
Peptides Derived from Growth Factors to Treat Alzheimer’s Disease

 and
and
Abstract
:1. Introduction
2. Current Strategies Targeting AD Development
3. Growth Factors in Brain Function and AD
3.1. Neurotrophins
3.1.1. Structure
3.1.2. Neurotrophin Receptors and Signal Transduction
3.1.3. Effects of Neurotrophins on the CNS Cells
- NGF
- BDNF
3.1.4. Effects of Neurotrophins on AD Hallmarks
- NGF
- BDNF
3.2. The Bone Morphogenetic Protein (BMP)
3.2.1. Pro-BMP and Mature BMP Complexes
3.2.2. BMP Receptors and Signal Transduction
3.2.3. Effect of BMP on CNS
3.2.4. Effect of BMP on AD Hallmarks
3.3. FGF and Other Growth Factors
3.3.1. FGF
- Receptors and signal transduction
- Effect of FGF on CNS and AD hallmarks
3.3.2. Other Growth Factors
4. Peptides Derived from Growth Factors
4.1. Peptides Derived from Neurotrophins
4.1.1. NGF
- Peptides derived from mNGF L1-L4 loops
- Linear peptides derived from the mNGF N-terminal region
- Peptides designed by combining sequences from mNGF loops L1 and L4 and the N-terminal region
- Effect of NGF-derived peptides in the context of neurodegenerative diseases
4.1.2. BDNF
- Linear peptides derived from mBDNF
- Cyclic dimeric peptides derived from mBDNF loops L2 and L4
- Peptides combining different regions of mBDNF
- Effect of BDNF-derived peptides in the context of brain trauma and diseases
4.2. Peptides Derived from BMP
4.2.1. Peptides Derived from the Knuckle Epitope
4.2.2. Peptide Derived from the Wrist Epitope
4.3. Peptides Derived from FGF and Other Factors
4.3.1. FGF-2
- Peptide derived from FGF-2
4.3.2. Other Growth Factors
- Peptides derived from IGF
- Peptides derived from CNTF
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV2-NGF | Adenoviral vector to deliver NGF |
| ActR-1A | Type 1A activin receptor |
| ActRIIA | Type II activin receptor |
| ActRIIB | Type IIB activin receptor |
| AD | Alzheimer’s disease |
| ADAM17 | A disintegrin and metalloproteinase 17 |
| ALK | Activin receptor-like kinases receptor |
| AMH/MIS | Anti-Müllerian hormone/Müllerian inhibiting substance |
| AMHRII | Anti-Mullerian hormone receptor type II |
| AMPA | Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate |
| APOE | Apolipoprotein E |
| APOE4 | Apolipoprotein E4 |
| APP | Amyloid-β precursor protein |
| AVVS2-NGF | Adeno-associated virus serotype 2 delivering NGF |
| Aβ | Amyloid-β |
| BAMBI | BMP and activin membrane-bound inhibitor |
| BBB | Blood–brain barrier |
| BDNF | Brain-derived neurotrophic factor (mature form: mBDNF) |
| BFCNs | Cholinergic neurons of the basal forebrain |
| BMP | Bone morphogenetic protein (mature form) |
| BMPR-1A | Type 1A BMP receptor |
| BMPR-1B | Type 1B BMP receptor |
| BMPRII | Type II BMP receptor |
| CAM | Calmodulin kinase |
| CDC42 | Cell division control protein 42 homolog |
| ChAT | Acetylcholine synthesis enzyme |
| CNS | Central nervous system |
| CNTF | Ciliary neurotrophic factor |
| CR | Cysteine clusters |
| CREB | cAMP response element-binding protein |
| DAG | Diacylglycerol |
| DMSO | Dimethyl sulfoxide |
| dpp | Drosophila decapentaplegic |
| ERK1/2 | Extracellular signal-regulated kinase |
| FDA | Food and Drug Administration |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| FRS2 | Fibroblast growth factor receptor substrate 2 |
| Gab1 | Grb2-associated binder-1 |
| GDF | Growth differentiation factors |
| GDNF | Glial derived neurotrophic factor |
| GFAP | Glial fibrillary acidic protein |
| GPCRs | G-protein-coupled receptors |
| Grb2 | Growth factor receptor-bound protein 2 |
| GSK3β | Glycogen synthase kinase-3-β |
| GWAS | Genome-wide association |
| HMW | High molecular weight |
| Ig-C2 | Immunoglobulin-like C2 type domains |
| IGF | Insulin-like growth factor |
| IP3 | Inositol 1,4,5-trisphosphate |
| I-Smad | Inhibitory Smad: Smad6/7 |
| JNK | cJun N-terminal kinase |
| Lhx8 | LIM homeobox 8 |
| LMW | Low molecular weight |
| LOAD | Late-onset Alzheimer’s disease |
| LRR1-3 | Leucine-rich 24-residue motifs |
| LTP | Long-term potentiation |
| MAGE | Melanoma-associated antigen |
| MAP-2 | Microtubule associated protein 2 |
| MAPK | Mitogen-activated protein kinase |
| MMP-9 | Matric metalloprotease |
| NeuN | Neuronal nuclear protein |
| NF-kB | Nuclear factor kB |
| NG2 | Neuron-glial antigen 2 |
| NGF | Nerve growth factor (mature form: mNGF) |
| NMDA | N-methyl-D-aspartate |
| NMDAR | NMDA receptor |
| NRAGE | Neurotrophin receptor-interacting MAGE protein |
| NSE | Neuron specific enolase |
| NT-3 | Neurotrophin-3 |
| NT-4/5 | Neurotrophin-4/5 |
| p75NTR | p75 neurotrophin receptor |
| PI3K/AKT | Phosphoinositide 3-kinase/protein kinase B |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PKC | Protein kinase C |
| PLCγ | Phospholipase Cγ |
| PPM1A | Protein phosphatase magnesium-dependent 1A |
| RAF | Rapidly accelerated fibrosarcoma |
| RhoGDI1 | RhoGDP dissociation inhibitor 1 |
| Shc | Src homology 2 domain containing |
| Smad | Small mothers against decapentaplegic |
| SOS | Salt overly sensitive |
| STAT | Signal transducer of activators of transcription |
| TAB1/2/3 | TAK1-binding protein 1/2/3 |
| TAK1 | Transforming growth factor β-activated kinase 1 |
| TF | Transcription factor |
| Tg | Transgenic |
| TGF-β | Transforming growth factor-β |
| TRAF | TNF receptor associated factor |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| Trk | Tropomyosin receptor kinase |
| Trk-FL | Trk full length |
| VAChT | Vericular acetylcholine transporter |
| WHO | World Health Organization |
| WT | Wild type |
| XIAP | X-linked inhibitor of apoptosis |
References
- Alzheimer’s Association Report. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Comas-Herrera, A.; Knapp, M.; Guerchet, M.; Karagiannidou, M. World Alzheimer Report 2016; Alzheimer’s Disease International (ADI): London, UK, 2016. [Google Scholar]
- Calderon-Garciduenas, A.L.; Duyckaerts, C. Alzheimer disease. Handb. Clin. Neurol. 2017, 145, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A. The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1273. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Behbahani, H.; Eriksdotter, M. Innovative Therapy for Alzheimer’s Disease-With Focus on Biodelivery of NGF. Front. Neurosci. 2019, 13, 38. [Google Scholar] [CrossRef] [Green Version]
- Bartus, R.T.; Dean, R.L., 3rd; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by α-, β- and γ-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011, 1415, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Xia, W.; Zhang, Y.; Wang, C. Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 137. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Zheng, H.; Cheng, B.; Li, Y.; Li, X.; Chen, X.; Zhang, Y.W. TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front. Aging Neurosci. 2018, 10, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Kloske, C.M.; Wilcock, D.M. The Important Interface Between Apolipoprotein E and Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 2020, 11, 754. [Google Scholar] [CrossRef]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Vidal, C.; Zhang, L. An Analysis of the Neurological and Molecular Alterations Underlying the Pathogenesis of Alzheimer’s Disease. Cells 2021, 10, 546. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Shaikh, S.; Verma, A.; Siddiqui, S.; Ahmad, S.S.; Rizvi, S.M.; Shakil, S.; Biswas, D.; Singh, D.; Siddiqui, M.H.; Shakil, S.; et al. Current acetylcholinesterase-inhibitors: A neuroinformatics perspective. CNS Neurol. Disord. Drug. Targets 2014, 13, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Deardorff, W.J.; Feen, E.; Grossberg, G.T. The Use of Cholinesterase Inhibitors Across All Stages of Alzheimer’s Disease. Drugs Aging 2015, 32, 537–547. [Google Scholar] [CrossRef]
- Kang, Y.J.; Diep, Y.N.; Tran, M.; Cho, H. Therapeutic Targeting Strategies for Early- to Late-Staged Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9591. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- De Strooper, B. Lessons from a failed gamma-secretase Alzheimer trial. Cell 2014, 159, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef]
- Caselli, R.J.; Knopman, D.S.; Bu, G. An agnostic reevaluation of the amyloid cascade hypothesis of Alzheimer’s disease pathogenesis: The role of APP homeostasis. Alzheimers Dement. 2020, 16, 1582–1590. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.; Avila, J.; Scholl, M.; Kovacs, G.G.; Kovari, E.; Skrabana, R.; Evans, L.D.; Kontsekova, E.; Malawska, B.; de Silva, R.; et al. A walk through tau therapeutic strategies. Acta Neuropathol. Commun. 2019, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levälahti, E.; Ahtiluoto, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nistico, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Im, H.; Kang, Y.J.; Kim, Y.; Shin, J.H.; Won, W.; Lim, J.; Ju, Y.; Park, Y.M.; Kim, S.; et al. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H2O2(-) production. Nat. Neurosci. 2020, 23, 1555–1566. [Google Scholar] [CrossRef]
