
In Utero Exposure to Δ9-Tetrahydrocannabinol Leads to Postnatal Catch-Up Growth and Dysmetabolism in the Adult Rat Liver



Abstract
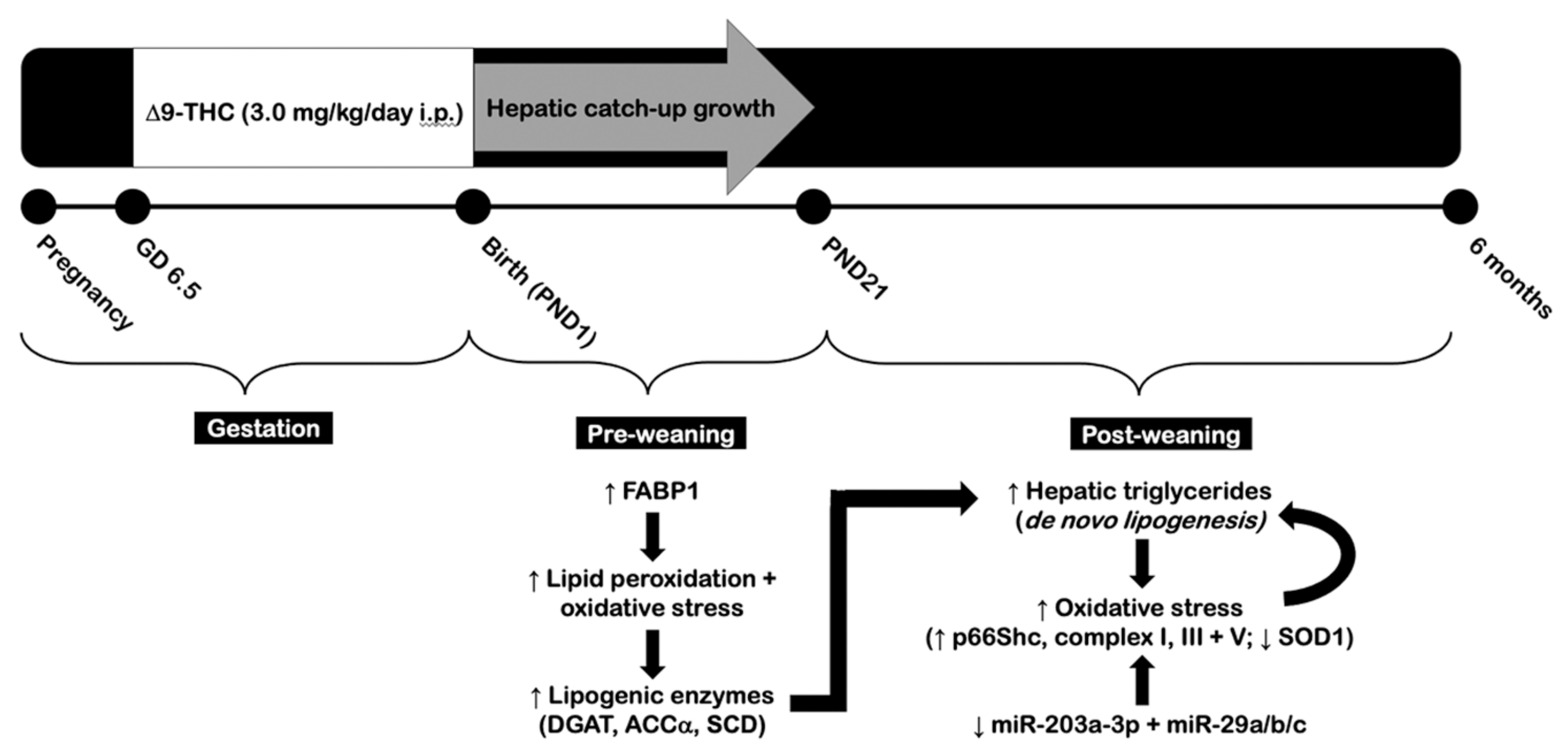
:1. Introduction
2. Results
2.1. Gestational Exposure to Δ9-THC Leads to Hepatic Catch-Up Growth by Three Weeks of Age
2.2. Adult Δ9-THC-Exposed Offspring Exhibit Elevated Visceral Adiposity and Hepatic Dyslipidemia Following Gestational Exposure to Δ9-THC
2.3. Elevated Hepatic Triglyceride Levels Coincide with Increased Diglycerol Acyltransferase and p66Shc Protein Levels in the Livers of Adult Male Δ9-THC-Exposed Offspring
2.4. Gestational Exposure to Δ9-THC Does Not Affect Protein Levels of Enzymes Involved in Aerobic Metabolism at Six Months of Age
2.5. Gestational Exposure to Δ9-THC Leads to Altered Hepatic Protein Levels of Superoxide Dismutase 1 in Adult Male Offspring
2.6. Mitochondrial Electron Transport Chain Complexes Are Increased in Male Offspring at Six Months and Three Weeks of Age Following Gestational Exposure to Δ9-THC
2.7. Adult Male Offspring Exposed to Gestational Δ9-THC Exhibit Decreased Hepatic Transcript Levels of miR-203a-3p and miR-29a/b/c
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Handling
4.2. Hepatic Triglyceride Measurements
4.3. RNA Isolation and Quantitative Real-Time PCR Analysis
4.4. Protein Extraction and Western Immunoblot
4.5. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Health Canada. Canadian Cannabis Survey 2019—Summary. Government of Canada. 2019. Available online: https://www.canada.ca/en/health-canada/services/publications/drugs-health-products/canadian-cannabis-survey-2019-summary.html (accessed on 7 July 2021).
