
Insulin Receptor Isoforms Differently Regulate Cell Proliferation and Apoptosis in the Ligand-Occupied and Unoccupied State

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
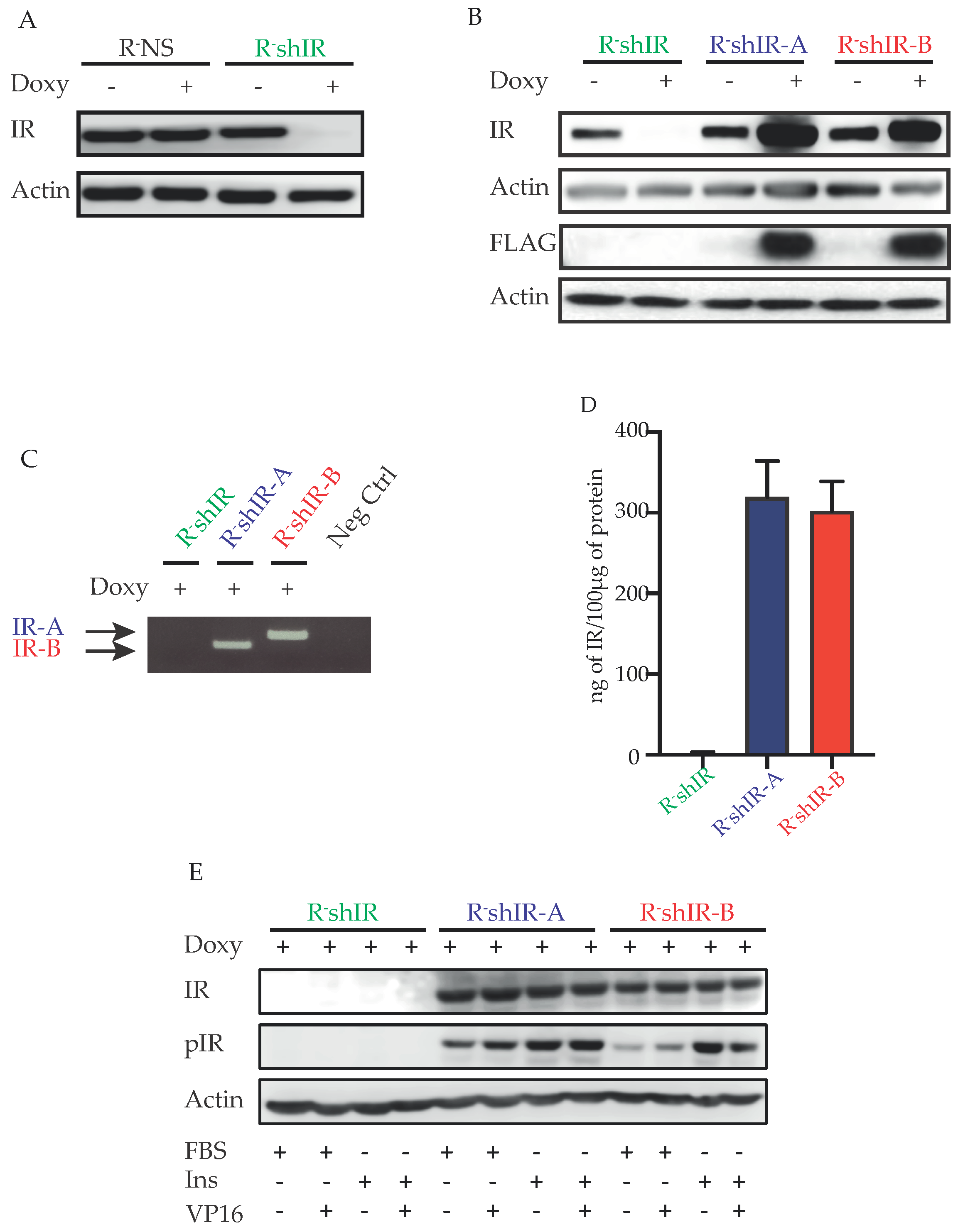
2.1. Characteristics of the Generated Cell Models
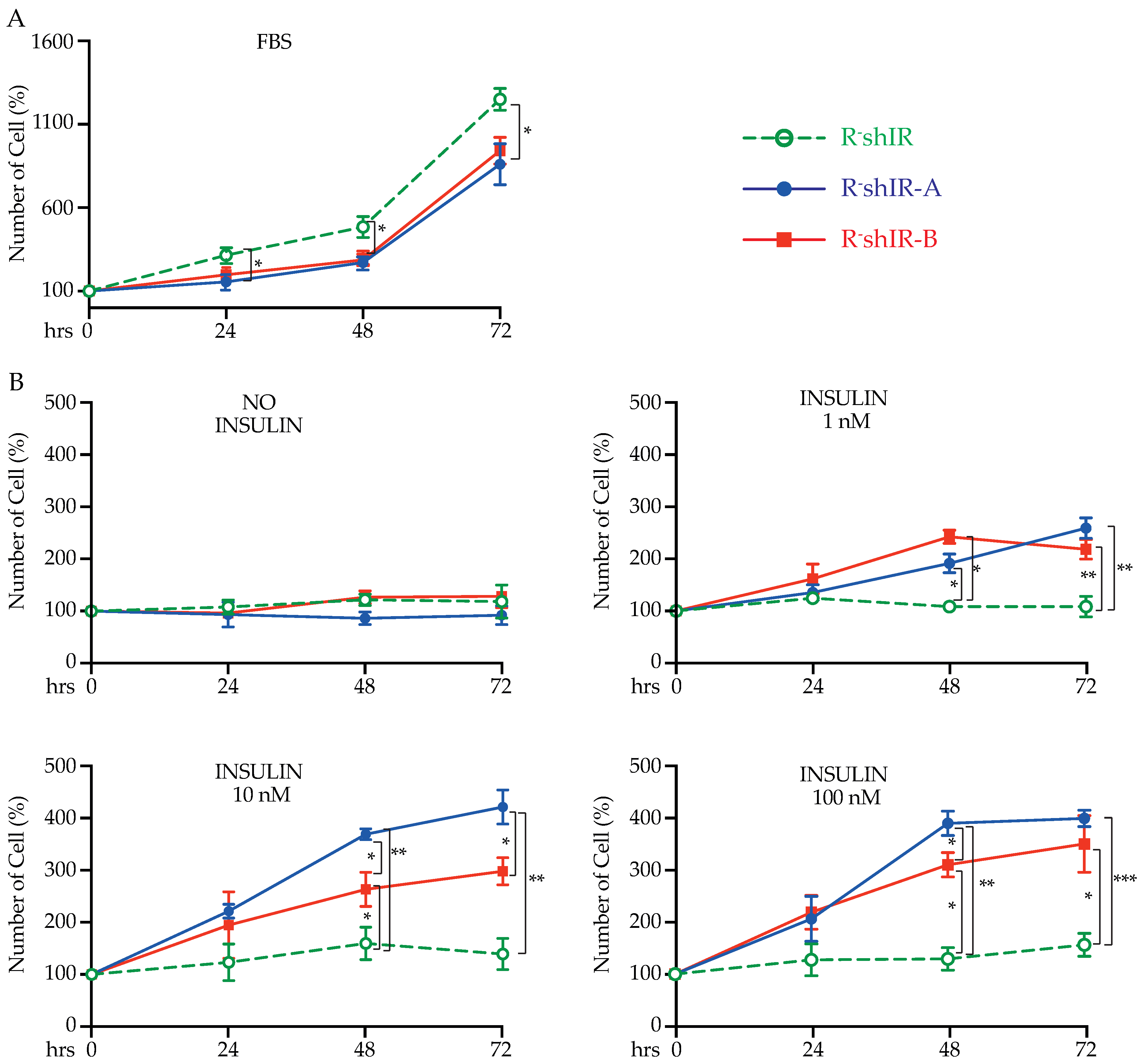
2.2. IR-A and IR-B Differently Affect Insulin-Dependent but Not Insulin-Independent Cell Proliferation
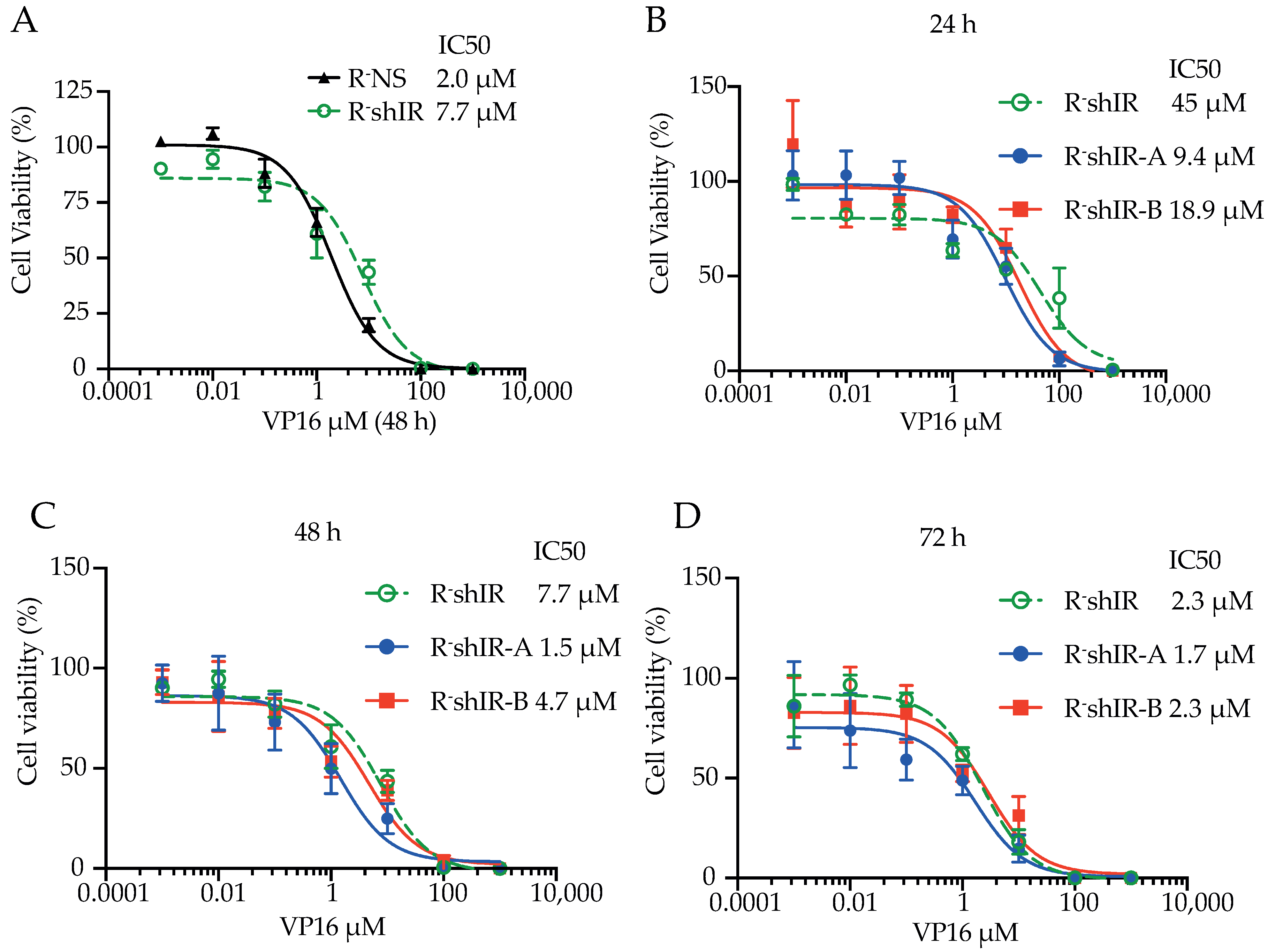
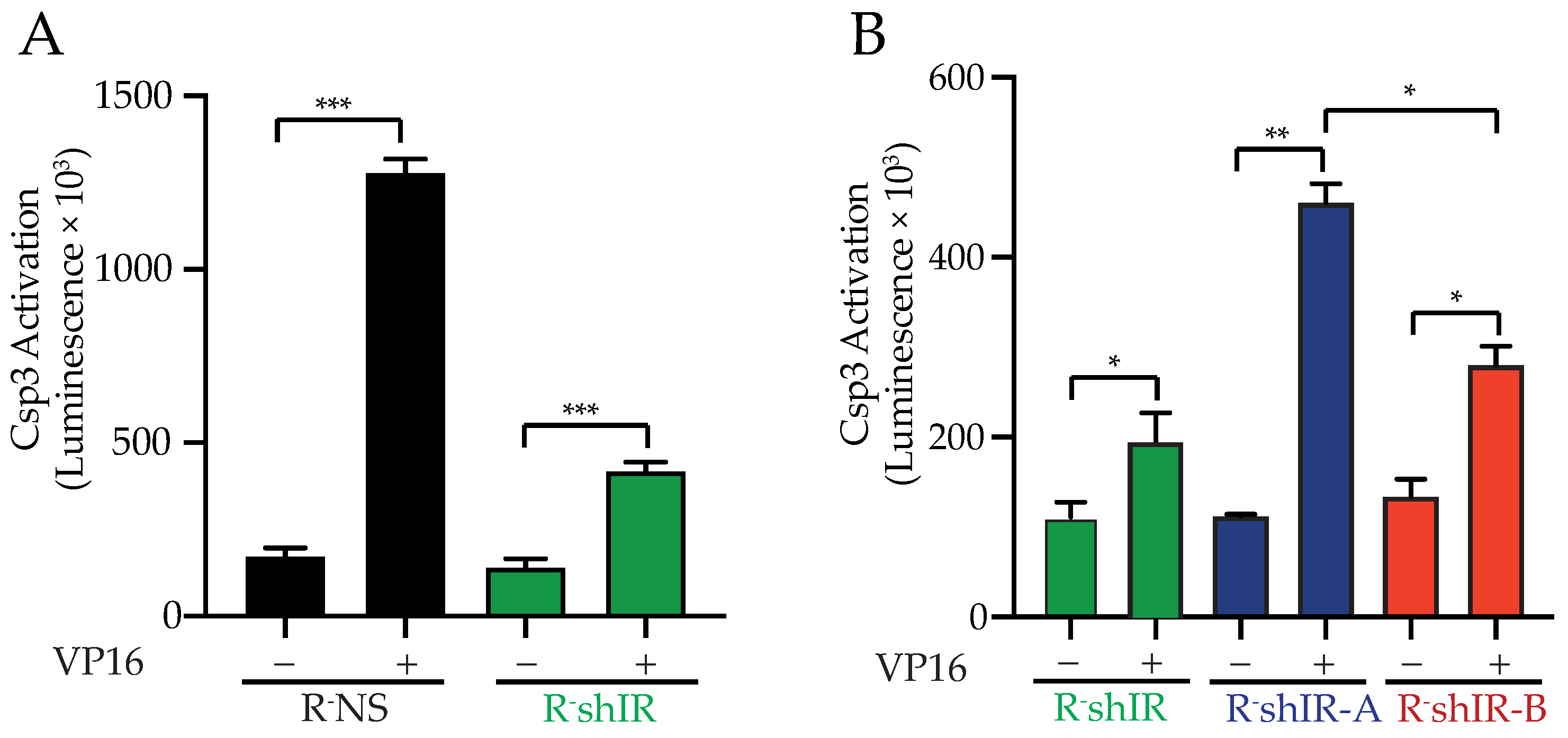
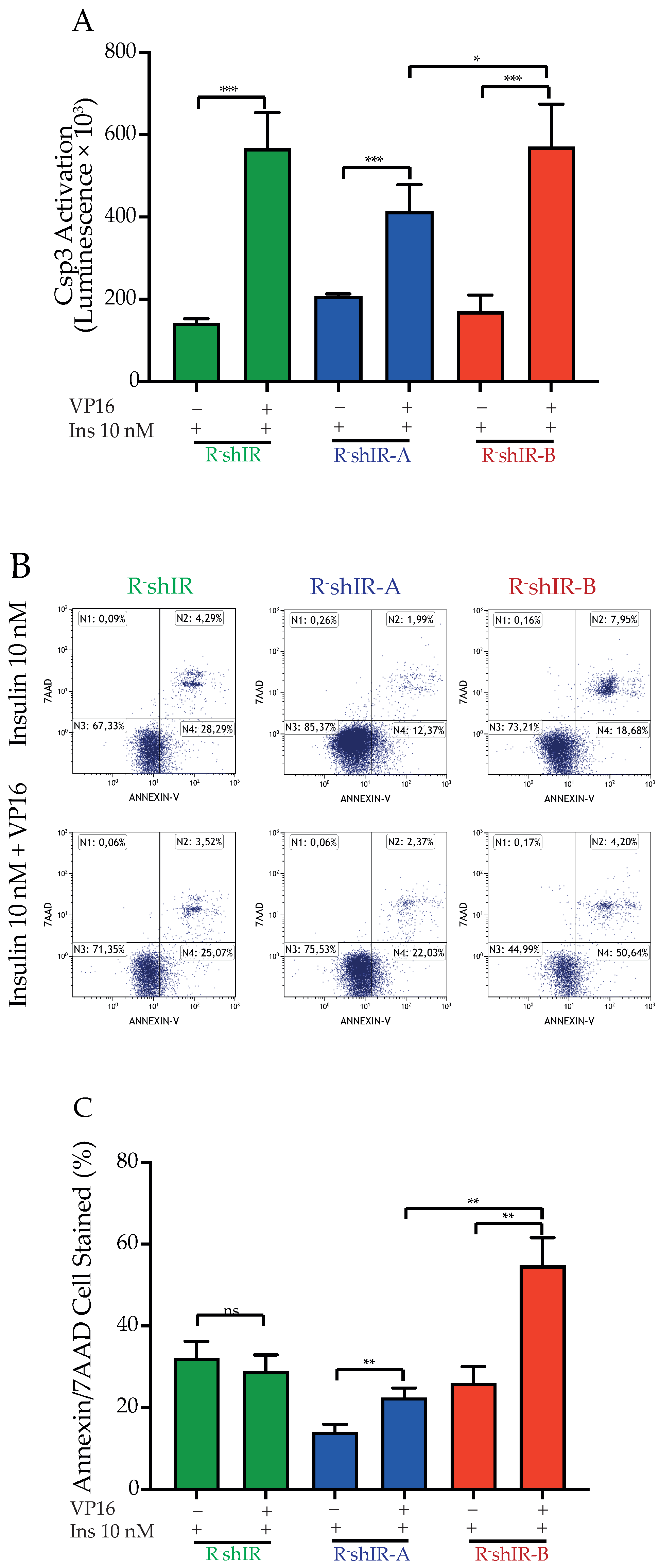
2.3. IR-A and IR-B Differently Affect Apoptosis in the Presence and Absence of Insulin
3. Discussion
4. Materials and Methods
4.1. IGF-1 Receptor Deprived Cells
4.2. Generation and Transduction of Lentiviral Vectors
4.3. Generation of R-NS, R-shIR, R-shIR-A and R-shIR-B Cell Lines and IR Measurement
4.4. Immunoblot
4.5. RNA Extraction and RT-PCR
4.6. Cell Proliferation, Caspase 3 Activation and Apoptosis Measurements