- Yang, A.; Kantor, B.; Chiba-Falek, O. APOE: The New Frontier in the Development of a Therapeutic Target towards Precision Medicine in Late-Onset Alzheimer’s. Int. J. Mol. Sci. 2021, 22, 1244. [Google Scholar] [CrossRef]
- Allen, S.J.; Watson, J.J.; Dawbarn, D. The neurotrophins and their role in Alzheimer’s disease. Curr. Neuropharmacol. 2011, 9, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Fang, Y.; Lian, Y.; Chen, Y.; Wu, T.; Zheng, Y.; Zong, H.; Sun, L.; Zhang, R.; Wang, Z.; et al. Brain-derived neurotrophic factor ameliorates learning deficits in a rat model of Alzheimer’s disease induced by abeta1-42. PLoS ONE 2015, 10, e0122415. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, R.; Garcia, A.A.; Braschi, C.; Capsoni, S.; Maffei, L.; Berardi, N.; Cattaneo, A. Intranasal administration of nerve growth factor (NGF) rescues recognition memory deficits in AD11 anti-NGF transgenic mice. Proc. Natl. Acad. Sci. USA 2005, 102, 3811–3816. [Google Scholar] [CrossRef] [Green Version]
- Rafii, M.S.; Tuszynski, M.H.; Thomas, R.G.; Barba, D.; Brewer, J.B.; Rissman, R.A.; Siffert, J.; Aisen, P.S.; Team, A.N.S. Adeno-Associated Viral Vector (Serotype 2)-Nerve Growth Factor for Patients With Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 834–841. [Google Scholar] [CrossRef]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Hallböök, F. Evolution of the vertebrate neurotrophin and Trk receptor gene families. Curr. Opin. Neurobiol. 1999, 9, 616–621. [Google Scholar] [CrossRef]
- Aloe, L. Rita Levi-Montalcini: The discovery of nerve growth factor and modern neurobiology. Trends Cell Biol. 2004, 14, 395–399. [Google Scholar] [CrossRef]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef]
- Pruunsild, P.; Kazantseva, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007, 90, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Baj, G.; Enrico, T. BDNF splice variants from the second promoter cluster support cell survival of differentiated neuroblastoma upon cytotoxic stress. J. Cell Sci. 2009, 122, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Chiaruttini, C.; Sonego, M.; Baj, G.; Simonato, M.; Tongiorgi, E. BDNF mRNA splice variants display activity-dependent targeting to distinct hippocampal laminae. Mol. Cell Neurosci. 2008, 37, 11–19. [Google Scholar] [CrossRef]
- Tuvikene, J.; Pruunsild, P.; Orav, E.; Esvald, E.E.; Timmusk, T. AP-1 Transcription Factors Mediate BDNF-Positive Feedback Loop in Cortical Neurons. J. Neurosci. 2016, 36, 1290–1305. [Google Scholar] [CrossRef]
- Hong, E.J.; McCord, A.E.; Greenberg, M.E. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron 2008, 60, 610–624. [Google Scholar] [CrossRef] [Green Version]
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef]
- Bruno, M.A.; Cuello, A.C. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 6735–6740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, S.; Jacobs, M.L.; Do Carmo, S.; Cuello, A.C. Compromise of cortical proNGF maturation causes selective retrograde atrophy in cholinergic nucleus basalis neurons. Neurobiol. Aging 2018, 67, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Iulita, M.F.; Bistue Millon, M.B.; Pentz, R.; Aguilar, L.F.; Do Carmo, S.; Allard, S.; Michalski, B.; Wilson, E.N.; Ducatenzeiler, A.; Bruno, M.A.; et al. Differential deregulation of NGF and BDNF neurotrophins in a transgenic rat model of Alzheimer’s disease. Neurobiol. Dis. 2017, 108, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C.; Pentz, R.; Hall, H. The Brain NGF Metabolic Pathway in Health and in Alzheimer’s Pathology. Front. Neurosci. 2019, 13, 62. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, A.; Gray, A.; Berman, C.; Dull, T.J. Human β-nerve growth factor gene sequence highly homologous to that of mouse. Nature 1983, 303, 821–825. [Google Scholar] [CrossRef]
- Lewin, G.R.; Barde, Y.A. Physiology of the neurotrophins. Annu. Rev. Neurosci. 1996, 19, 289–317. [Google Scholar] [CrossRef]
- McDonald, N.Q.; Lapatto, R.; Rust, J.M.; Gunning, J.; Wlodawer, A.; Blundell, T.L. New protein fold revealed by a 2.3-Å resolution crystal structure of nerve growth factor. Nature 1991, 354, 411–414. [Google Scholar] [CrossRef]
- Gudasheva, T.A.; Povarnina, P.Y.; Tarasiuk, A.V.; Seredenin, S.B. Low-molecular mimetics of nerve growth factor and brain-derived neurotrophic factor: Design and pharmacological properties. Med. Res. Rev. 2020. [Google Scholar] [CrossRef]
- Mcdonald, N.Q.; Lapatto, R.; Murray-Rust, J.; Gunning, J.; Wlodawer, A.; Blundell, T.L. New protein fold revealed by a 2.3 angstrom resolution crystal structure of nerve growth factor. Nature 1993. [Google Scholar] [CrossRef]
- He, X.; Garcia, K.C. Structure of the extracellular segment of human TRKA in complex with nerve growth factor. 2006. [Google Scholar] [CrossRef]
- Wehrman, T.; He, X.; Raab, B.; Dukipatti, A.; Blau, H.; Garcia, K.C. Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 2007, 53, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [Green Version]
- Nykjaer, A.; Lee, R.; Teng, K.K.; Jansen, P.; Madsen, P.; Nielsen, M.S.; Jacobsen, C.; Kliemannel, M.; Schwarz, E.; Willnow, T.E.; et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature 2004, 427, 843–848. [Google Scholar] [CrossRef]
- Yan, Q.; Radeke, M.J.; Matheson, C.R.; Talvenheimo, J.; Welcher, A.A.; Felnstein, S.C. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J. Comp. Neurol. 1997, 378, 135–157. [Google Scholar] [CrossRef]
- Chao, M.V.; Hempstead, B.L. p75 and Trk: A two-receptor system. Trends Neurosci. 1995, 18, 321–326. [Google Scholar] [CrossRef]
- Mahadeo, D.; Kaplan, L.; Chao, M.V.; Hempstead, B.L. High affinity nerve growth factor binding displays a faster rate of association than p140trk binding. Implications for multi-subunit polypeptide receptors. J. Biol. Chem. 1994, 269, 6884–6891. [Google Scholar] [CrossRef]
- Wiesmann, C.; Ultsch, M.H.; Bass, S.H.; de Vos, A.M. Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 1999, 401, 184–188. [Google Scholar] [CrossRef]
- Barbacid, M. The Trk family of neurotrophin receptors. J. Neurobiol. 1994, 25, 1386–1403. [Google Scholar] [CrossRef]
- Molloy, N.H.; Read, D.E.; Gorman, A.M. Nerve growth factor in cancer cell death and survival. Cancers 2011, 3, 510–530. [Google Scholar] [CrossRef] [Green Version]
- Meeker, R.B.; Williams, K.S. The p75 neurotrophin receptor: At the crossroad of neural repair and death. Neural. Regen. Res. 2015, 10, 721–725. [Google Scholar] [CrossRef]
- Pramanik, S.; Sulistio, Y.A.; Heese, K. Neurotrophin Signaling and Stem Cells-Implications for Neurodegenerative Diseases and Stem Cell Therapy. Mol. Neurobiol. 2017, 54, 7401–7459. [Google Scholar] [CrossRef] [PubMed]
- Sasi, M.; Vignoli, B.; Canossa, M.; Blum, R. Neurobiology of local and intercellular BDNF signaling. Pflugers. Arch. 2017, 469, 593–610. [Google Scholar] [CrossRef] [Green Version]
- Amidfar, M.; de Oliveira, J.; Kucharska, E.; Budni, J.; Kim, Y.K. The role of CREB and BDNF in neurobiology and treatment of Alzheimer’s disease. Life Sci. 2020, 257, 118020. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor, BDNF.; Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- Notaras, M.; van den Buuse, M. Brain-Derived Neurotrophic Factor (BDNF): Novel Insights into Regulation and Genetic Variation. Neuroscientist 2019, 25, 434–454. [Google Scholar] [CrossRef]
- Tomioka, T.; Shimazaki, T.; Yamauchi, T.; Oki, T.; Ohgoh, M.; Okano, H. LIM homeobox 8 (Lhx8) is a key regulator of the cholinergic neuronal function via a tropomyosin receptor kinase A (TrkA)-mediated positive feedback loop. J. Biol. Chem. 2014, 289, 1000–1010. [Google Scholar] [CrossRef] [Green Version]
- Minichiello, L.; Calella, A.M.; Medina, D.L.; Bonhoeffer, T.; Klein, R.; Korte, M. Mechanism of TrkB-Mediated Hippocampal Long-Term Potentiation. Neuron 2002, 36, 121–137. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.S.; Rajagopal, R.; Kim, A.H.; Chang, P.C.; Chao, M.V. Activation of Trk neurotrophin receptor signaling by pituitary adenylate cyclase-activating polypeptides. J. Biol. Chem. 2002, 277, 9096–9102. [Google Scholar] [CrossRef] [Green Version]
- Takei, N.; Torres, E.; Yuhara, A.; Jongsma, H.; Otto, C.; Korhonen, L.; Abiru, Y.; Skoglosa, Y.; Schutz, G.; Hatanaka, H.; et al. Pituitary adenylate cyclase-activating polypeptide promotes the survival of basal forebrain cholinergic neurons in vitro and in vivo: Comparison with effects of nerve growth factor. Eur. J. Neurosci. 2000, 12, 2273–2280. [Google Scholar] [CrossRef]
- Ohira, K.; Kumanogoh, H.; Sahara, Y.; Homma, K.J.; Hirai, H.; Nakamura, S.; Hayashi, M. A truncated tropomyosin-related kinase B receptor, T1, regulates glial cell morphology via Rho GDP dissociation inhibitor 1. J. Neurosci. 2005, 25, 1343–1353. [Google Scholar] [CrossRef]
- Ohira, K.; Homma, K.J.; Hirai, H.; Nakamura, S.; Hayashi, M. TrkB-T1 regulates the RhoA signaling and actin cytoskeleton in glioma cells. Biochem. Biophys. Res. Commun. 2006, 342, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Fenner, B.M. Truncated TrkB: Beyond a dominant negative receptor. Cytokine Growth. Factor Rev. 2012, 23, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Matyas, J.J.; Renn, C.L.; Faden, A.I.; Dorsey, S.G.; Wu, J. Function and Mechanisms of Truncated BDNF Receptor TrkB.T1 in Neuropathic Pain. Cells 2020, 9, 1194. [Google Scholar] [CrossRef] [PubMed]