- Committee on Obstetric Practice. Marijuana Use during Pregnancy and Lactation. Obstet. Gynecol. 2017, 130, e205–e209. [Google Scholar] [CrossRef] [PubMed]
- Westfall, R.E.; Janssen, P.A.; Lucas, P.; Capler, R. Survey of Medicinal Cannabis Use among Childbearing Women: Patterns of Its Use in Pregnancy and Retroactive Self-Assessment of Its Efficacy against “Morning Sickness”. Complement. Ther. Clin. Pract. 2006, 12, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Tarr, J.A.; Holland, C.L.; De Genna, N.M.; Richardson, G.A.; Rodriguez, K.L.; Sheeder, J.; Kraemer, K.L.; Day, N.L.; Rubio, D.; et al. Beliefs and Attitudes Regarding Prenatal Marijuana Use: Perspectives of Pregnant Women Who Report Use. Drug Alcohol Depend. 2019, 196, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.; Dakkak, H.; Gilliland, J.; Seabrook, J.A. Predictors of Drug Use during Pregnancy: The Relative Effects of Socioeconomic, Demographic, and Mental Health Risk Factors. J. Neonatal-Perinat. Med. 2019, 12, 179–187. [Google Scholar] [CrossRef] [PubMed]
- English, D.R.; Hulse, G.K.; Milne, E.; Holman, C.D.J.; Bower, C.I. Maternal Cannabis Use and Birth Weight: A Meta-Analysis. Addiction 1997, 92, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.K.L.; Rosales, C.B.; Center, K.E.; Nuñez, A.; Gibson, S.J.; Christ, C.; Ehiri, J.E. Prenatal Exposure to Cannabis and Maternal and Child Health Outcomes: A Systematic Review and Meta-Analysis. BMJ Open 2016, 6, e009986. [Google Scholar] [CrossRef] [Green Version]
- Conner, S.N.; Bedell, V.; Lipsey, K.; Macones, G.A.; Cahill, A.G.; Tuuli, M.G. Maternal Marijuana Use and Adverse Neonatal Outcomes: A Systematic Review and Meta-Analysis. Obstet. Gynecol. 2016, 128, 713–723. [Google Scholar] [CrossRef]
- Michalski, C.A.; Hung, R.J.; Seeto, R.A.; Dennis, C.L.; Brooks, J.D.; Henderson, J.; Levitan, R.; Lye, S.J.; Matthews, S.G.; Knight, J.A. Association between Maternal Cannabis Use and Birth Outcomes: An Observational Study. BMC Pregnancy Childbirth 2020, 20. [Google Scholar] [CrossRef]
- Silvestri, C.; Di Marzo, V. The Endocannabinoid System in Energy Homeostasis and the Etiopathology of Metabolic Disorders. Cell Metab. 2013, 17, 475–490. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, J.F.; Lépicier, P.; Lamontagne, D. Contribution of Endocannabinoids in the Endothelial Protection Afforded by Ischemic Preconditioning in the Isolated Rat Heart. Life Sci. 2003, 72, 1859–1870. [Google Scholar] [CrossRef] [Green Version]
- Malenczyk, K.; Keimpema, E.; Piscitelli, F.; Calvigioni, D.; Björklund, P.; Mackie, K.; Di Marzo, V.; Hökfelt, T.G.M.; Dobrzyn, A.; Harkany, T. Fetal Endocannabinoids Orchestrate the Organization of Pancreatic Islet Microarchitecture. Proc. Natl. Acad. Sci. USA 2015, 112, E6185–E6194. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Dey, S.K. Endocannabinoid Signaling in Female Reproduction. ACS Chem. Neurosci. 2012, 3, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez-López, M.T.; Arco, R.; Decara, J.; Vázquez, M.; Blanco, R.N.; Alén, F.; Suárez, J.; De Heras, R.G.; De Fonseca, F.R. Exposure to a Highly Caloric Palatable Diet during the Perinatal Period Affects the Expression of the Endogenous Cannabinoid System in the Brain, Liver and Adipose Tissue of Adult Rat Offspring. PLoS ONE 2016, 11, e0165432. [Google Scholar] [CrossRef] [Green Version]
- Maia, J.; Midão, L.; Cunha, S.C.; Almada, M.; Fonseca, B.M.; Braga, J.; Gonçalves, D.; Teixeira, N.; Correia-da-Silva, G. Effects of Cannabis Tetrahydrocannabinol on Endocannabinoid Homeostasis in Human Placenta. Arch. Toxicol. 2019, 93, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Fried, P.A. Short and Long-Term Effects of Pre-Natal Cannabis Inhalation upon Rat Offspring. Psychopharmacology 1976, 50, 285–291. [Google Scholar] [CrossRef]
- Harbison, R.D.; Mantilla-Plata, B. Prenatal toxicity, maternal distribution and placental transfer of tetrahydrocannabinol. J. Pharmacol. Exp. Ther. 1972, 180, 446–453. [Google Scholar]
- Hurd, Y.L.; Wang, X.; Anderson, V.; Beck, O.; Minkoff, H.; Dow-Edwards, D. Marijuana Impairs Growth in Mid-Gestation Fetuses. Neurotoxicol. Teratol. 2005, 27, 221–229. [Google Scholar] [CrossRef]
