.; Jayalekshmi, D.; Pillai, M.R. Lung cancer and human papilloma viruses (HPVs): Examining the molecular evidence. J. Oncol. 2012, 2012, 750270. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, M.; Mostafaei, S.; Aghaei, A.; Hosseini, N.; Darabi, H.; Nouri, M.; Etemadi, A.; Neill, A.O.; Nahand, J.S.; Mirzaei, H.; et al. The association between HPV gene expression, inflammatory agents and cellular genes involved in EMT in lung cancer tissue. BMC Cancer 2020, 20, 916. [Google Scholar] [CrossRef]
- Kheir, F.; Zhao, M.; Strong, M.J.; Yu, Y.; Nanbo, A.; Flemington, E.K.; Morris, G.F.; Reiss, K.; Li, L.; Lin, Z. Detection of Epstein-Barr virus infection in non-small cell lung cancer. Cancers 2019, 11, 759. [Google Scholar] [CrossRef] [Green Version]
- Söderberg-Nauclér, C. Does cytomegalovirus play a causative role in the development of various inflammatory diseases and cancer? J. Internal Med. 2006, 259, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Varsano, S.; Rashkovsky, L.; Shapiro, H.; Ophir, D.; Mark-Bentankur, T. Human lung cancer cell lines express cell membrane complement inhibitory proteins and are extremely resistant to complement-mediated lysis; a comparison with normal human respiratory epithelium in vitro, and an insight into mechanism(s) of resistance. Clin. Exp. Immunol. 1998, 113, 173–182. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Hernández-Ramírez, R.U.; Shiels, M.S.; Dubrow, R.; Engels, E.A. Cancer risk in HIV-infected people in the USA from 1996 to 2012: A population-based, registry-linkage study. Lancet HIV 2017, 4, e495–e504. [Google Scholar] [CrossRef]
- de Groot, P.M.; Wu, C.C.; Carter, B.W.; Munden, R.F. The epidemiology of lung cancer. Transl. Lung Cancer Res. 2018, 7, 220–233. [Google Scholar] [CrossRef]
- Engels, E.A.; Brock, M.V.; Chen, J.; Hooker, C.M.; Gillison, M.; Moore, R.D. Elevated incidence of lung cancer among HIV-infected individuals. J. Clin. Oncol. 2006, 24, 1383–1388. [Google Scholar] [CrossRef]
- Sigel, K.; Wisnivesky, J.; Crothers, K.; Gordon, K.; Brown, S.T.; Rimland, D.; Rodriguez-Barradas, M.C.; Gibert, C.; Goetz, M.B.; Bedimo, R.; et al. Immunological and infectious risk factors for lung cancer in US veterans with HIV: A longitudinal cohort study. Lancet HIV 2017, 4, e67–e73. [Google Scholar] [CrossRef] [Green Version]
- Sigel, K.; Makinson, A.; Thaler, J. Lung cancer in persons with HIV. Curr. Opin. HIV AIDS 2017, 12, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Bearz, A.; Vaccher, E.; Talamini, R.; Berretta, M.; Tirelli, U. Comment on ‘Lung cancer in the Swiss HIV Cohort Study: Role of smoking, immunodeficiency and pulmonary infection’. Brit. J. Cancer 2012, 106, 1899–1900. [Google Scholar] [CrossRef] [Green Version]
- Mdodo, R.; Frazier, E.L.; Dube, S.R.; Mattson, C.L.; Sutton, M.Y.; Brooks, J.T.; Skarbinski, J. Cigarette smoking prevalence among adults with HIV compared with the general adult population in the United States: Cross-sectional surveys. Ann. Intern. Med. 2015, 162, 335–344. [Google Scholar] [CrossRef]
- Serrão, R.; Piñero, C.; Velez, J.; Coutinho, D.; Maltez, F.; Lino, S.; Sarmento, E.C.R.; Tavares, A.P.; Pacheco, P.; Lopes, M.J.; et al. Non-AIDS-related comorbidities in people living with HIV-1 aged 50 years and older: The AGING POSITIVE study. Int. J. Infect. Dis. 2019, 79, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Bichara, B.; Routy, J.P.; Ezer, N.; Costiniuk, C.T. Primary lung cancer diagnoses in people living with HIV in a large clinical centre in Montreal, Canada over 3 decades. AIDS Care 2020, 32, 979–983. [Google Scholar] [CrossRef]