- Charalampopoulos, I.; Vicario, A.; Pediaditakis, I.; Gravanis, A.; Simi, A.; Ibanez, C.F. Genetic dissection of neurotrophin signaling through the p75 neurotrophin receptor. Cell Rep. 2012, 2, 1563–1570. [Google Scholar] [CrossRef] [Green Version]
- Kowianski, P.; Lietzau, G.; Czuba, E.; Waskow, M.; Steliga, A.; Morys, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Ieraci, A.; Teng, H.; Dall, H.; Meng, C.X.; Herrera, D.G.; Nykjaer, A.; Hempstead, B.L.; Lee, F.S. Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J. Neurosci. 2005, 25, 6156–6166. [Google Scholar] [CrossRef]
- Counts, S.E.; Mufson, E.J. The Role of Nerve Growth Factor Receptors in Cholinergic Basal Forebrain Degeneration in Prodromal Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2005, 64, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005, 25, 5455–5463. [Google Scholar] [CrossRef]
- Almeida, R.D.; Manadas, B.J.; Melo, C.V.; Gomes, J.R.; Mendes, C.S.; Graos, M.M.; Carvalho, R.F.; Carvalho, A.P.; Duarte, C.B. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005, 12, 1329–1343. [Google Scholar] [CrossRef] [Green Version]
- Leal, G.; Afonso, P.M.; Salazar, I.L.; Duarte, C.B. Regulation of hippocampal synaptic plasticity by BDNF. Brain Res. 2015, 1621, 82–101. [Google Scholar] [CrossRef]
- Lärkfors, L.; Ebendal, T.; Whittemore, S.R.; Persson, H.; Hoffer, B.; Olson, L. Decreased level of nerve growth factor (NGF) and its messenger RNA in the aged rat brain. Mol. Brain Res. 1987, 3, 55–60. [Google Scholar] [CrossRef]
- Goedert, M.; Fine, A.; Hunt, S.P.; Ullrich, A. Nerve growth factor mRNA in peripheral and central rat tissues and in the human central nervous system: Lesion effects in the rat brain and levels in Alzheimer’s disease. Mol. Brain Res. 1986, 1, 85–92. [Google Scholar] [CrossRef]
- Solari, N.; Hangya, B. Cholinergic modulation of spatial learning, memory and navigation. Eur. J. Neurosci. 2018, 48, 2199–2230. [Google Scholar] [CrossRef] [Green Version]
- Gielow, M.R.; Zaborszky, L. The Input-Output Relationship of the Cholinergic Basal Forebrain. Cell Rep. 2017, 18, 1817–1830. [Google Scholar] [CrossRef]
- Cuello, A.C. Chapter 32: Trophic responses of forebrain cholinergic neurons: A discussion. In Progress in Brain Research; Cuello, A.C., Ed.; Elsevier: Amsterdam, The Netherlands, 1993; pp. 265–277. [Google Scholar]
- Berse, B.; Lopez-Coviella, I.; Blusztajn, J.K. Activation of TrkA by nerve growth factor upregulates expression of the cholinergic gene locus but attenuates the response to ciliary neurotrophic growth factor. Biochem. J. 1999, 342 Pt 2, 301–308. [Google Scholar] [CrossRef]
- Fagan, A.M.; Garber, M.; Barbacid, M.; Silos-Santiago, I.; Holtzman, D.M. A Role for TrkA during Maturation of Striatal and Basal Forebrain Cholinergic NeuronsIn Vivo. J. Neurosci. 1997, 17, 7644–7654. [Google Scholar] [CrossRef] [Green Version]
- Crowley, C.; Spencer, S.D.; Nishimura, M.C.; Chen, K.S.; Pitts-Meek, S.; Armaninl, M.P.; Ling, L.H.; McMahon, S.B.; Shelton, D.L.; Levinson, A.D.; et al. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell 1994, 76, 1001–1011. [Google Scholar] [CrossRef]
- Eu, W.Z.; Chen, Y.J.; Chen, W.T.; Wu, K.Y.; Tsai, C.Y.; Cheng, S.J.; Carter, R.N.; Huang, G.J. The effect of nerve growth factor on supporting spatial memory depends upon hippocampal cholinergic innervation. Transl. Psychiatry 2021, 11, 162. [Google Scholar] [CrossRef]
- Chen, K.S.; Nishimura, M.C.; Armanini, M.P.; Crowley, C.; Spencer, S.D.; Phillips, H.S. Disruption of a Single Allele of the Nerve Growth Factor Gene Results in Atrophy of Basal Forebrain Cholinergic Neurons and Memory Deficits. J. Neurosci. 1997, 17, 7288–7296. [Google Scholar] [CrossRef]
- Allard, S.; Leon, W.C.; Pakavathkumar, P.; Bruno, M.A.; Ribeiro-da-Silva, A.; Cuello, A.C. Impact of the NGF maturation and degradation pathway on the cortical cholinergic system phenotype. J. Neurosci. 2012, 32, 2002–2012. [Google Scholar] [CrossRef]
- Kopec, B.M.; Zhao, L.; Rosa-Molinar, E.; Siahaan, T.J. Non-invasive Brain Delivery and Efficacy of BDNF in APP/PS1 Transgenic Mice as a Model of Alzheimer’s Disease. Med. Res. Arch. 2020, 8. [Google Scholar] [CrossRef]
- Braschi, C.; Capsoni, S.; Narducci, R.; Poli, A.; Sansevero, G.; Brandi, R.; Maffei, L.; Cattaneo, A.; Berardi, N. Intranasal delivery of BDNF rescues memory deficits in AD11 mice and reduces brain microgliosis. Aging Clin. Exp. Res. 2020. [Google Scholar] [CrossRef]
- Crews, L.; Adame, A.; Patrick, C.; Delaney, A.; Pham, E.; Rockenstein, E.; Hansen, L.; Masliah, E. Increased BMP6 levels in the brains of Alzheimer’s disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J. Neurosci. 2010, 30, 12252–12262. [Google Scholar] [CrossRef]
- Burke, R.M.; Norman, T.A.; Haydar, T.F.; Slack, B.E.; Leeman, S.E.; Blusztajn, J.K.; Mellott, T.J. BMP9 ameliorates amyloidosis and the cholinergic defect in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, 19567–19572. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Xiong, L.; Wan, W.; Duan, L.; Bai, X.; Zu, H. Intranasal BMP9 Ameliorates Alzheimer Disease-Like Pathology and Cognitive Deficits in APP/PS1 Transgenic Mice. Front. Mol. Neurosci. 2017, 10, 32. [Google Scholar] [CrossRef]
- Xia, L.; Zhu, X.; Zhao, Y.; Yang, G.; Zuo, X.; Xie, P.; Chen, C.; Han, Q. Genome-wide RNA sequencing analysis reveals that IGF-2 attenuates memory decline, oxidative stress and amyloid plaques in an Alzheimer’s disease mouse model (AD) by activating the PI3K/AKT/CREB signaling pathway. Int. Psychogeriatr. 2019, 1–13. [Google Scholar] [CrossRef]
- Mellott, T.J.; Pender, S.M.; Burke, R.M.; Langley, E.A.; Blusztajn, J.K. IGF2 ameliorates amyloidosis, increases cholinergic marker expression and raises BMP9 and neurotrophin levels in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PLoS ONE 2014, 9, e94287. [Google Scholar] [CrossRef]
- Kiyota, T.; Ingraham, K.L.; Jacobsen, M.T.; Xiong, H.; Ikezu, T. FGF2 gene transfer restores hippocampal functions in mouse models of Alzheimer’s disease and has therapeutic implications for neurocognitive disorders. Proc. Natl. Acad. Sci. USA 2011, 108, E1339-48. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, Z.; Cheng, Y.; Kardami, E.; Loh, Y.P. Low and High Molecular Weight FGF-2 Have Differential Effects on Astrocyte Proliferation, but Are Both Protective Against Abeta-Induced Cytotoxicity. Front. Mol. Neurosci. 2019, 12, 328. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wang, Y.; Chen, S.T.; Chen, Y.J.; Shen, J.; Yao, W.B.; Gao, X.D.; Chen, S. Modulation of the Astrocyte-Neuron Lactate Shuttle System contributes to Neuroprotective action of Fibroblast Growth Factor 21. Theranostics 2020, 10, 8430–8445. [Google Scholar] [CrossRef]
- Patterson, S.L.; Abel, T.; Deuel, T.A.S.; Martin, K.C.; Rose, J.C.; Kandel, E.R. Recombinant BDNF Rescues Deficits in Basal Synaptic Transmission and Hippocampal LTP in BDNF Knockout Mice. Neuron 1996, 16, 1137–1145. [Google Scholar] [CrossRef] [Green Version]
- Seoane, A.; Tinsley, C.J.; Brown, M.W. Interfering with perirhinal brain-derived neurotrophic factor expression impairs recognition memory in rats. Hippocampus 2011, 21, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altar, C.A.; Cai, N.; Bliven, T.; Juhasz, M.; Conner, J.M.; Acheson, A.L.; Lindsay, R.M.; Wiegand, S.J. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997, 389, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Yamada, K.; Olariu, A.; Nawa, H.; Nabeshima, T. Involvement of Brain-Derived Neurotrophic Factor in Spatial Memory Formation and Maintenance in a Radial Arm Maze Test in Rats. J. Neurosci. 2000, 20, 7116–7121. [Google Scholar] [CrossRef] [Green Version]
- Gorski, J.A.; Balogh, S.A.; Wehner, J.M.; Jones, K.R. Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience 2003, 121, 341–354. [Google Scholar] [CrossRef]
- Carvalho, A.L.; Caldeira, M.V.; Santos, S.D.; Duarte, C.B. Role of the brain-derived neurotrophic factor at glutamatergic synapses. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S310–S324. [Google Scholar] [CrossRef] [Green Version]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Abeta Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J. Neurosci. 2015, 35, 7538–7551. [Google Scholar] [CrossRef]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE isoforms differentially regulates cleavage and secretion of BDNF. Mol. Brain 2017, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014, 7, 796–806. [Google Scholar] [CrossRef] [Green Version]
- Fahnestock, M.; Scott, S.A.; Jetté, N.; Weingartner, J.A.; Crutcher, K.A. Nerve growth factor mRNA and protein levels measured in the same tissue from normal and Alzheimer’s disease parietal cortex. Mol. Brain Res. 1996, 42, 175–178. [Google Scholar] [CrossRef]