- Benevenuto, S.G.; Domenico, M.D.; Martins, M.A.G.; Costa, N.S.; de Souza, A.R.L.; Costa, J.L.; Tavares, M.F.M.; Dolhnikoff, M.; Veras, M.M. Recreational Use of Marijuana during Pregnancy and Negative Gestational and Fetal Outcomes: An Experimental Study in Mice. Toxicology 2017, 376, 94–101. [Google Scholar] [CrossRef]
- Chang, X.; Bian, Y.; He, Q.; Yao, J.; Zhu, J.; Wu, J.; Wang, K.; Duan, T. Suppression of STAT3 Signaling by Δ 9 -Tetrahydrocannabinol (THC) Induces Trophoblast Dysfunction. Cell. Physiol. Biochem. 2017, 42, 537–550. [Google Scholar] [CrossRef]
- Natale, B.V.; Gustin, K.N.; Lee, K.; Holloway, A.C.; Laviolette, S.R.; Natale, D.R.C.; Hardy, D.B. Δ9-Tetrahydrocannabinol Exposure during Rat Pregnancy Leads to Symmetrical Fetal Growth Restriction and Labyrinth-Specific Vascular Defects in the Placenta. Sci. Rep. 2020, 10, 544. [Google Scholar] [CrossRef]
- James, P.T.; Rigby, N.; Leach, R. The Obesity Epidemic, Metabolic Syndrome and Future Prevention Strategies. Eur. J. Cardiovasc. Prev. Rehabil. 2004, 11, 3–8. [Google Scholar] [CrossRef]
- Eriksson, J.; Forsén, T.; Tuomilehto, J.; Osmond, C.; Barker, D. Size at Birth, Childhood Growth and Obesity in Adult Life. Int. J. Obes. 2001, 25, 735–740. [Google Scholar] [CrossRef] [Green Version]
- Suomela, E.; Oikonen, M.; Pitkänen, N.; Ahola-Olli, A.; Virtanen, J.; Parkkola, R.; Jokinen, E.; Laitinen, T.; Hutri-Kähönen, N.; Kähönen, M.; et al. Childhood Predictors of Adult Fatty Liver. The Cardiovascular Risk in Young Finns Study. J. Hepatol. 2016, 65, 784–790. [Google Scholar] [CrossRef] [Green Version]
- Nobili, V.; Marcellini, M.; Marchesini, G.; Vanni, E.; Manco, M.; Villani, A.; Bugianesi, E. Intrauterine Growth Retardation, Insulin Resistance, and Nonalcoholic Fatty Liver Disease in Children. Diabetes Care 2007, 10, 2638–2640. [Google Scholar] [CrossRef] [Green Version]
- Ravelli, G.P.; Stein, Z.A.; Susser, M.W. Obesity in Young Men after Famine Exposure in Utero and Early Infancy. N. Engl. J. Med. 1976, 295, 349–353. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, W.; Zhang, X.; Mu, R.; Zhai, Y.; Kong, L.; Chen, C. Impact of Famine during Pregnancy and Infancy on Health in Adulthood. Obes. Rev. 2008, 9, 95–99. [Google Scholar] [CrossRef]
- Faienza, M.F.; Brunetti, G.; Ventura, A.; D’Aniello, M.; Pepe, T.; Giordano, P.; Monteduro, M.; Cavallo, L. Nonalcoholic Fatty Liver Disease in Prepubertal Children Born Small for Gestational Age: Influence of Rapid Weight Catch-Up Growth. HRP 2013, 79, 103–109. [Google Scholar] [CrossRef]
- Barker, D.J.; Martyn, C.N.; Osmond, C.; Hales, C.N.; Fall, C.H. Growth in Utero and Serum Cholesterol Concentrations in Adult Life. BMJ 1993, 307, 1524–1527. [Google Scholar] [CrossRef] [Green Version]
- Ma, N.; Nicholson, C.J.; Wong, M.; Holloway, A.C.; Hardy, D.B. Fetal and Neonatal Exposure to Nicotine Leads to Augmented Hepatic and Circulating Triglycerides in Adult Male Offspring Due to Increased Expression of Fatty Acid Synthase. Toxicol. Appl. Pharmacol. 2013, 275, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sohi, G.; Marchand, K.; Revesz, A.; Arany, E.; Hardy, D.B. Maternal Protein Restriction Elevates Cholesterol in Adult Rat Offspring Due to Repressive Changes in Histone Modifications at the Cholesterol 7alpha-Hydroxylase Promoter. Mol. Endocrinol. 2011, 25, 785–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen-Urstad, A.P.; Semenkovich, C.F. Fatty Acid Synthase and Liver Triglyceride Metabolism: Housekeeper or Messenger? Biochim. Biophys. Acta 2012, 1821, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H. Regulation of Mammalian Acetyl-Coenzyme A Carboxylase. Annu. Rev. Nutr. 1997, 17, 77–99. [Google Scholar] [CrossRef]
- Miyazaki, M.; Ntambi, J.M. Role of Stearoyl-Coenzyme A Desaturase in Lipid Metabolism. Prostaglandins Leukot. Essent. Fat. Acids 2003, 68, 113–121. [Google Scholar] [CrossRef]
- Cases, S.; Smith, S.J.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J.; et al. Identification of a Gene Encoding an Acyl CoA:Diacylglycerol Acyltransferase, a Key Enzyme in Triacylglycerol Synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.J.; Cases, S.; Jensen, D.R.; Chen, H.C.; Sande, E.; Tow, B.; Sanan, D.A.; Raber, J.; Eckel, R.H.; Farese, R.V. Obesity Resistance and Multiple Mechanisms of Triglyceride Synthesis in Mice Lacking Dgat. Nat. Genet. 2000, 25, 87–90. [Google Scholar] [CrossRef]