4.7. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boucher, J.; Macotela, Y.; Bezy, O.; Mori, M.A.; Kriauciunas, K.; Kahn, C.R. A Kinase-Independent Role for Unoccupied Insulin and IGF-1 Receptors in the Control of Apoptosis. Sci. Signal. 2010, 3, ra87. [Google Scholar] [CrossRef] [Green Version]
- Delcros, J.-G.; Mehlen, P. Les récepteurs à dépendance: Carrefours entre vie et mort. Bull Cancer 2013, 100, 1261–1274. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Seino, S.; Seino, M.; Nishi, S.; Bell, G.I. Structure of the human insulin receptor gene and characterization of its promoter. Proc. Natl. Acad. Sci. USA 1989, 86, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin Receptor Isoform A, a Newly Recognized, High-Affinity Insulin-Like Growth Factor II Receptor in Fetal and Cancer Cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef] [Green Version]
- Avnet, S.; Perut, F.; Salerno, M.; Sciacca, L.; Baldini, N. Insulin receptor isoforms are differently expressed during human osteoblastogenesis. Differentiation 2012, 83, 242–248. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/Insulin-like Growth Factor I Hybrid Receptors Have Different Biological Characteristics Depending on the Insulin Receptor Isoform Involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef] [Green Version]
- Sciacca, L.; Cassarino, M.F.; Genua, M.; Vigneri, P.; Pennisi, M.G.; Malandrino, P.; Squatrito, S.; Pezzino, V.; Vigneri, R. Biological Effects of Insulin and Its Analogs on Cancer Cells With Different Insulin Family Receptor Expression. J. Cell. Physiol. 2014, 229, 1817–1821. [Google Scholar] [CrossRef]
- Nevado, C.; Benito, M.; Valverde, A.M. Role of Insulin Receptor and Balance in Insulin Receptor Isoforms A and B in Regulation of Apoptosis in Simian Virus 40-immortalized Neonatal Hepatocytes. Mol. Biol. Cell 2008, 19, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Sciacca, L.; Cassarino, M.F.; Genua, M.; Pandini, G.; Le Moli, R.; Squatrito, S.; Vigneri, R.; Moli, R. Insulin analogues differently activate insulin receptor isoforms and post-receptor signalling. Diabetologia 2010, 53, 1743–1753. [Google Scholar] [CrossRef] [Green Version]
- Alvino, C.L.; Ong, S.C.; McNeil, K.A.; Delaine, C.; Booker, G.W.; Wallace, J.C.; Forbes, B.E. Understanding the Mechanism of Insulin and Insulin-Like Growth Factor (IGF) Receptor Activation by IGF-II. PLoS ONE 2011, 6, e27488. [Google Scholar] [CrossRef] [Green Version]