- Jetté, N.; Cole, M.S.; Fahnestock, M. NGF mRNA is not decreased in frontal cortex from Alzheimer’s Disease patients. Mol. Brain Res. 1994, 25, 242–250. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Malek-Ahmadi, M.H.; Alldred, M.J.; Che, S.; Elarova, I.; Chen, Y.; Jeanneteau, F.; Kranz, T.M.; Chao, M.V.; Counts, S.E.; et al. Selective decline of neurotrophin and neurotrophin receptor genes within CA1 pyramidal neurons and hippocampus proper: Correlation with cognitive performance and neuropathology in mild cognitive impairment and Alzheimer’s disease. Hippocampus 2019, 29, 422–439. [Google Scholar] [CrossRef]
- Iulita, M.F.; Cuello, A.C. Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends Pharmacol. Sci. 2014, 35, 338–348. [Google Scholar] [CrossRef]
- Fahnestock, M.; Michalski, B.; Xu, B.; Coughlin, M.D. The Precursor Pro-Nerve Growth Factor Is the Predominant Form of Nerve Growth Factor in Brain and Is Increased in Alzheimer’s Disease. Mol. Cell. Neurosci. 2001, 18, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J. Neuropathol. Exp. Neurol. 2004, 63, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, M.A.; Mufson, E.J.; Wuu, J.; Cuello, A.C. Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J. Neuropathol. Exp. Neurol. 2009, 68, 1309–1318. [Google Scholar] [CrossRef]
- Mufson, E.J.; He, B.; Nadeem, M.; Perez, S.E.; Counts, S.E.; Leurgans, S.; Fritz, J.; Lah, J.; Ginsberg, S.D.; Wuu, J.; et al. Hippocampal ProNGF Signaling Pathways and β-Amyloid Levels in Mild Cognitive Impairment and Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2012, 71, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Fabbro, S.; Seeds, N.W. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J. Neurochem. 2009, 109, 303–315. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Che, S.; Wuu, J.; Counts, S.E.; Mufson, E.J. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J. Neurochem. 2006, 97, 475–487. [Google Scholar] [CrossRef]
- Podlesniy, P.; Kichev, A.; Pedraza, C.; Saurat, J.; Encinas, M.; Perez, B.; Ferrer, I.; Espinet, C. Pro-NGF from Alzheimer’s disease and normal human brain displays distinctive abilities to induce processing and nuclear translocation of intracellular domain of p75NTR and apoptosis. Am. J. Pathol. 2006, 169, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Volosin, M.; Song, W.; Almeida, R.D.; Kaplan, D.R.; Hempstead, B.L.; Friedman, W.J. Interaction of survival and death signaling in basal forebrain neurons: Roles of neurotrophins and proneurotrophins. J. Neurosci. 2006, 26, 7756–7766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canu, N.; Amadoro, G.; Triaca, V.; Latina, V.; Sposato, V.; Corsetti, V.; Severini, C.; Ciotti, M.T.; Calissano, P. The Intersection of NGF/TrkA Signaling and Amyloid Precursor Protein Processing in Alzheimer’s Disease Neuropathology. Int. J. Mol. Sci. 2017, 18, 1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocco, M.L.; Soligo, M.; Manni, L.; Aloe, L. Nerve Growth Factor: Early Studies and Recent Clinical Trials. Curr. Neuropharmacol. 2018, 16, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Poduslo, J.F.; Curran, G.L. Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Mol. Brain Res. 1996, 36, 280–286. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; Tong, G.; et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 2005, 11, 551–555. [Google Scholar] [CrossRef]
- Petty, B.G.; Cornblath, D.R.; Adornato, B.T.; Chaudhry, V.; Flexner, C.; Wachsman, M.; Sinicropi, D.; Burton, L.E.; Peroutka, S.J. The effect of systemically administered recombinant human nerve growth factor in healthy human subjects. Ann. Neurol. 1994, 36, 244–246. [Google Scholar] [CrossRef]
- Hefti, F.; Dravid, A.; Hartikka, J. Chronic intraventricular injections of nerve growth factor elevate hippocampal choline acetyltransferase activity in adult rats with partial septo-hippocampal lesions. Brain Res. 1984, 293, 305–311. [Google Scholar] [CrossRef]
- Koliatsos, V.E.; Nauta, H.J.; Clatterbuck, R.E.; Holtzman, D.M.; Mobley, W.C.; Price, D.L. Mouse nerve growth factor prevents degeneration of axotomized basal forebrain cholinergic neurons in the monkey. J. Neurosci. 1990, 10, 3801–3813. [Google Scholar] [CrossRef] [Green Version]
- Koliatsos, V.E.; Clatterbuck, R.E.; Nauta, H.J.; Knusel, B.; Burton, L.E.; Hefti, F.F.; Mobley, W.C.; Price, D.L. Human nerve growth factor prevents degeneration of basal forebrain cholinergic neurons in primates. Ann. Neurol. 1991, 30, 831–840. [Google Scholar] [CrossRef]
- Jönhagen, M.E.; Nordberg, A.; Amberla, K.; Bäckman, L.; Ebendal, T.; Meyerson, B.; Olson, L.; Seiger, Å.; Shigeta, M.; Theodorsson, E.; et al. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 1998, 9, 246–257. [Google Scholar] [CrossRef]
- Lv, Q.; Lan, W.; Sun, W.; Ye, R.; Fan, X.; Ma, M.; Yin, Q.; Jiang, Y.; Xu, G.; Dai, J.; et al. Intranasal nerve growth factor attenuates tau phosphorylation in brain after traumatic brain injury in rats. J. Neurol. Sci. 2014, 345, 48–55. [Google Scholar] [CrossRef]
- Tirassa, P. The nerve growth factor administrated as eye drops activates mature and precursor cells in subventricular zone of adult rats. Arch. Ital. Biol. 2011, 149, 205–213. [Google Scholar] [CrossRef]
- Rafii, M.S.; Baumann, T.L.; Bakay, R.A.; Ostrove, J.M.; Siffert, J.; Fleisher, A.S.; Herzog, C.D.; Barba, D.; Pay, M.; Salmon, D.P.; et al. A phase1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimers Dement. 2014, 10, 571–581. [Google Scholar] [CrossRef]
- Eyjolfsdottir, H.; Eriksdotter, M.; Linderoth, B.; Lind, G.; Juliusson, B.; Kusk, P.; Almkvist, O.; Andreasen, N.; Blennow, K.; Ferreira, D.; et al. Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: Application of a second-generation encapsulated cell biodelivery device. Alzheimers Res. Ther. 2016, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Lee, J.K.; Lee, H.; Carter, J.E.; Chang, J.W.; Oh, W.; Yang, Y.S.; Suh, J.G.; Lee, B.H.; Jin, H.K.; et al. Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol. Aging 2012, 33, 588–602. [Google Scholar] [CrossRef]
- Phillips, H.S.; Hains, J.M.; Armanini, M.; Laramee, G.R.; Johnson, S.A.; Winslow, J.W. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 1991, 7, 695–702. [Google Scholar] [CrossRef]
- Ferrer, I.; Marin, C.; Rey, M.J.; Ribalta, T.; Goutan, E.; Blanco, R.; Tolosa, E.; Marti, E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol. 1999, 58, 729–739. [Google Scholar] [CrossRef]
- Holsinger, R.M.D.; Schnarr, J.; Henry, P.; Castelo, V.T.; Fahnestock, M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: Decreased levels in Alzheimer’s disease. Mol. Brain Res. 2000, 76, 347–354. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef]
- Buchman, A.S.; Yu, L.; Boyle, P.A.; Schneider, J.A.; De Jager, P.L.; Bennett, D.A. Higher brain BDNF gene expression is associated with slower cognitive decline in older adults. Neurology 2016, 86, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, S.D.; Malek-Ahmadi, M.H.; Alldred, M.J.; Chen, Y.; Chen, K.; Chao, M.V.; Counts, S.E.; Mufson, E.J. Brain-derived neurotrophic factor (BDNF) and TrkB hippocampal gene expression are putative predictors of neuritic plaque and neurofibrillary tangle pathology. Neurobiol. Dis. 2019, 132, 104540. [Google Scholar] [CrossRef] [PubMed]
- Arango-Lievano, M.; Peguet, C.; Catteau, M.; Parmentier, M.-L.; Wu, S.; Chao, M.V.; Ginsberg, S.D.; Jeanneteau, F. Deletion of Neurotrophin Signaling through the Glucocorticoid Receptor Pathway Causes Tau Neuropathology. Sci. Rep. 2016, 6, 37231. [Google Scholar] [CrossRef] [PubMed]
- Poon, W.W.; Blurton-Jones, M.; Tu, C.H.; Feinberg, L.M.; Chabrier, M.A.; Harris, J.W.; Jeon, N.L.; Cotman, C.W. beta-Amyloid impairs axonal BDNF retrograde trafficking. Neurobiol. Aging 2011, 32, 821–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanqueiro, S.R.; Ramalho, R.M.; Rodrigues, T.M.; Lopes, L.V.; Sebastiao, A.M.; Diogenes, M.J. Inhibition of NMDA Receptors Prevents the Loss of BDNF Function Induced by Amyloid beta. Front. Pharmacol. 2018, 9, 237. [Google Scholar] [CrossRef]
- Jeronimo-Santos, A.; Vaz, S.H.; Parreira, S.; Rapaz-Lerias, S.; Caetano, A.P.; Buee-Scherrer, V.; Castren, E.; Valente, C.A.; Blum, D.; Sebastiao, A.M.; et al. Dysregulation of TrkB Receptors and BDNF Function by Amyloid-beta Peptide is Mediated by Calpain. Cereb. Cortex. 2015, 25, 3107–3121. [Google Scholar] [CrossRef] [Green Version]
- Bharani, K.L.; Ledreux, A.; Gilmore, A.; Carroll, S.L.; Granholm, A.-C. Serum pro-BDNF levels correlate with phospho-tau staining in Alzheimer’s disease. Neurobiol. Aging 2020, 87, 49–59. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Banks, W.A.; Fasold, M.B.; Bluth, J.; Kastin, A.J. Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology 1998, 37, 1553–1561. [Google Scholar] [CrossRef]
- Olah, M.; Patrick, E.; Villani, A.-C.; Xu, J.; White, C.C.; Ryan, K.J.; Piehowski, P.; Kapasi, A.; Nejad, P.; Cimpean, M.; et al. A transcriptomic atlas of aged human microglia. Nat. Commun. 2018, 9, 539. [Google Scholar] [CrossRef] [Green Version]