- Deodati, A.; Argemí, J.; Germani, D.; Puglianiello, A.; Alisi, A.; De Stefanis, C.; Ferrero, R.; Nobili, V.; Aragón, T.; Cianfarani, S. The Exposure to Uteroplacental Insufficiency is Associated with Activation of Unfolded Protein Response in Postnatal Life. PLoS ONE 2018, 13, e0198490. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Wolfe, D.; Han, G.; French, S.W.; Ross, M.G.; Desai, M. Early Onset of Fatty Liver in Growth-Restricted Rat Fetuses and Newborns. Congenit. Anom. 2011, 51, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Ji, S.; Jia, P.; Zhang, H.; Wang, T.; Song, Z.; Zhang, L.; Wang, T. Resveratrol Improves Hepatic Redox Status and Lipid Balance of Neonates with Intrauterine Growth Retardation in a Piglet Model. BioMed Res. Int. 2020, 2020, 7402645. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Li, D.; Yin, Y.; Wang, X.; Li, P.; Dangott, L.J.; Hu, W.; Wu, G. Intrauterine Growth Restriction Affects the Proteomes of the Small Intestine, Liver, and Skeletal Muscle in Newborn Pigs. J. Nutr. 2008, 138, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, A.; Rajasekaran, N.S.; Valburg, C.; Ganapathy, E.; Bindra, S.; Freije, W.A. Maternal Perinatal Calorie Restriction Temporally Regulates the Hepatic Autophagy and Redox Status in Male Rat. Free. Radic. Biol. Med. 2019, 130, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.; Rebelato, H.J.; Amaral, M.E.C.; Resende, T.M.; Silva, E.V.C.; Esquisatto, M.A.M.; Catisti, R. Effect of Maternal Protein Restriction on Liver Metabolism in Rat Offspring. J. Physiol. Sci. 2014, 64, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Theys, N.; Clippe, A.; Bouckenooghe, T.; Reusens, B.; Remacle, C. Early Low Protein Diet Aggravates Unbalance between Antioxidant Enzymes Leading to Islet Dysfunction. PLoS ONE 2009, 4, e6110. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Kim, S.K.; Kim, M.S.; Cho, E.Y.; Lee, J.H.; Lee, K.-U.; Pak, Y.K.; Lee, H.K. Fetal and Early Postnatal Protein Malnutrition Cause Long-Term Changes in Rat Liver and Muscle Mitochondria. J. Nutr. 2003, 133, 3085–3090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oke, S.L.; Sohi, G.; Hardy, D.B. Perinatal Protein Restriction with Postnatal Catch-up Growth Leads to Elevated P66Shc and Mitochondrial Dysfunction in the Adult Rat Liver. Reproduction 2020, 159, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron Transfer between Cytochrome c and P66Shc Generates Reactive Oxygen Species That Trigger Mitochondrial Apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Berniakovich, I.; Trinei, M.; Stendardo, M.; Migliaccio, E.; Minucci, S.; Bernardi, P.; Pelicci, P.G.; Giorgio, M. P66Shc-Generated Oxidative Signal Promotes Fat Accumulation. J. Biol. Chem. 2008, 283, 34283–34293. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K.; Teratani, T.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Nishiyama, K.; Mataki, N.; Irie, R.; Minamino, T.; et al. P53/P66Shc-Mediated Signaling Contributes to the Progression of Non-Alcoholic Steatohepatitis in Humans and Mice. J. Hepatol. 2012, 57, 837–843. [Google Scholar] [CrossRef]
- Luyckx, V.A.; Compston, C.A.; Simmen, T.; Mueller, T.F. Accelerated Senescence in Kidneys of Low-Birth-Weight Rats after Catch-up Growth. Am. J. Physiol. Ren. Physiol. 2009, 297, F1697–F1705. [Google Scholar] [CrossRef] [Green Version]
- Raez-Villanueva, S.; Debnath, A.; Hardy, D.B.; Holloway, A.C. Prenatal Nicotine Exposure Leads to Decreased Histone H3 Lysine 9 (H3K9) Methylation and Increased P66shc Expression in the Neonatal Pancreas. J. Dev. Orig. Health Dis. 2021, 28, 1–5. [Google Scholar] [CrossRef]
- Sohi, G.; Revesz, A.; Ramkumar, J.; Hardy, D.B. Higher Hepatic MiR-29 Expression in Undernourished Male Rats during the Postnatal Period Targets the Long-Term Repression of IGF-1. Endocrinology 2015, 156, 3069–3076. [Google Scholar] [CrossRef]