- Heidegger, I.; Ofer, P.; Doppler, W.; Rotter, V.; Klocker, H.; Massoner, P. Diverse Functions of IGF/Insulin Signaling in Malignant and Noncancerous Prostate Cells: Proliferation in Cancer Cells and Differentiation in Noncancerous Cells. Endocrinology 2012, 153, 4633–4643. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-F.; Zhang, G.; Zhao, L.-J.; Qi, W.-J.; Li, X.-P.; Wang, J.-L.; Wei, L.-H. Overexpression of the Insulin Receptor Isoform A Promotes Endometrial Carcinoma Cell Growth. PLoS ONE 2013, 8, e69001. [Google Scholar] [CrossRef]
- Singh, S.K.; Brito, C.; Tan, Q.; De Leon, M.; De Leon, D.D. Differential expression and signaling activation of insulin receptor isoforms A and B: A link between breast cancer and diabetes. Growth Factors 2011, 29, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Aljada, A.; Saleh, A.M.; Al-Aqeel, S.M.; Shamsa, H.B.; Al-Bawab, A.; Al Dubayee, M.; Ahmed, A.A. Quantification of insulin receptor mRNA splice variants as a diagnostic tumor marker in breast cancer. Cancer Biomark. 2015, 15, 653–661. [Google Scholar] [CrossRef]