- Felsky, D.; Roostaei, T.; Nho, K.; Risacher, S.L.; Bradshaw, E.M.; Petyuk, V.; Schneider, J.A.; Saykin, A.; Bennett, D.A.; De Jager, P.L. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat. Commun. 2019, 10, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jann, J.; Gascon, S.; Roux, S.; Faucheux, N. Influence of the TGF-beta Superfamily on Osteoclasts/Osteoblasts Balance in Physiological and Pathological Bone Conditions. Int. J. Mol. Sci. 2020, 21, 7537. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, D.M. The TGF-beta superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994, 8, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.A.; Zhao, Q.; Baker, K.A.; Naik, C.; Chen, C.; Pukac, L.; Singh, M.; Tsareva, T.; Parice, Y.; Mahoney, A.; et al. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J. Biol. Chem. 2005, 280, 25111–25118. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Chen, D. The BMP signaling and in vivo bone formation. Gene 2005, 357, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Hofmann, C.; Bronckers, A.L.; Sohocki, M.; Bradley, A.; Karsenty, G. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. 1995, 9, 2808–2820. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.Q. Consequences of knocking out BMP signaling in the mouse. Genesis 2003, 35, 43–56. [Google Scholar] [CrossRef]
- Bakrania, P.; Efthymiou, M.; Klein, J.C.; Salt, A.; Bunyan, D.J.; Wyatt, A.; Ponting, C.P.; Martin, A.; Williams, S.; Lindley, V.; et al. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: Overlap between the BMP4 and hedgehog signaling pathways. Am. J. Hum. Genet. 2008, 82, 304–319. [Google Scholar] [CrossRef] [Green Version]
- Bidart, M.; Ricard, N.; Levet, S.; Samson, M.; Mallet, C.; David, L.; Subileau, M.; Tillet, E.; Feige, J.J.; Bailly, S. BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell Mol. Life Sci. 2012, 69, 313–324. [Google Scholar] [CrossRef]
- Wohl, A.P.; Troilo, H.; Collins, R.F.; Baldock, C.; Sengle, G. Extracellular Regulation of Bone Morphogenetic Protein Activity by the Microfibril Component Fibrillin-1. J. Biol. Chem. 2016, 291, 12732–12746. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.-H.; Miyazono, K.; ten Dijke, P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Lowik, C.W.; ten Dijke, P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 2007, 120, 964–972. [Google Scholar] [CrossRef] [Green Version]
- Yadin, D.; Knaus, P.; Mueller, T.D. Structural insights into BMP receptors: Specificity, activation and inhibition. Cytokine Growth. Factor Rev. 2016, 27, 13–34. [Google Scholar] [CrossRef]
- Okadome, T.; Yamashita, H.; Franzén, P.; Morén, A.; Heldin, C.H.; Miyazono, K. Distinct roles of the intracellular domains of transforming growth factor-beta type I and type II receptors in signal transduction. J. Biol. Chem. 1994, 269, 30753–30756. [Google Scholar] [CrossRef]
- Hinck, A.P. Structural studies of the TGF-betas and their receptors—Insights into evolution of the TGF-beta superfamily. FEBS Lett. 2012, 586, 1860–1870. [Google Scholar] [CrossRef] [Green Version]
- Mi, L.Z.; Brown, C.T.; Gao, Y.; Tian, Y.; Le, V.Q.; Walz, T.; Springer, T.A. Structure of bone morphogenetic protein 9 procomplex. Proc. Natl. Acad. Sci. USA 2015, 112, 3710–3715. [Google Scholar] [CrossRef] [Green Version]
- Salmon, R.M.; Guo, J.; Wood, J.H.; Tong, Z.; Beech, J.S.; Lawera, A.; Yu, M.; Grainger, D.J.; Reckless, J.; Morrell, N.W.; et al. Molecular basis of ALK1-mediated signalling by BMP9/BMP10 and their prodomain-bound forms. Nat. Commun. 2020, 11, 1621. [Google Scholar] [CrossRef] [Green Version]
- Charytoniuk, D.A.; Traiffort, E.; Pinard, E.; Issertial, O.; Seylaz, J.; Ruat, M. Distribution of bone morphogenetic protein and bone morphogenetic protein receptor transcripts in the rodent nervous system and up-regulation of bone morphogenetic protein receptor type II in hippocampal dentate gyrus in a rat model of global cerebral ischemia. Neuroscience 2000, 100, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Mira, H.; Andreu, Z.; Suh, H.; Lie, D.C.; Jessberger, S.; Consiglio, A.; San Emeterio, J.; Hortiguela, R.; Marques-Torrejon, M.A.; Nakashima, K.; et al. Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell Stem. Cell 2010, 7, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Gipson, G.R.; Goebel, E.J.; Hart, K.N.; Kappes, E.C.; Kattamuri, C.; McCoy, J.C.; Thompson, T.B. Structural perspective of BMP ligands and signaling. Bone 2020, 140, 115549. [Google Scholar] [CrossRef]
- Nohe, A.; Hassel, S.; Ehrlich, M.; Neubauer, F.; Sebald, W.; Henis, Y.I.; Knaus, P. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J. Biol. Chem. 2002, 277, 5330–5338. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. Endocytosis and trafficking of BMP receptors: Regulatory mechanisms for fine-tuning the signaling response in different cellular contexts. Cytokine Growth. Factor Rev. 2016, 27, 35–42. [Google Scholar] [CrossRef]
- Higashi, T.; Tanaka, S.; Iida, T.; Okabe, S. Synapse Elimination Triggered by BMP4 Exocytosis and Presynaptic BMP Receptor Activation. Cell Rep. 2018, 22, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Lauzon, M.A.; Daviau, A.; Marcos, B.; Faucheux, N. Growth factor treatment to overcome Alzheimer’s dysfunctional signaling. Cell Signal. 2015, 27, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Onichtchouk, D.; Chen, Y.G.; Dosch, R.; Gawantka, V.; Delius, H.; Massague, J.; Niehrs, C. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 1999, 401, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-beta Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Coviella, I.; Follettie, M.T.; Mellott, T.J.; Kovacheva, V.P.; Slack, B.E.; Diesl, V.; Berse, B.; Thies, R.S.; Blusztajn, J.K. Bone morphogenetic protein 9 induces the transcriptome of basal forebrain cholinergic neurons. Proc. Natl. Acad. Sci. USA 2005, 102, 6984–6989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnitzler, A.C.; Lopez-Coviella, I.; Blusztajn, J.K. Differential modulation of nerve growth factor receptor (p75) and cholinergic gene expression in purified p75-expressing and non-expressing basal forebrain neurons by BMP9. Brain Res. 2008, 1246, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Schnitzler, A.C.; Mellott, T.J.; Lopez-Coviella, I.; Tallini, Y.N.; Kotlikoff, M.I.; Follettie, M.T.; Blusztajn, J.K. BMP9 (bone morphogenetic protein 9) induces NGF as an autocrine/paracrine cholinergic trophic factor in developing basal forebrain neurons. J. Neurosci. 2010, 30, 8221–8228. [Google Scholar] [CrossRef] [Green Version]
- Dinh Duong, T.A.; Hoshiba, Y.; Saito, K.; Kawasaki, K.; Ichikawa, Y.; Matsumoto, N.; Shinmyo, Y.; Kawasaki, H. FGF Signaling Directs the Cell Fate Switch from Neurons to Astrocytes in the Developing Mouse Cerebral Cortex. J. Neurosci. 2019, 39, 6081–6094. [Google Scholar] [CrossRef] [Green Version]
- Guillemot, F.; Zimmer, C. From cradle to grave: The multiple roles of fibroblast growth factors in neural development. Neuron 2011, 71, 574–588. [Google Scholar] [CrossRef] [Green Version]
- Mikawa, S.; Wang, C.; Sato, K. Bone morphogenetic protein-4 expression in the adult rat brain. J. Comp. Neurol. 2006, 499, 613–625. [Google Scholar] [CrossRef] [Green Version]
- Wagner, D.O.; Sieber, C.; Bhushan, R.; Borgermann, J.H.; Graf, D.; Knaus, P. BMPs: From bone to body morphogenetic proteins. Sci. Signal. 2010, 3, mr1. [Google Scholar] [CrossRef]
- Sato, T.; Mikawa, S.; Sato, K. BMP2 expression in the adult rat brain. J. Comp. Neurol. 2010, 518, 4513–4530. [Google Scholar] [CrossRef] [Green Version]
- Kusakawa, Y.; Mikawa, S.; Sato, K. BMP5 expression in the adult rat brain. Neuroscience 2015, 284, 972–987. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Mikawa, S.; Ogawa, C.; Masumoto, K.; Katou, F.; Sato, K. BMP6 expression in the adult rat central nervous system. J. Chem. Neuroanat. 2019, 98, 41–54. [Google Scholar] [CrossRef]
- López-Coviella, I.; Berse, B.; Krauss, R.; Thies, R.S.; Blusztajn, J.K. Induction and Maintenance of the Neuronal Cholinergic Phenotype in the Central Nervous System by BMP-9. Science 2000, 289, 313–316. [Google Scholar] [CrossRef]
- Chen, H.L.; Lein, P.J.; Wang, J.Y.; Gash, D.; Hoffer, B.J.; Chiang, Y.H. Expression of bone morphogenetic proteins in the brain during normal aging and in 6-hydroxydopamine-lesioned animals. Brain Res. 2003, 994, 81–90. [Google Scholar] [CrossRef]
- Hart, C.G.; Karimi-Abdolrezaee, S. Bone morphogenetic proteins: New insights into their roles and mechanisms in CNS development, pathology and repair. Exp. Neurol. 2020, 334, 113455. [Google Scholar] [CrossRef]
- Bond, A.M.; Bhalala, O.G.; Kessler, J.A. The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev. Neurobiol. 2012, 72, 1068–1084. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Hu, Y. Bone morphogenetic protein 9 serves a protective role in response to ischemicreperfusion in the brain by promoting ERK activation. Mol. Med. Rep. 2018, 17, 2845–2852. [Google Scholar] [CrossRef]