- Barra, N.G.; Lisyansky, M.; Vanduzer, T.A.; Raha, S.; Holloway, A.C.; Hardy, D.B. Maternal Nicotine Exposure Leads to Decreased Cardiac Protein Disulfide Isomerase and Impaired Mitochondrial Function in Male Rat Offspring. J. Appl. Toxicol. 2017, 37, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Mechanisms of Hepatic Lipid Accumulation in Non-Alcoholic Fatty Liver Disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Lone, A.; Harris, R.A.; Singh, O.; Betts, D.H.; Cumming, R.C. P66Shc Activation Promotes Increased Oxidative Phosphorylation and Renders CNS Cells More Vulnerable to Amyloid Beta Toxicity. Sci. Rep. 2018, 8, 17081. [Google Scholar] [CrossRef]
- Brand, M.D. The Sites and Topology of Mitochondrial Superoxide Production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhao, Y.; Zhao, H.; Zhou, J.; Feng, D.; Tang, F.; Li, Y.; Lv, L.; Chen, Z.; Ma, X.; et al. Inhibition of P66Shc Oxidative Signaling via CA-Induced Upregulation of MiR-203a-3p Alleviates Liver Fibrosis Progression. Mol. Ther. Nucleic Acids 2020, 21, 751–763. [Google Scholar] [CrossRef]
- Musa, M.G.; Kagura, J.; Pisa, P.T.; Norris, S.A. Relationship between Early Growth and CVD Risk Factors in Adolescents. J. Dev. Orig. Health Dis. 2015, 7, 132–143. [Google Scholar] [CrossRef]
- Victora, C.G.; Barros, F.C.; Lima, R.C.; Behague, D.P.; Gon alves, H.; Horta, B.L.; Gigante, D.P.; Vaughan, J.P. The Pelotas Birth Cohort Study, Rio Grande Do Sul, Brazil, 1982–2001. Cad Saude Publica. 2003, 19, 1241–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perng, W.; Hajj, H.; Belfort, M.B.; Rifas-Shiman, S.L.; Kramer, M.S.; Gillman, M.W.; Oken, E. Birth Size, Early Life Weight Gain, and Midchildhood Cardiometabolic Health. J. Pediatr. 2016, 173, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.J.; Holloway, A.C.; Zeng, Z.H.; Lim, G.E.; Petrik, J.J.; Foster, W.G.; Lee, R.M. Prenatal Exposure to Nicotine Causes Postnatal Obesity and Altered Perivascular Adipose Tissue Function. Obes. Res. 2005, 13, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Arany, E.; van Beek, J.P.; Chamson-Reig, A.; Thyssen, S.; Hill, D.J.; Yang, K. Adipose Tissue Gene Expression Profiling Reveals Distinct Molecular Pathways That Define Visceral Adiposity in Offspring of Maternal Protein-Restricted Rats. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E663–E673. [Google Scholar] [CrossRef] [PubMed]
- Riediger, N.D.; Clara, I. Prevalence of Metabolic Syndrome in the Canadian Adult Population. CMAJ 2011, 183, E1127–E1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillies, R.; Lee, K.; Vanin, S.; Laviolette, S.R.; Holloway, A.C.; Arany, E.; Hardy, D.B. Maternal Exposure to Δ9-Tetrahydrocannabinol Impairs Female Offspring Glucose Homeostasis and Endocrine Pancreatic Development in the Rat. Reprod. Toxicol. 2020, 94, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Della Torre, S.; Stell, A.; Cook, J.; Brown, M.; Maggi, A. Tetradian Oscillation of Estrogen Receptor α Is Necessary to Prevent Liver Lipid Deposition. Proc. Natl. Acad. Sci. USA 2012, 109, 11806–11811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.J.; Zhao, Z.; Glidewell-Kenney, C.; Lazic, M.; Chambon, P.; Krust, A.; Weiss, J.; Clegg, D.J.; Dunaif, A.; Jameson, J.L.; et al. Genetic Rescue of Nonclassical ERα Signaling Normalizes Energy Balance in Obese Erα-Null Mutant Mice. J. Clin. Investig. 2011, 121, 604–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of Hepatic Steatosis in an Urban Population in the United States: Impact of Ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Kojima, T.; Takeda, N.; Nakagawa, T.; Taniguchi, H.; Fujii, K.; Omatsu, T.; Nakajima, T.; Sarui, H.; Shimazaki, M.; et al. The Metabolic Syndrome as a Predictor of Nonalcoholic Fatty Liver Disease. Ann. Intern. Med. 2005, 143, 722–728. [Google Scholar] [CrossRef]
- Suzuki, A.; Angulo, P.; Lymp, J.; St Sauver, J.; Muto, A.; Okada, T.; Lindor, K. Chronological Development of Elevated Aminotransferases in a Nonalcoholic Population. Hepatology 2005, 41, 64–71. [Google Scholar] [CrossRef]
- Tsuneto, A.; Hida, A.; Sera, N.; Imaizumi, M.; Ichimaru, S.; Nakashima, E.; Seto, S.; Maemura, K.; Akahoshi, M. Fatty Liver Incidence and Predictive Variables. Hypertens. Res. 2010, 33, 638–643. [Google Scholar] [CrossRef] [Green Version]