- De Flora, S.; La Maestra, S. Epidemiology of cancers of infectious origin and prevention strategies. J. Prev. Med. Hyg. 2015, 56, E15–E20. [Google Scholar]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.M.; Kim, E.H. Human papillomavirus infection and risk of lung cancer in never-smokers and women: An ‘adaptive’ meta-analysis. Epidemiol. Health 2015, 37, e2015052. [Google Scholar] [CrossRef]
- Syrjänen, K. Detection of human papillomavirus in lung cancer: Systematic review and meta-analysis. Anticancer Res. 2012, 32, 3235–3250. [Google Scholar]
- Zhai, K.; Ding, J.; Shi, H.Z. HPV and lung cancer risk: A meta-analysis. J. Clin. Virol. 2015, 63, 84–90. [Google Scholar] [CrossRef]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Chen, K.Y.; Wu, S.M.; Liu, J.C.; Lee, K.Y. Effect of annual influenza vaccination on reducing lung cancer in patients with chronic obstructive pulmonary disease from a population-based cohort study. Medicine 2019, 98, e18035. [Google Scholar] [CrossRef]
- Lung, M.L.; Lam, W.K.; So, S.Y.; Lam, W.P.; Chan, K.H.; Ng, M.H. Evidence that respiratory tract is major reservoir for Epstein-Barr virus. Lancet 1985, 1, 889–892. [Google Scholar] [CrossRef]
- Duan, W.; Gao, L.; Druhan, L.J.; Zhu, W.G.; Morrison, C.; Otterson, G.A.; Villalona-Calero, M.A. Expression of Pirh2, a newly identified ubiquitin protein ligase, in lung cancer. J. Nat. Cancer Instit. 2004, 96, 1718–1721. [Google Scholar] [CrossRef] [Green Version]
- Freeman, R.B., Jr. The ‘indirect’ effects of cytomegalovirus infection. Am. J. Transplant. 2009, 9, 2453–2458. [Google Scholar] [CrossRef]
- Pereg, D.; Lishner, M. Non-steroidal anti-inflammatory drugs for the prevention and treatment of cancer. J. Internal. Med. 2005, 258, 115–123. [Google Scholar] [CrossRef]
- Zhu, H.; Cong, J.P.; Yu, D.; Bresnahan, W.A.; Shenk, T.E. Inhibition of cyclooxygenase 2 blocks human cytomegalovirus replication. Proc. Natl. Acad. Sci. USA 2002, 99, 3932–3937. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Coquard, L.; Pasquereau, S.; Russo, L.; Valmary-Degano, S.; Borg, C.; Pothier, P.; Herbein, G. Tumor control by human cytomegalovirus in a murine model of hepatocellular carcinoma. Mol. Ther. Oncolytics 2016, 3, 16012. [Google Scholar] [CrossRef]
- Weng, C.F.; Chen, L.J.; Lin, C.W.; Chen, H.M.; Lee, H.H.; Ling, T.Y.; Hsiao, F.Y. Association between the risk of lung cancer and influenza: A population-based nested case-control study. Int. J. Infect. Dis. 2019, 88, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rima, B.K.; Duprex, W.P. Molecular mechanisms of measles virus persistence. Virus Res. 2005, 111, 132–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Cortay, J.C.; Logan, I.R.; Sapountzi, V.; Robson, C.N.; Gerlier, D. Inhibition of ubiquitination and stabilization of human ubiquitin E3 ligase PIRH2 by measles virus phosphoprotein. J. Virol. 2005, 79, 11824–11836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.R.; Jacobson, B.A.; Belgum, H.; Raza, A.; Sadiq, A.; Drees, J.; Wang, H.; Jay-Dixon, J.; Etchison, R.; Federspiel, M.J.; et al. Measles vaccine strains for virotherapy of non-small-cell lung carcinoma. J. Thoracic. Oncol. 2014, 9, 1101–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.; Guan, W.; Chen, R.; Wang, W.; Li, J.; Xu, K.; Li, C.; Ai, Q.; Lu, W.; Liang, H.; et al. Cancer patients in SARS-CoV-2 infection: A nationwide analysis in China. Lancet Oncol. 2020, 21, 335–337. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Res. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Gupta, I.; Rizeq, B.; Elkord, E.; Vranic, S.; Al Moustafa, A.E. SARS-CoV-2 infection and lung cancer: Potential therapeutic modalities. Cancers 2020, 12, 2186. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F.; Watz, H. Chronic obstructive pulmonary disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal microbiota promote lung cancer development via γδ T cells. Cell 2019, 176, 998–1013.e16. [Google Scholar] [CrossRef] [Green Version]