- Lopez-Coviella, I.; Mellott, T.J.; Schnitzler, A.C.; Blusztajn, J.K. BMP9 protects septal neurons from axotomy-evoked loss of cholinergic phenotype. PLoS ONE 2011, 6, e21166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousef, H.; Morgenthaler, A.; Schlesinger, C.; Bugaj, L.; Conboy, I.M.; Schaffer, D.V. Age-Associated Increase in BMP Signaling Inhibits Hippocampal Neurogenesis. Stem Cells. 2015, 33, 1577–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Tang, J.; Xu, H.; Fan, X.; Bai, Y.; Yang, L. Decreased hippocampal cell proliferation correlates with increased expression of BMP4 in the APPswe/PS1DeltaE9 mouse model of Alzheimer’s disease. Hippocampus 2008, 18, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.L.; Benayoun, L.; Tilton, K.; Mellott, T.J.; Seshadri, S.; Blusztajn, J.K.; Delalle, I. Immunohistochemical Analysis of Activin Receptor-Like Kinase 1 (ACVRL1/ALK1) Expression in the Rat and Human Hippocampus: Decline in CA3 During Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 63, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Klimaschewski, L.; Claus, P. Fibroblast Growth Factor Signalling in the Diseased Nervous System. Mol. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, E.; Touriol, C.; Boutonnet, C.; Gensac, M.C.; Vagner, S.; Prats, H.; Prats, A.C. A new 34-kilodalton isoform of human fibroblast growth factor 2 is cap dependently synthesized by using a non-AUG start codon and behaves as a survival factor. Mol. Cell Biol. 1999, 19, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Touriol, C.; Bornes, S.; Bonnal, S.; Audigier, S.; Prats, H.; Prats, A.-C.; Vagner, S. Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biol. Cell 2003, 95, 169–178. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.A.; Eren-Kocak, E.; Inui, E.G.; Watson, S.J.; Akil, H. Dysregulated fibroblast growth factor (FGF) signaling in neurological and psychiatric disorders. Semin. Cell Dev. Biol. 2016, 53, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Asai, T.; Wanaka, A.; Kato, H.; Masana, Y.; Seo, M.; Tohyama, M. Differential expression of two members of FGF receptor gene family, FGFR-1 and FGFR-2 mRNA, in the adult rat central nervous system. Mol. Brain Res. 1993, 17, 174–178. [Google Scholar] [CrossRef]
- Miyake, A.; Hattori, Y.; Ohta, M.; Itoh, N. Rat oligodendrocytes and astrocytes preferentially express fibroblast growth factor receptor-2 and -3 mRNAs. J. Neurosci. Res. 1996, 45, 534–541. [Google Scholar] [CrossRef]
- Latko, M.; Czyrek, A.; Porebska, N.; Kucinska, M.; Otlewski, J.; Zakrzewska, M.; Opalinski, L. Cross-Talk between Fibroblast Growth Factor Receptors and Other Cell Surface Proteins. Cells 2019, 8, 455. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, N.; Shinmyo, Y.; Ichikawa, Y.; Kawasaki, H. Gyrification of the cerebral cortex requires FGF signaling in the mammalian brain. Elife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.; Unsicker, K.; von Bohlen und Halbach, O. Fibroblast growth factor-2 deficiency causes defects in adult hippocampal neurogenesis, which are not rescued by exogenous fibroblast growth factor-2. J. Neurosci. Res. 2011, 89, 1605–1617. [Google Scholar] [CrossRef]
- Gonzalez, A.M.; Berry, M.; Maher, P.A.; Logan, A.; Baird, A. A comprehensive analysis of the distribution of FGF-2 and FGFR1 in the rat brain. Brain Res. 1995, 701, 201–226. [Google Scholar] [CrossRef]
- Katsouri, L.; Ashraf, A.; Birch, A.M.; Lee, K.K.; Mirzaei, N.; Sastre, M. Systemic administration of fibroblast growth factor-2 (FGF2) reduces BACE1 expression and amyloid pathology in APP23 mice. Neurobiol. Aging 2015, 36, 821–831. [Google Scholar] [CrossRef]
- Feng, C.; Zhang, C.; Shao, X.; Liu, Q.; Qian, Y.; Feng, L.; Chen, J.; Zha, Y.; Zhang, Q.; Jiang, X. Enhancement of nose-to-brain delivery of basic fibroblast growth factor for improving rat memory impairments induced by co-injection of beta-amyloid and ibotenic acid into the bilateral hippocampus. Int. J. Pharm. 2012, 423, 226–234. [Google Scholar] [CrossRef]
- Pasquin, S.; Sharma, M.; Gauchat, J.F. Ciliary neurotrophic factor (CNTF): New facets of an old molecule for treating neurodegenerative and metabolic syndrome pathologies. Cytokine Growth. Factor Rev. 2015, 26, 507–515. [Google Scholar] [CrossRef]
- Rollero, A.; Murialdo, G.; Fonzi, S.; Garrone, S.; Gianelli, M.V.; Gazzerro, E.; Barreca, A.; Polleri, A. Relationship between cognitive function, growth hormone and insulin-like growth factor I plasma levels in aged subjects. Neuropsychobiology 1998, 38, 73–79. [Google Scholar] [CrossRef]
- Aleman, A.; Verhaar, H.J.; De Haan, E.H.; De Vries, W.R.; Samson, M.M.; Drent, M.L.; Van der Veen, E.A.; Koppeschaar, H.P. Insulin-like growth factor-I and cognitive function in healthy older men. J. Clin. Endocrinol. Metab. 1999, 84, 471–475. [Google Scholar] [CrossRef]
- Tumati, S.; Burger, H.; Martens, S.; van der Schouw, Y.T.; Aleman, A. Association between Cognition and Serum Insulin-Like Growth Factor-1 in Middle-Aged & Older Men: An 8 Year Follow-Up Study. PLoS ONE 2016, 11, e0154450. [Google Scholar] [CrossRef]
- Carro, E.; Trejo, J.L.; Gomez-Isla, T.; LeRoith, D.; Torres-Aleman, I. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat. Med. 2002, 8, 1390–1397. [Google Scholar] [CrossRef]
- Carro, E.; Trejo, J.L.; Gerber, A.; Loetscher, H.; Torrado, J.; Metzger, F.; Torres-Aleman, I. Therapeutic actions of insulin-like growth factor I on APP/PS2 mice with severe brain amyloidosis. Neurobiol. Aging 2006, 27, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- Lanz, T.A.; Salatto, C.T.; Semproni, A.R.; Marconi, M.; Brown, T.M.; Richter, K.E.G.; Schmidt, K.; Nelson, F.R.; Schachter, J.B. Peripheral elevation of IGF-1 fails to alter Aβ clearance in multiple in vivo models. Biochem. Pharmacol. 2008, 75, 1093–1103. [Google Scholar] [CrossRef]
- Sohrabi, M.; Floden, A.M.; Manocha, G.D.; Klug, M.G.; Combs, C.K. IGF-1R Inhibitor Ameliorates Neuroinflammation in an Alzheimer’s Disease Transgenic Mouse Model. Front. Cell Neurosci. 2020, 14, 200. [Google Scholar] [CrossRef]
- Sevigny, J.J.; Ryan, J.M.; van Dyck, C.H.; Peng, Y.; Lines, C.R.; Nessly, M.L. Growth hormone secretagogue MK-677: No clinical effect on AD progression in a randomized trial. Neurology 2008, 71, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Antipova, T.A.; Gudasheva, T.A.; Seredenin, S.B. In Vitro Study of Neuroprotective Properties of GK-2, a New Original Nerve Growth Factor Mimetic. Bull. Exp. Biol. Med. 2011, 150, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Gudasheva, T.A.; Antipova, T.A.; Seredenin, S.B. Novel low-molecular-weight mimetics of the nerve growth factor. Dokl. Biochem. Biophys. 2010, 434, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Naletova, I.; Satriano, C.; Pietropaolo, A.; Giani, F.; Pandini, G.; Triaca, V.; Amadoro, G.; Latina, V.; Calissano, P.; Travaglia, A.; et al. The Copper(II)-Assisted Connection between NGF and BDNF by Means of Nerve Growth Factor-Mimicking Short Peptides. Cells 2019, 8, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaar, M.; Zhai, S.; Panova, I.; Fine, R.E.; Eisenhauer, P.B.; Blusztajn, J.K.; Lopez-Coviella, I.; Gilchrest, B.A. A cyclic peptide that binds p75(NTR) protects neurones from beta amyloid (1-40)-induced cell death. Neuropathol. Appl. Neurobiol. 2007, 33, 533–543. [Google Scholar] [CrossRef]
- Povarnina, P.Y.; Vorontsova, O.N.; Gudasheva, T.A.; Ostrovskaya, R.U.; Seredenin, S.B. Original Nerve Growth Factor Mimetic Dipeptide GK-2 Restores Impaired Cognitive Functions in Rat Models of Alzheimer’s Disease. Acta Nat. 2013, 5, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Gudasheva, T.A.; Povarnina, P.Y.; Volkova, A.A.; Kruglov, S.V.; Antipova, T.A.; Seredenin, S.B. A Nerve Growth Factor Dipeptide Mimetic Stimulates Neurogenesis and Synaptogenesis in the Hippocampus and Striatum of Adult Rats with Focal Cerebral Ischemia. Acta Nat. 2019, 11, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colangelo, A.M.; Bianco, M.R.; Vitagliano, L.; Cavaliere, C.; Cirillo, G.; De Gioia, L.; Diana, D.; Colombo, D.; Redaelli, C.; Zaccaro, L.; et al. A new nerve growth factor-mimetic peptide active on neuropathic pain in rats. J. Neurosci. 2008, 28, 2698–2709. [Google Scholar] [CrossRef] [Green Version]
- Cardenas-Aguayo, M.d.C.; Kazim, S.F.; Grundke-Iqbal, I.; Iqbal, K. Neurogenic and neurotrophic effects of BDNF peptides in mouse hippocampal primary neuronal cell cultures. PLoS ONE 2013, 8, e53596. [Google Scholar] [CrossRef]
- Gudasheva, T.A.; Tallerova, A.V.; Mezhlumyan, A.G.; Antipova, T.A.; Logvinov, I.O.; Firsova, Y.N.; Povarnina, P.Y.; Seredenin, S.B. Low-Molecular Weight BDNF Mimetic, Dimeric Dipeptide GSB-106, Reverses Depressive Symptoms in Mouse Chronic Social Defeat Stress. Biomolecules 2021, 11, 252. [Google Scholar] [CrossRef]
- Lauzon, M.A.; Drevelle, O.; Faucheux, N. Peptides derived from the knuckle epitope of BMP-9 induce the cholinergic differentiation and inactivate GSk3beta in human SH-SY5Y neuroblastoma cells. Sci. Rep. 2017, 7, 4695. [Google Scholar] [CrossRef] [Green Version]
- Lauzon, M.A.; Faucheux, N. A small peptide derived from BMP-9 can increase the effect of bFGF and NGF on SH-SY5Y cells differentiation. Mol. Cell Neurosci. 2018, 88, 83–92. [Google Scholar] [CrossRef]