- Sung, K.-C.; Kim, B.-S.; Cho, Y.-K.; Park, D.; Woo, S.; Kim, S.; Wild, S.H.; Byrne, C.D. Predicting Incident Fatty Liver Using Simple Cardio-Metabolic Risk Factors at Baseline. BMC Gastroenterol. 2012, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Pramfalk, C.; Pavlides, M.; Banerjee, R.; McNeil, C.A.; Neubauer, S.; Karpe, F.; Hodson, L. Sex-Specific Differences in Hepatic Fat Oxidation and Synthesis May Explain the Higher Propensity for NAFLD in Men. J. Clin. Endocrinol. Metab. 2015, 100, 4425–4433. [Google Scholar] [CrossRef] [Green Version]
- Tran, C.; Jacot-Descombes, D.; Lecoultre, V.; Fielding, B.A.; Carrel, G.; Lê, K.-A.; Schneiter, P.; Bortolotti, M.; Frayn, K.N.; Tappy, L. Sex Differences in Lipid and Glucose Kinetics after Ingestion of an Acute Oral Fructose Load. Br. J. Nutr. 2010, 104, 1139–1147. [Google Scholar] [CrossRef] [Green Version]
- Leamy, A.K.; Hasenour, C.M.; Egnatchik, R.A.; Trenary, I.A.; Yao, C.-H.; Patti, G.J.; Shiota, M.; Young, J.D. Knockdown of Triglyceride Synthesis Does Not Enhance Palmitate Lipotoxicity or Prevent Oleate-Mediated Rescue in Rat Hepatocytes. Biochim. Biophys. Acta 2016, 1861, 1005–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmes, M.W.; Kaczocha, M.; Berger, W.T.; Leung, K.; Ralph, B.P.; Wang, L.; Sweeney, J.M.; Miyauchi, J.T.; Tsirka, S.E.; Ojima, I.; et al. Fatty Acid-Binding Proteins (FABPs) Are Intracellular Carriers for Δ9-Tetrahydrocannabinol (THC) and Cannabidiol (CBD)*. J. Biol. Chem. 2015, 290, 8711–8721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmes, M.W.; Prentis, L.E.; McGoldrick, L.L.; Giuliano, C.J.; Sweeney, J.M.; Joseph, O.M.; Che, J.; Carbonetti, G.S.; Studholme, K.; Deutsch, D.G.; et al. FABP1 Controls Hepatic Transport and Biotransformation of Δ 9 -THC. Sci. Rep. 2019, 9, 7588. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Egawa, M.; Takeuchi, T.; Yamashita, H.; Kusudo, T. Silencing of FABP1 Ameliorates Hepatic Steatosis, Inflammation, and Oxidative Stress in Mice with Nonalcoholic Fatty Liver Disease. FEBS Open Bio 2017, 7, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Steatohepatitis. Free. Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Free. Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Eccleston, H.B.; Bailey, S.M. Mitochondrial Dysfunction and Oxidative Stress in the Pathogenesis of Alcohol- and Obesity-Induced Fatty Liver Diseases. Free. Radic. Biol. Med. 2008, 44, 1259–1272. [Google Scholar] [CrossRef] [Green Version]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial Adaptations and Dysfunctions in Nonalcoholic Fatty Liver Disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Seo, E.; Kang, H.; Choi, H.; Choi, W.; Jun, H.S. Reactive Oxygen Species-Induced Changes in Glucose and Lipid Metabolism Contribute to the Accumulation of Cholesterol in the Liver during Aging. Aging Cell 2019, 18. [Google Scholar] [CrossRef] [Green Version]
- Lockman, K.A.; Baren, J.P.; Pemberton, C.J.; Baghdadi, H.; Burgess, K.E.; Plevris-Papaioannou, N.; Lee, P.; Howie, F.; Beckett, G.; Pryde, A.; et al. Oxidative Stress Rather than Triglyceride Accumulation Is a Determinant of Mitochondrial Dysfunction in in Vitro Models of Hepatic Cellular Steatosis. Liver Int. 2012, 32, 1079–1092. [Google Scholar] [CrossRef]
- Migliaccio, E.; Giogio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The P66(Shc) Adaptor Protein Controls Oxidative Stress Response and Life Span in Mammals. Nature 1999, 402, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Migliaccio, E.; Moroni, M.; Contursi, C.; Raker, V.A.; Piccini, D.; Martin-Padura, I.; Pelliccia, G.; Trinei, M.; Bono, M.; et al. The Life Span Determinant P66Shc Localizes to Mitochondria Where It Associates with Mitochondrial Heat Shock Protein 70 and Regulates Trans-Membrane Potential. J. Biol. Chem. 2004, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinei, M.; Migliaccio, E.; Bernardi, P.; Paolucci, F.; Pelicci, P.; Giorgio, M. p66Shc, Mitochondria, and the Generation of Reactive Oxygen Species. Methods Enzymol. 2013, 528, 99–110. [Google Scholar] [PubMed]