- Lanoix, J.P.; Pluquet, E.; Lescure, F.X.; Bentayeb, H.; Lecuyer, E.; Boutemy, M.; Dumont, P.; Jounieaux, V.; Schmit, J.L.; Dayen, C.; et al. Bacterial infection profiles in lung cancer patients with febrile neutropenia. BMC Infect. Dis. 2011, 11, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganzel, C.; Silverman, B.; Chemtob, D.; Ben Shoham, A.; Wiener-Well, Y. The risk of tuberculosis in cancer patients is greatest in lymphoma and myelodysplastic syndrome/myeloproliferative neoplasm: A large population-based cohort study. Leuk Lymphoma 2019, 60, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I.; Goo, J.M.; Kim, H.Y.; Song, J.W.; Im, J.G. Coexisting bronchogenic carcinoma and pulmonary tuberculosis in the same lobe: Radiologic findings and clinical significance. Korea J. Radiol. 2001, 2, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Elkington, P.T.; Bateman, A.C.; Thomas, G.J.; Ottensmeier, C.H. Implications of tuberculosis reactivation after immune checkpoint inhibition. Am. J. Resp. Crit. Care Med. 2018, 198, 1451–1453. [Google Scholar] [CrossRef]
- Barber, D.L.; Mayer-Barber, K.D.; Feng, C.G.; Sharpe, A.H.; Sher, A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J. Immunol. 2011, 186, 1598–1607. [Google Scholar] [CrossRef] [Green Version]
- Langan, E.A.; Graetz, V.; Allerheiligen, J.; Zillikens, D.; Rupp, J.; Terheyden, P. Immune checkpoint inhibitors and tuberculosis: An old disease in a new context. Lancet. Oncol. 2020, 21, e55–e65. [Google Scholar] [CrossRef]
- Fujita, K.; Terashima, T.; Mio, T. Anti-PD1 Antibody Treatment and the Development of Acute Pulmonary Tuberculosis. J. Thorac. Oncol. 2016, 11, 2238–2240. [Google Scholar] [CrossRef] [PubMed]
- Anastasopoulou, A.; Ziogas, D.C.; Samarkos, M.; Kirkwood, J.M.; Gogas, H. Reactivation of tuberculosis in cancer patients following administration of immune checkpoint inhibitors: Current evidence and clinical practice recommendations. J. Immunother. Cancer 2019, 7, 239. [Google Scholar] [CrossRef] [Green Version]
- Picchi, H.; Mateus, C.; Chouaid, C.; Besse, B.; Marabelle, A.; Michot, J.M.; Champiat, S.; Voisin, A.L.; Lambotte, O. Infectious complications associated with the use of immune checkpoint inhibitors in oncology: Reactivation of tuberculosis after anti PD-1 treatment. Clin. Microbiol. Infect. 2018, 24, 216–218. [Google Scholar] [CrossRef] [Green Version]
- Takata, S.; Koh, G.; Han, Y.; Yoshida, H.; Shiroyama, T.; Takada, H.; Masuhiro, K.; Nasu, S.; Morita, S.; Tanaka, A.; et al. Paradoxical response in a patient with non-small cell lung cancer who received nivolumab followed by anti-Mycobacterium tuberculosis agents. J. Infect. Chemother. 2019, 25, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.O.; Jung, S.S.; Kim, S.Y.; Kim, T.Y.; Shin, D.W.; Lee, J.H.; Lee, Y.H. Inhibition of Lewis lung carcinoma growth by Toxoplasma gondii through induction of Th1 immune responses and inhibition of angiogenesis. J. Korean Med. Sci. 2007, 22, S38–S46. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhao, Y.; Okwan-Duodu, D.; Basho, R.; Cui, X. COVID-19 in cancer patients: Risk, clinical features, and management. Cancer Biol. Med. 2020, 17, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Moujaess, E.; Kourie, H.R.; Ghosn, M. Cancer patients and research during COVID-19 pandemic: A systematic review of current evidence. Crit. Rev. Oncol. Hematol. 2020, 150, 102972. [Google Scholar] [CrossRef]