- Rampazzo, E.; Dettin, M.; Maule, F.; Scabello, A.; Calvanese, L.; D’Auria, G.; Falcigno, L.; Porcu, E.; Zamuner, A.; Della Puppa, A.; et al. A synthetic BMP-2 mimicking peptide induces glioblastoma stem cell differentiation. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2282–2292. [Google Scholar] [CrossRef]
- Xiong, S.; Ma, M.; Xu, Y.; Wei, F.; Gu, Q.; He, X.; Xu, X. Protective effects of peptide FK18 against neuro-excitotoxicity in SH-SY5Y cells. Exp. Ther. Med. 2021, 21, 451. [Google Scholar] [CrossRef]
- Gudasheva, T.A.; Povarnina, P.Y.; Antipova, T.A.; Firsova, Y.N.; Konstantinopolsky, M.A.; Seredenin, S.B. Dimeric dipeptide mimetics of the nerve growth factor Loop 4 and Loop 1 activate TRKA with different patterns of intracellular signal transduction. J. Biomed. Sci. 2015, 22, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, J.M.; Morton, C.J.; Zwar, R.A.; Murray, S.S.; O’Leary, P.D.; Hughes, R.A. Design of a conformationally defined and proteolytically stable circular mimetic of brain-derived neurotrophic factor. J. Biol. Chem. 2008, 283, 33375–33383. [Google Scholar] [CrossRef] [Green Version]
- Gudasheva, T.A.; Logvinov, I.O.; Antipova, T.A.; Seredenin, S.B. Brain-derived neurotrophic factor loop 4 dipeptide mimetic GSB-106 activates TrkB, Erk, and Akt and promotes neuronal survival in vitro. Dokl. Biochem. Biophys. 2013, 451, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.M.; Vu, T.K.; Mobley, W.C. The in vitro biological effect of nerve growth factor is inhibited by synthetic peptides. Cell Regul. 1990, 1, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, C.F.; Ebendal, T.; Barbany, G.; Murray-Rust, J.; Blundell, T.L.; Persson, H. Disruption of the low affinity receptor-binding site in NGF allows neuronal survival and differentiation by binding to the trk gene product. Cell 1992, 69, 329–341. [Google Scholar] [CrossRef]
- McDonald, N.Q.; Lapatto, R.; Murray-Rust, J.; Blundell, T.L. X-ray crystallographic studies on murine nerve growth factor. J. Cell Sci. Suppl. 1990, 13, 19–30. [Google Scholar] [CrossRef] [Green Version]
- LeSauteur, L.; Wei, L.; Gibbs, B.F.; Saragovi, H.U. Small peptide mimics of nerve growth factor bind TrkA receptors and affect biological responses. J. Biol. Chem. 1995, 270, 6564–6569. [Google Scholar] [CrossRef] [Green Version]
- Maliartchouk, S.; Debeir, T.; Beglova, N.; Cuello, A.C.; Gehring, K.; Saragovi, H.U. Genuine monovalent ligands of TrkA nerve growth factor receptors reveal a novel pharmacological mechanism of action. J. Biol. Chem. 2000, 275, 9946–9956. [Google Scholar] [CrossRef] [Green Version]
- Longo, F.M.; Manthorpe, M.; Xie, Y.M.; Varon, S. Synthetic NGF peptide derivatives prevent neuronal death via a p75 receptor-dependent mechanism. J. Neurosci. Res. 1997, 48, 1–17. [Google Scholar] [CrossRef]
- Gudasheva, T.A.; Tarasiuk, A.V.; Sazonova, N.M.; Pomogaibo, S.V.; Shumskiy, A.N.; Logvinov, I.O.; Nikolaev, S.V.; Povarnina, P.Y.; Konstantinopolsky, M.A.; Antipova, T.A.; et al. Design, synthesis, and neuroprotective effects of a dimeric dipeptide mimetic of the third loop of the nerve growth factor. Russ. J. Bioorganic Chem. 2017, 43, 235–247. [Google Scholar] [CrossRef]
- Woo, S.B.; Neet, K.E. Characterization of histidine residues essential for receptor binding and activity of nerve growth factor. J. Biol. Chem. 1996, 271, 24433–24441. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.; Laramee, G.R.; Schmelzer, C.H.; Burton, L.E.; Winslow, J.W. Mutagenesis identifies amino-terminal residues of nerve growth factor necessary for Trk receptor binding and biological activity. J. Biol. Chem. 1994, 269, 27679–27686. [Google Scholar] [CrossRef]
- Berrera, M.; Cattaneo, A.; Carloni, P. Molecular simulation of the binding of nerve growth factor peptide mimics to the receptor tyrosine kinase A. Biophys. J. 2006, 91, 2063–2071. [Google Scholar] [CrossRef] [Green Version]
- Travaglia, A.; Arena, G.; Fattorusso, R.; Isernia, C.; La Mendola, D.; Malgieri, G.; Nicoletti, V.G.; Rizzarelli, E. The inorganic perspective of nerve growth factor: Interactions of Cu2+ and Zn2+ with the N-terminus fragment of nerve growth factor encompassing the recognition domain of the TrkA receptor. Chemistry 2011, 17, 3726–3738. [Google Scholar] [CrossRef] [PubMed]
- Travaglia, A.; Pietropaolo, A.; Di Martino, R.; Nicoletti, V.G.; La Mendola, D.; Calissano, P.; Rizzarelli, E. A small linear peptide encompassing the NGF N-terminus partly mimics the biological activities of the entire neurotrophin in PC12 cells. ACS Chem. Neurosci. 2015, 6, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Pandini, G.; Satriano, C.; Pietropaolo, A.; Giani, F.; Travaglia, A.; La Mendola, D.; Nicoletti, V.G.; Rizzarelli, E. The Inorganic Side of NGF: Copper(II) and Zinc(II) Affect the NGF Mimicking Signaling of the N-Terminus Peptides Encompassing the Recognition Domain of TrkA Receptor. Front. Neurosci. 2016, 10, 569. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? Acc. Chem. Res. 2019, 52, 2026–2035. [Google Scholar] [CrossRef]
- Stelmashook, E.V.; Genrikhs, E.E.; Novikova, S.V.; Barskov, I.V.; Gudasheva, T.A.; Seredenin, S.B.; Khaspekov, L.G.; Isaev, N.K. Behavioral effect of dipeptide NGF mimetic GK-2 in an in vivo model of rat traumatic brain injury and its neuroprotective and regenerative properties in vitro. Int. J. Neurosci. 2015, 125, 375–379. [Google Scholar] [CrossRef]
- Povarnina, P.Y.; Gudasheva, T.A.; Vorontsova, O.N.; Bondarenko, N.A.; Seredenin, S.B. Antiparkinsonian Properties of a Nerve Growth Factor Dipeptide Mimetic GK-2 in in Vivo Experiments. Bull. Exp. Biol. Med. 2011, 151, 690. [Google Scholar] [CrossRef]
- Soumen, R.; Johnston, A.H.; Moin, S.T.; Dudas, J.; Newman, T.A.; Hausott, B.; Schrott-Fischer, A.; Glueckert, R. Activation of TrkB receptors by NGFbeta mimetic peptide conjugated polymersome nanoparticles. Nanomedicine 2012, 8, 271–274. [Google Scholar] [CrossRef]
- O’Leary, P.D.; Hughes, R.A. Structure-activity relationships of conformationally constrained peptide analogues of loop 2 of brain-derived neurotrophic factor. J. Neurochem. 1998, 70, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, C.F.; Ebendal, T.; Persson, H. Chimeric molecules with multiple neurotrophic activities reveal structural elements determining the specificities of NGF and BDNF. EMBO J. 1991, 10, 2105–2110. [Google Scholar] [CrossRef]
- Ibáñez, C.F.; Ilag, L.L.; Murray-Rust, J.; Persson, H. An extended surface of binding to Trk tyrosine kinase receptors in NGF and BDNF allows the engineering of a multifunctional pan-neurotrophin. EMBO J. 1993, 12, 2281–2293. [Google Scholar] [CrossRef]
- O’Leary, P.D.; Hughes, R.A. Design of potent peptide mimetics of brain-derived neurotrophic factor. J. Biol. Chem. 2003, 278, 25738–25744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, J.M.; Hughes, R.A. Modified low molecular weight cyclic peptides as mimetics of BDNF with improved potency, proteolytic stability and transmembrane passage in vitro. Bioorg. Med. Chem. 2009, 17, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Zainullina, L.F.; Vakhitova, Y.V.; Lusta, A.Y.; Gudasheva, T.A.; Seredenin, S.B. Dimeric mimetic of BDNF loop 4 promotes survival of serum-deprived cell through TrkB-dependent apoptosis suppression. Sci. Rep. 2021, 11, 7781. [Google Scholar] [CrossRef] [PubMed]
- Fobian, K.; Owczarek, S.; Budtz, C.; Bock, E.; Berezin, V.; Pedersen, M.V. Peptides derived from the solvent-exposed loops 3 and 4 of BDNF bind TrkB and p75(NTR) receptors and stimulate neurite outgrowth and survival. J. Neurosci. Res. 2010, 88, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Seredenin, S.B.; Voronina, T.A.; Gudasheva, T.A.; Garibova, T.L.; Molodavkin, G.M.; Litvinova, S.A.; Elizarova, E.A.; Poseva, V.I. Antidepressant Effect of Dimeric Dipeptide GSB-106, an Original Low-Molecular-Weight Mimetic of BDNF. Acta Nat. 2013, 5, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Gudasheva, T.A.; Povarnina, P.; Logvinov, I.O.; Antipova, T.A.; Seredenin, S.B. Mimetics of brain-derived neurotrophic factor loops 1 and 4 are active in a model of ischemic stroke in rats. Drug Des. Devel. Ther. 2016, 10, 3545–3553. [Google Scholar] [CrossRef] [Green Version]
- Kolyvanov, G.B.; Zherdev, V.P.; Gribakina, O.G.; Bochkov, P.O.; Shevchenko, R.V.; Litvin, A.A.; Blynskaya, E.V.; Bueva, V.V. Comparative Preclinical Pharmacokinetics and Bioavailability of Antidepressant GSB-106 Tablet Form. Bull. Exp. Biol. Med. 2019, 167, 637–640. [Google Scholar] [CrossRef]
- Povarnina, P.Y.; Garibova, T.L.; Gudasheva, T.A.; Seredenin, S.B. Antidepressant Effect of an Orally Administered Dipeptide Mimetic of the Brain-Derived Neurotrophic Factor. Acta Nat. 2018, 10, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Suzuki, Y.; Ogata, S.; Ohtsuki, C.; Tanihara, M. Accelerated bone repair with the use of a synthetic BMP-2-derived peptide and bone-marrow stromal cells. J. Biomed. Mater. Res. A 2005, 72, 77–82. [Google Scholar] [CrossRef]