- Firmin, S.; Elmhiri, G.; Crepin, D.; Souidi, M.; Taouis, M.; Abdennebi-Najar, L. Formula Derived Maillard Reaction Products in Post-Weaning Intrauterine Growth-Restricted Piglets Induce Developmental Programming of Hepatic Oxidative Stress Independently of MicroRNA-21 and MicroRNA-155. J. Dev. Orig. Health Dis. 2018, 9, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Zinkhan, E.K.; Yu, B.; McKnight, R. Uteroplacental Insufficiency Impairs Cholesterol Elimination in Adult Female Growth-Restricted Rat Offspring Fed a High-Fat Diet. Reprod. Sci. 2019, 26, 1173–1180. [Google Scholar] [CrossRef]
- Saget, S.; Cong, R.; Decourtye, L.; Endale, M.-L.; Martinerie, L.; Girardet, C.; Perret, C.; Clemessy, M.; Leneuve, P.; Dinard, L.; et al. Changes in Circulating MiRNA19a-3p Precede Insulin Resistance Programmed by Intra-Uterine Growth Retardation in Mice. Mol. Metab. 2020, 42, 101083. [Google Scholar] [CrossRef]
- Roderburg, C.; Urban, G.-W.; Bettermann, K.; Vucur, M.; Zimmermann, H.; Schmidt, S.; Janssen, J.; Koppe, C.; Knolle, P.; Castoldi, M.; et al. Micro-RNA Profiling Reveals a Role for MiR-29 in Human and Murine Liver Fibrosis. Hepatology 2011, 53, 209–218. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Wang, F.-S.; Yang, Y.-L.; Huang, Y.-H. MicroRNA-29a Suppresses CD36 to Ameliorate High Fat Diet-Induced Steatohepatitis and Liver Fibrosis in Mice. Cells 2019, 8, 1298. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-L.; Kuo, H.-C.; Wang, F.-S.; Huang, Y.-H. MicroRNA-29a Disrupts DNMT3b to Ameliorate Diet-Induced Non-Alcoholic Steatohepatitis in Mice. Int. J. Mol. Sci. 2019, 20, 1499. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Laviolette, S.R.; Hardy, D.B. Exposure to Δ9-Tetrahydrocannabinol during Rat Pregnancy Leads to Impaired Cardiac Dysfunction in Postnatal Life. Pediatr. Res. 2021, 1–8. [Google Scholar] [CrossRef]
- Grundy, D. Principles and Standards for Reporting Animal Experiments in the Journal of Physiology and Experimental Physiology. Exp. Physiol. 2015, 100, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Karanges, E.; Spiro, A.; Wong, A.; Spencer, J.; Huynh, T.; Gunasekaran, N.; Karl, T.; Long, L.E.; Huang, X.-F.; et al. Cannabidiol Potentiates Δ9-Tetrahydrocannabinol (THC) Behavioural Effects and Alters THC Pharmacokinetics during Acute and Chronic Treatment in Adolescent Rats. Psychopharmacology 2011, 218, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Falcon, M.; Pichini, S.; Joya, J.; Pujadas, M.; Sanchez, A.; Vall, O.; García Algar, O.; Luna, A.; de la Torre, R.; Rotolo, M.C.; et al. Maternal Hair Testing for the Assessment of Fetal Exposure to Drug of Abuse during Early Pregnancy: Comparison with Testing in Placental and Fetal Remains. Forensic Sci. Int. 2012, 218, 92–96. [Google Scholar] [CrossRef]
- Schwope, D.M.; Karschner, E.L.; Gorelick, D.A.; Huestis, M.A. Identification of Recent Cannabis Use: Whole-Blood and Plasma Free and Glucuronidated Cannabinoid Pharmacokinetics Following Controlled Smoked Cannabis Administration. Clin. Chem. 2011, 57, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Mato, S.; Chevaleyre, V.; Robbe, D.; Pazos, A.; Castillo, P.E.; Manzoni, O.J. A Single In-Vivo Exposure to Δ9THC Blocks Endocannabinoid-Mediated Synaptic Plasticity. Nat. Neurosci. 2004, 7, 585–586. [Google Scholar] [CrossRef]
- Tortoriello, G.; Morris, C.V.; Alpar, A.; Fuzik, J.; Shirran, S.L.; Calvigioni, D.; Keimpema, E.; Botting, C.H.; Reinecke, K.; Herdegen, T.; et al. Miswiring the Brain: Δ9-Tetrahydrocannabinol Disrupts Cortical Development by Inducing an SCG10/Stathmin-2 Degradation Pathway. EMBO J. 2014, 33, 668–685. [Google Scholar] [CrossRef]
- Dinieri, J.A.; Hurd, Y.L. Rat Models of Prenatal and Adolescent Cannabis Exposure. Methods Mol. Biol. 2012, 829, 231–242. [Google Scholar] [CrossRef]
- Hayes, E.K.; Lechowicz, A.; Petrik, J.J.; Storozhuk, Y.; Paez-Parent, S.; Dai, Q.; Samjoo, I.A.; Mansell, M.; Gruslin, A.; Holloway, A.C.; et al. Adverse Fetal and Neonatal Outcomes Associated with a Life-Long High Fat Diet: Role of Altered Development of the Placental Vasculature. PLoS ONE 2012, 7, e33370. [Google Scholar] [CrossRef] [Green Version]