- Yang, K.; Sheng, Y.; Huang, C.; Jin, Y.; Xiong, N.; Jiang, K.; Lu, H.; Liu, J.; Yang, J.; Dong, Y.; et al. Clinical characteristics, outcomes, and risk factors for mortality in patients with cancer and COVID-19 in Hubei, China: A multicentre, retrospective, cohort study. Lancet Oncol. 2020, 21, 904–913. [Google Scholar] [CrossRef]
- Dai, M.; Liu, D.; Liu, M.; Zhou, F.; Li, G.; Chen, Z.; Zhang, Z.; You, H.; Wu, M.; Zheng, Q.; et al. Patients with Cancer Appear More Vulnerable to SARS-CoV-2: A Multicenter Study during the COVID-19 Outbreak. Cancer Discov. 2020, 10, 783–791. [Google Scholar] [PubMed]
- Riordan, J.F. Angiotensin-I-converting enzyme and its relatives. Genome Biol. 2003, 4, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.; Hu, W.; Niu, L.; Liu, H.; Xu, H.; Xiao, S.Y. Pulmonary Pathology of Early-Phase 2019 Novel Coronavirus (COVID-19) Pneumonia in Two Patients With Lung Cancer. J. Thorac. Oncol. 2020, 15, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Ruck, C.; Reikie, B.A.; Marchant, A.; Kollmann, T.R.; Kakkar, F. Linking Susceptibility to Infectious Diseases to Immune System Abnormalities among HIV-Exposed Uninfected Infants. Front. Immunol. 2016, 7, 310. [Google Scholar] [CrossRef] [Green Version]
- Cunha, B.M.; Mota, L.M.; Pileggi, G.S.; Safe, I.P.; Lacerda, M.V. HIV/AIDS and rheumatoid arthritis. Autoimmun. Rev. 2015, 14, 396–400. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Bacteria Inducing Lung Cancer | Effect(s) on the Immune System | Reference(s) |
|---|---|---|
| Chlamydophila pneumoniae | Induction of TNF-α, IL-1β, and IL-6 | [40,41] |
| IL-10 induction | [42,43] | |
| Release of CHSP-60 protein | [44] | |
| Mycobacterium tuberculosis | Activation of neutrophils and production of reactive oxygen species | [45,46] |
| Release of TNF-α, INF-γ, IL-1, IL-2, and IL-12 | [47,48] | |
| Increase in levels of TGF-β, IL-4, IL-10, IL-3, and IL-13 | [47,48] | |
| Increased levels of IL-17 and THFα | [49,50] | |
| Secretion of IFN-γ and TNF-α | [51] | |
| Cryptococcus sp. | Activation of Th1/Th17 immune responses | [52] |
| Helicobacter pylori | Induced IL-6 and IL-8 production | [53,54] |
| Overexpression of Toll-like receptors (TLRs) | [55,56] |
| Virus Inducing Lung Cancer | Effect(s) on the Immune System | Reference(s) |
|---|---|---|
| Human immunodeficiency virus | CD4 count | [88] |
| Human papilloma virus | Activation of the mitogenic signaling | [89] |
| Increase TNF-α and reactive oxygen-nitrogen species (RONS) | [90] | |
| Activation of p53, IL-6, IL-10, pRb, EGFR, HIF-1α, Mcl-1, Bcl-2VEGF, and cIAP-2 | [2] | |
| Epstein–Barr virus | Increase immune cell infiltration | [91] |
| Cytomegalovirus | Prevention of activated NK and T cells | [92] |
| Influenza virus | Promotion of systemic CD8+ T cell-mediated antitumor immunity | [32] |
| Measles virus | Overexpression of CD46 | [93] |
| SARS-CoV-2 virus | Increase IL-6, IL-7, TNF-α, CCL2, CCL3, and CXCL10 | [94] |
| Secretion of mature IL-1β and/or IL-18 | [94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budisan, L.; Zanoaga, O.; Braicu, C.; Pirlog, R.; Covaliu, B.; Esanu, V.; Korban, S.S.; Berindan-Neagoe, I. Links between Infections, Lung Cancer, and the Immune System. Int. J. Mol. Sci. 2021, 22, 9394. https://doi.org/10.3390/ijms22179394
Budisan L, Zanoaga O, Braicu C, Pirlog R, Covaliu B, Esanu V, Korban SS, Berindan-Neagoe I. Links between Infections, Lung Cancer, and the Immune System. International Journal of Molecular Sciences. 2021; 22(17):9394. https://doi.org/10.3390/ijms22179394
Chicago/Turabian StyleBudisan, Liviuta, Oana Zanoaga, Cornelia Braicu, Radu Pirlog, Bogdan Covaliu, Victor Esanu, Schuyler S. Korban, and Ioana Berindan-Neagoe. 2021. "Links between Infections, Lung Cancer, and the Immune System" International Journal of Molecular Sciences 22, no. 17: 9394. https://doi.org/10.3390/ijms22179394