- Andres, S.F.; Simmons, J.G.; Mah, A.T.; Santoro, M.A.; Van Landeghem, L.; Lund, P.K. Insulin receptor isoform switching in intestinal stem cells, progenitors, differentiated lineages and tumors: Evidence that IR-B limits proliferation. J. Cell Sci. 2013, 126, 5645–5656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigneri, P.; Frasca, F.; Sciacca, L.; Frittitta, L.; Vigneri, R. Obesity and cancer. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 1–7. [Google Scholar] [CrossRef]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massimino, M.; Consoli, M.L.; Mesuraca, M.; Stagno, F.; Tirrò, E.; Stella, S.; Pennisi, M.S.; Romano, C.; Buffa, P.; Bond, H.M.; et al. IRF5 is a target of BCR-ABL kinase activity and reduces CML cell proliferation. Carcinogenesis 2014, 35, 1132–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirrò, E.; Massimino, M.; Romano, C.; Pennisi, M.S.; Stella, S.; Vitale, S.R.; Fidilio, A.; Manzella, L.; Parrinello, N.L.; Stagno, F.; et al. Chk1 Inhibition Restores Inotuzumab Ozogamicin Citotoxicity in CD22-Positive Cells Expressing Mutant p53. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massimino, M.; Sciacca, L.; Parrinello, N.L.; Scalisi, N.M.; Belfiore, A.; Vigneri, R.; Vigneri, P. Insulin Receptor Isoforms Differently Regulate Cell Proliferation and Apoptosis in the Ligand-Occupied and Unoccupied State. Int. J. Mol. Sci. 2021, 22, 8729. https://doi.org/10.3390/ijms22168729
Massimino M, Sciacca L, Parrinello NL, Scalisi NM, Belfiore A, Vigneri R, Vigneri P. Insulin Receptor Isoforms Differently Regulate Cell Proliferation and Apoptosis in the Ligand-Occupied and Unoccupied State. International Journal of Molecular Sciences. 2021; 22(16):8729. https://doi.org/10.3390/ijms22168729
Chicago/Turabian StyleMassimino, Michele, Laura Sciacca, Nunziatina Laura Parrinello, Nunzio Massimo Scalisi, Antonino Belfiore, Riccardo Vigneri, and Paolo Vigneri. 2021. "Insulin Receptor Isoforms Differently Regulate Cell Proliferation and Apoptosis in the Ligand-Occupied and Unoccupied State" International Journal of Molecular Sciences 22, no. 16: 8729. https://doi.org/10.3390/ijms22168729