- Beamish, C.A.; Zhang, L.; Szlapinski, S.K.; Strutt, B.J.; Hill, D.J. An Increase in Immature β-Cells Lacking Glut2 Precedes the Expansion of β-Cell Mass in the Pregnant Mouse. PLoS ONE 2017, 12, e0182256. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Group | Sex | Birth | Three Weeks | Six Months | |
|---|---|---|---|---|---|
| Liver to Body Weight Ratio | Control | Male | 0.0458 a ± 0.0026 | 0.0319 a ± 0.0024 | |
| Female | 0.0445 a ± 0.0017 | 0.0354 a ± 0.0034 | |||
| Both sexes | 0.0392 # ± 0.0039 | 0.0451 # ± 0.0011 | 0.0333 # ± 0.0020 | ||
| Δ9-THC | Male | 0.0459 a ± 0.0010 | 0.0350 a ± 0.0017 | ||
| Female | 0.0437 a ± 0.0009 | 0.0351 a ± 0.0022 | |||
| Both sexes | 0.0293 ◆ ± 0.0018 | 0.0445 #± 0.0020 | 0.0351 # ± 0.0014 | ||
| Adipose to Body Weight Ratio | Control | Male | 0.01685 a ± 0.0015 | ||
| Female | 0.0202 a ± 0.0025 | ||||
| Both sexes | 0.01828 # ± 0.0014 | ||||
| Δ9-THC | Male | 0.0207 a ± 0.0020 | |||
| Female | 0.0234 a ± 0.0022 | ||||
| Both sexes | 0.0223 ◆ ± 0.0015 |
| Antibody Name | Source | Dilution | Company (Catalogue No.) |
|---|---|---|---|
| ACCα (H-76) | Rabbit polyclonal | 1:500 | Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA (sc-30212) |
| FAS (C20G5) | Rabbit monoclonal | 1:1000 | Cell Signaling Technology Inc., Danvers, MA, USA (#3180) |
| SCD (H300) | Rabbit polyclonal | 1:250 | Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA (sc-30081) |
| FABP1 (D2A3X) | Rabbit monoclonal | 1:1000 | Cell Signaling Technology Inc., Danvers, MA, USA (#13368) |
| DGAT1 | Rabbit polyclonal | 1:1000 | Novus Biologicals, Centennial, CO, USA (NB110-41487) |
| DGAT2 (4C1) | Mouse monoclonal | 1:1000 | Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA (sc-293211) |
| SHC | Mouse monoclonal | 1:1000 | BD BioSciences, San Jose, CA, USA (610879) |
| TFAM (D5C8) | Rabbit monoclonal | 1:1000 | Cell Signaling Technology Inc., Danvers, MA, USA (8076) |
| pSer(232) pyruvate dehydrogenase | Rabbit polyclonal | 1:1000 | EMD Millipore, Etobicoke, ON, Canada (AP1063) |
| Pyruvate dehydrogenase | Rabbit polyclonal | 1:1000 | Cell Signaling Technology Inc., Danvers, MA, USA (2784) |
| LDHa | Rabbit polyclonal | 1:1000 | Cell Signaling Technology Inc., Danvers, MA, USA (2012) |
| Citrate Synthase | Rabbit polyclonal | 1:1000 | Provided by Dr. S. Raha, McMaster University |
| Catalase (H-300) | Rabbit polyclonal | 1:1000 | Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA (sc-50508) |
| Superoxide dismutase (SOD)-1 (FL-154) | Rabbit polyclonal | 1:1000 | Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA (sc-11407) |
| Superoxide dismutase (SOD)-2 (FL-222) | Rabbit polyclonal | 1:1000 | Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA (sc-30080) |
| 4-hydroxynonenal | Mouse monoclonal | 1:1000 | R&D Systems, Oakville, ON, Canada (MAB3249) |
| OXPHOS rodent cocktail | Mouse monoclonal | 1:1000 | Abcam Inc., Toronto, ON, Canada (ab110413) |
| β-Actin Peroxidase | Mouse monoclonal | 1:25,000 | Sigma Aldrich Co., St. Louis, MO, USA (A3854-200UL) |
| Goat anti-rabbit IgG HRP-linked (H+L chain) | N/A | 1:10,000 | Cell Signaling Technology Inc., Danvers, MA, USA (7074P2) |
| Horse anti-mouse IgG HRP-linked (H+L chain) | N/A | 1:10,000 | Cell Signaling Technology Inc., Danvers, MA, USA (7076S) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oke, S.L.; Lee, K.; Papp, R.; Laviolette, S.R.; Hardy, D.B. In Utero Exposure to Δ9-Tetrahydrocannabinol Leads to Postnatal Catch-Up Growth and Dysmetabolism in the Adult Rat Liver. Int. J. Mol. Sci. 2021, 22, 7502. https://doi.org/10.3390/ijms22147502
Oke SL, Lee K, Papp R, Laviolette SR, Hardy DB. In Utero Exposure to Δ9-Tetrahydrocannabinol Leads to Postnatal Catch-Up Growth and Dysmetabolism in the Adult Rat Liver. International Journal of Molecular Sciences. 2021; 22(14):7502. https://doi.org/10.3390/ijms22147502
Chicago/Turabian StyleOke, Shelby L., Kendrick Lee, Rosemary Papp, Steven R. Laviolette, and Daniel B. Hardy. 2021. "In Utero Exposure to Δ9-Tetrahydrocannabinol Leads to Postnatal Catch-Up Growth and Dysmetabolism in the Adult Rat Liver" International Journal of Molecular Sciences 22, no. 14: 7502. https://doi.org/10.3390/ijms22147502