- Bergeron, E.; Marquis, M.E.; Chretien, I.; Faucheux, N. Differentiation of preosteoblasts using a delivery system with BMPs and bioactive glass microspheres. J. Mater. Sci. Mater. Med. 2007, 18, 255–263. [Google Scholar] [CrossRef]
- Beauvais, S.; Drevelle, O.; Lauzon, M.A.; Daviau, A.; Faucheux, N. Modulation of MAPK signalling by immobilized adhesive peptides: Effect on stem cell response to BMP-9-derived peptides. Acta Biomater. 2016, 31, 241–251. [Google Scholar] [CrossRef]
- Lauzon, M.A.; Marcos, B.; Faucheux, N. Characterization of alginate/chitosan-based nanoparticles and mathematical modeling of their SpBMP-9 release inducing neuronal differentiation of human SH-SY5Y cells. Carbohydr. Polym. 2018, 181, 801–811. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Baird, A.; Schubert, D.; Ling, N.; Guillemin, R. Receptor- and heparin-binding domains of basic fibroblast growth factor. Proc. Natl. Acad. Sci. USA 1988, 85, 2324–2328. [Google Scholar] [CrossRef] [Green Version]
- Xiong, S.; Xu, Y.; Ma, M.; Wang, H.; Wei, F.; Gu, Q.; Xu, X. Neuroprotective effects of a novel peptide, FK18, under oxygen-glucose deprivation in SH-SY5Y cells and retinal ischemia in rats via the Akt pathway. Neurochem. Int. 2017, 108, 78–90. [Google Scholar] [CrossRef]
- Plotnikov, A.N.; Schlessinger, J.; Hubbard, S.R.; Mohammadi, M. Structural Basis for FGF Receptor Dimerization and Activation. Cell 1999, 98, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Manfe, V.; Kochoyan, A.; Bock, E.; Berezin, V. Peptides derived from specific interaction sites of the fibroblast growth factor 2-FGF receptor complexes induce receptor activation and signaling. J. Neurochem. 2010, 114, 74–86. [Google Scholar] [CrossRef]
- Guan, J.; Waldvogel, H.J.; Faull, R.L.M.; Gluckman, P.D.; Williams, C.E. The effects of the N-terminal tripeptide of insulin-like growth factor-1, glycine-proline-glutamate in different regions following hypoxic-ischemic brain injury in adult rats. Neuroscience 1999, 89, 649–659. [Google Scholar] [CrossRef]
- Baker, A.M.; Batchelor, D.C.; Thomas, G.B.; Wen, J.Y.; Rafiee, M.; Lin, H.; Guan, J. Central penetration and stability of N-terminal tripeptide of insulin-like growth factor-I, glycine-proline-glutamate in adult rat. Neuropeptides 2005, 39, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Sara, V.R.; Carlsson-Skwirut, C.; Bergman, T.; Jörnvall, H.; Roberts, P.J.; Crawford, M.; Håkansson, L.N.; Civalero, I.; Nordberg, A. Identification of Gly-Pro-Glu (GPE), the aminoterminal tripeptide of insulin-like growth factor 1 which is truncated in brain, as a novel neuroactive peptide. Biochem. Biophys. Res. Commun. 1989, 165, 766–771. [Google Scholar] [CrossRef]
- Nilsson-Håkansson, L.; Civalero, I.; Zhang, X.; Carlsson-Skwirut, C.; Sara, V.R.; Nordberg, A. Effects of IGF-1, truncated IGF-1 and the tripeptide Gly-Pro-Glu on acetylcholine release from parietal cortex of rat brain. Neuroreport 1993, 4, 1111–1114. [Google Scholar]
- Saura, J.; Curatolo, L.; Williams, C.E.; Gatti, S.; Benatti, L.; Peeters, C.; Guan, J.; Dragunow, M.; Post, C.; Faull, R.L.; et al. Neuroprotective effects of Gly-Pro-Glu, the N- terminal tripeptide of IGF-1, in the hippocampus in vitro. NeuroReport 1999, 10, 161–164. [Google Scholar] [CrossRef]
- Marinelli, L.; Fornasari, E.; Di Stefano, A.; Turkez, H.; Arslan, M.E.; Eusepi, P.; Ciulla, M.; Cacciatore, I. (R)-alpha-Lipoyl-Gly-l-Pro-l-Glu dimethyl ester as dual acting agent for the treatment of Alzheimer’s disease. Neuropeptides 2017, 66, 52–58. [Google Scholar] [CrossRef]
- Marinelli, L.; Fornasari, E.; Di Stefano, A.; Turkez, H.; Genovese, S.; Epifano, F.; Di Biase, G.; Costantini, E.; D’Angelo, C.; Reale, M.; et al. Synthesis and biological evaluation of novel analogues of Gly-l-Pro-l-Glu (GPE) as neuroprotective agents. Bioorg. Med. Chem. Lett. 2019, 29, 194–198. [Google Scholar] [CrossRef]
- Blanchard, J.; Chohan, M.O.; Li, B.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I. Beneficial effect of a CNTF tetrapeptide on adult hippocampal neurogenesis, neuronal plasticity, and spatial memory in mice. J. Alzheimers Dis. 2010, 21, 1185–1195. [Google Scholar] [CrossRef]
- Chohan, M.O.; Li, B.; Blanchard, J.; Tung, Y.C.; Heaney, A.T.; Rabe, A.; Iqbal, K.; Grundke-Iqbal, I. Enhancement of dentate gyrus neurogenesis, dendritic and synaptic plasticity and memory by a neurotrophic peptide. Neurobiol. Aging 2011, 32, 1420–1434. [Google Scholar] [CrossRef]
- Blanchard, J.; Wanka, L.; Tung, Y.C.; Cardenas-Aguayo Mdel, C.; LaFerla, F.M.; Iqbal, K.; Grundke-Iqbal, I. Pharmacologic reversal of neurogenic and neuroplastic abnormalities and cognitive impairments without affecting Abeta and tau pathologies in 3xTg-AD mice. Acta Neuropathol. 2010, 120, 605–621. [Google Scholar] [CrossRef]
- Kazim, S.F.; Iqbal, K. Neurotrophic factor small-molecule mimetics mediated neuroregeneration and synaptic repair: Emerging therapeutic modality for Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 50. [Google Scholar] [CrossRef] [Green Version]
- Rockenstein, E.; Ubhi, K.; Doppler, E.; Novak, P.; Moessler, H.; Li, B.; Blanchard, J.; Grundke-Iqbal, I.; Iqbal, K.; Mante, M.; et al. Regional comparison of the neurogenic effects of CNTF-derived peptides and cerebrolysin in AbetaPP transgenic mice. J. Alzheimers Dis. 2011, 27, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Wanka, L.; Blanchard, J.; Liu, F.; Chohan, M.O.; Iqbal, K.; Grundke-Iqbal, I. Neurotrophic peptides incorporating adamantane improve learning and memory, promote neurogenesis and synaptic plasticity in mice. FEBS Lett. 2010, 584, 3359–3365. [Google Scholar] [CrossRef] [Green Version]
- Bolognin, S.; Buffelli, M.; Puolivali, J.; Iqbal, K. Rescue of cognitive-aging by administration of a neurogenic and/or neurotrophic compound. Neurobiol. Aging 2014, 35, 2134–2146. [Google Scholar] [CrossRef]





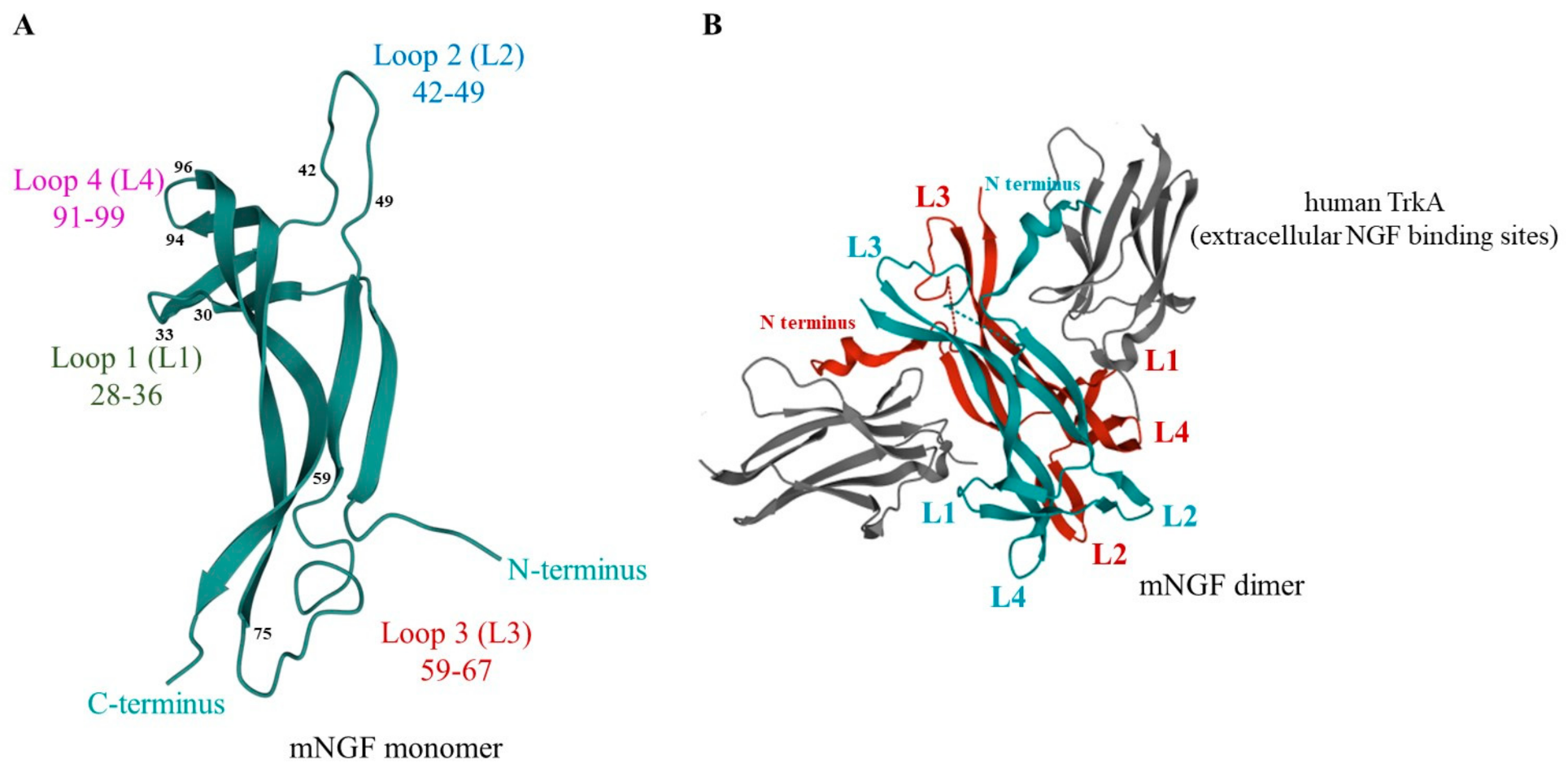{kind=link}
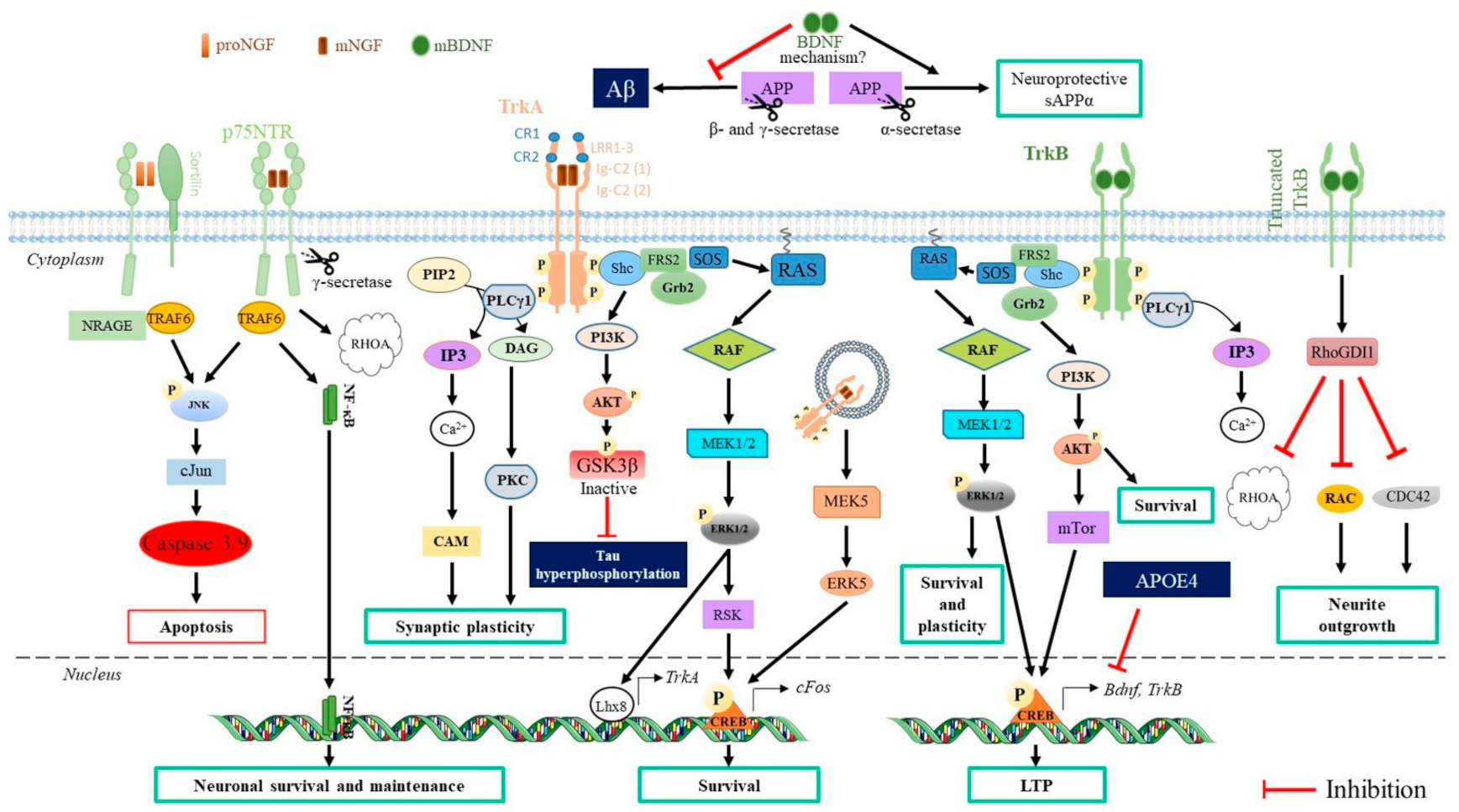{kind=link}
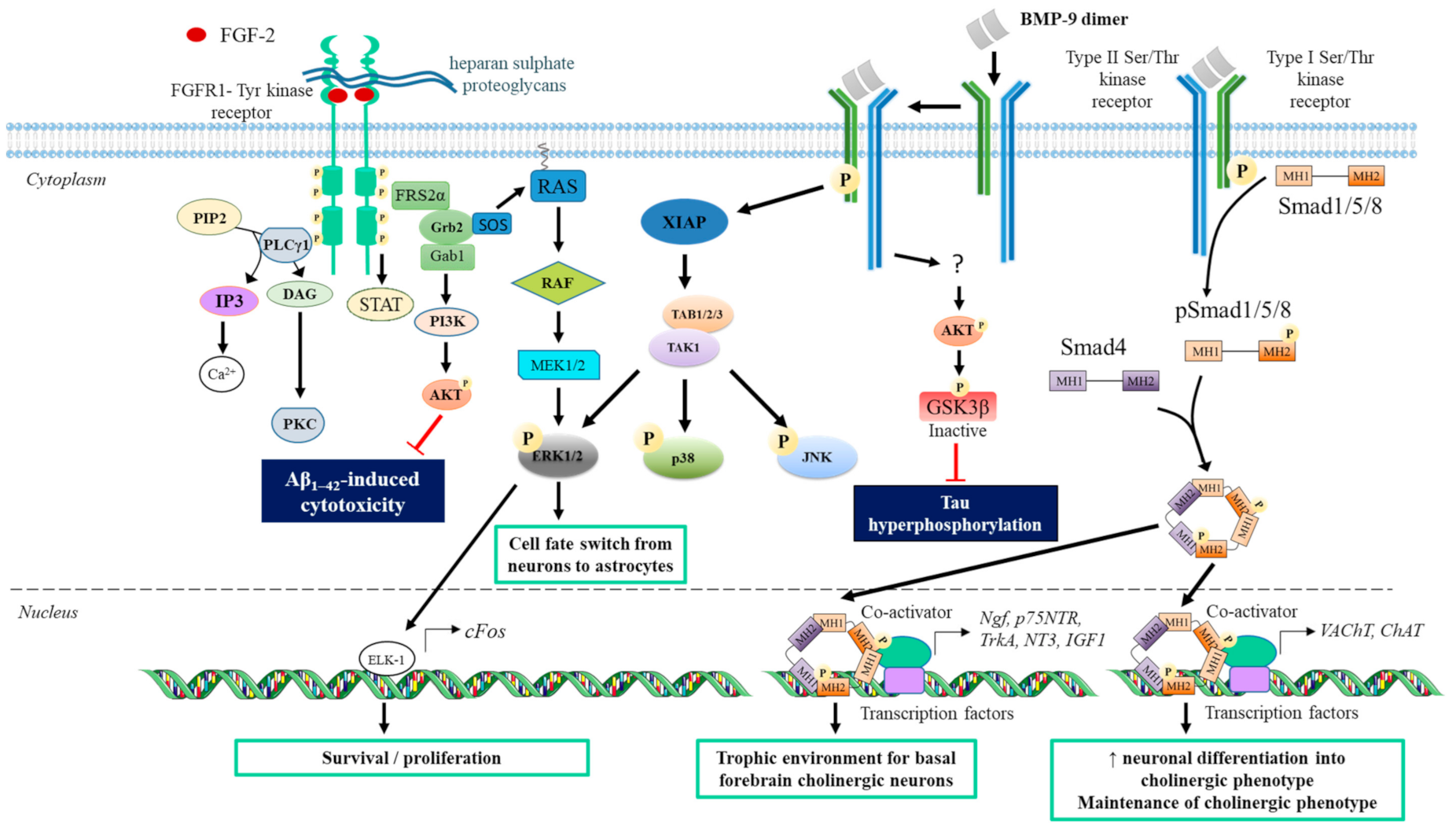{kind=link}
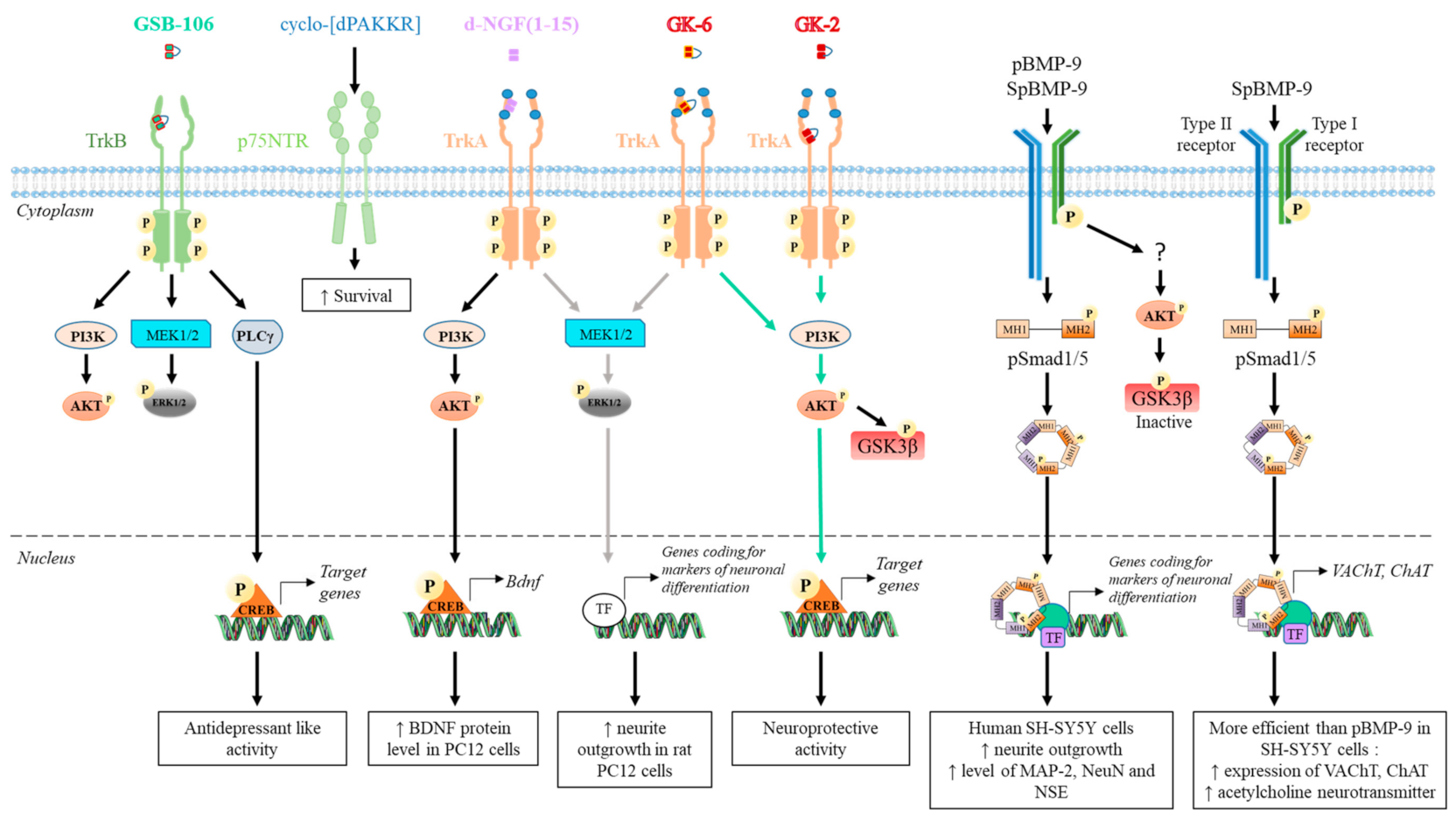{kind=link}
| Superfamily | Experimental Conditions | Effect on CNS Cells In Vitro or In vivo | Refs |
|---|---|---|---|
| Neurotrophin | |||
| NGF | Animal: Homozygous Ngf −/− knockout or WT mice C57BL/6 | Ngf cKO mice: | [100] |
| ↓ Hippocampal Ngf mRNA level | |||
| Treatment: Injection of viral vector for Ngf overexpression under stereotaxic guide | ↓ Adult hippocampal neurogenesis | ||
| ↓ Cholinergic fiber density in the hippocampus but not in the cortex | |||
| NGF restores hippocampal cholinergic fiber innervations and spatial memory. | |||
| BDNF | Animal: Female APP.PS1 transgenic mice | BDNF + ADTC5 compared to BDNF alone or vehicle: | [103] |
| ↑ Cognitive performance (Y-maze and new object recognition) | |||
| Treatment: BDNF at 5.7 nmol/kg, BDNF + ADTC5 (modulator to allow BDNF to pass BBB) at 10 µmol/kg or vehicle. Intravenous injection every 4 days for 8 injections | ↑ Degree of neuron-glial antigen 2 (NG2) receptor expression a marker for oligodendrocyte maturation | ||
| ↑ Hippocampus level of early growth response 1 (EGR1) and activity-related cytoskeleton-associated protein mRNA transcripts | |||
| No significant impact on Aβ plaque | |||
| Animal: AD11 anti-NGF mice (sporadic AD model), 6 months old | Rescue memory performance (object recognition and object context tests) | [104] | |
| Treatment: Intranasal delivery at 12.6, 42, and 420 pmol/administration, repeated 7 times for 15 days | No impact on Ab plaques, tau hyperphosphorylation and cholinergic deficit | ||
| ↓ CD11b-positive microglia in the hippocampus | |||
| BMP | |||
| BMP-6 | Cells: neuronal progenitor cells from adult rat (NPC) | Aβ1-42: ↑ BMP-6 level | [105] |
| Treatment: 50–100 ng/mL for 4 days with a refresh of medium containing BMP-6 at 2 days | BMP-6: ↓ Proliferation of NPC (dose dependent effect) | ||
| BMP-9 | Animal: APP.PS1/CHGFP (AD model) and WT/CHGFP (control) transgenic mice 5 and 10 months old | ↓ Number Aβ amyloid plaques in AD model | [106] |
| ↑ ChAT expression in APP.PS1/CHGFP and WT/CHGFP | |||
| ↑ Density of cholinergic fibers in APP.PS1/CHGFP and WT/CHGFP | |||
| Treatment: intraventricular infusion at 4 ng/h for 7 days | ↑ Hippocampal level of receptors TrkA and p75NTR in 5 months old mice but not in 10 months old mice | ||
| ↑ Hippocampal level of NGF in both mice (15–20%) | |||
| ↑ IGF-1 levels in 5 months APP.PS1/CHGFP | |||
| Animal: APP/PS1 mice (7 months) | Improve spatial and associative learning and memory (Morris water maze, contextual fear conditioning test) | [107] | |
| ↓ Aβ levels and number of plaques in AD model | |||
| Treatment: intranasal delivery of 50 ng/g/d for 30 days | ↓ Hyperphosphorylated tau in the cortex and hippocampus | ||
| ↓ Neuroinflammation (activated microglia and astrocytes) | |||
| ↑ Expression of low-density lipoprotein receptor-related protein 1 (LRP1), involved in the clearance of Ab | |||
| IGF | |||
| IGF-2 | Animal: Tg2576 AD mouse (10 months old) and WT C57BL/6 mouse (control) | ↓ Oxidative stress | [108] |
| ↑ Expression of PI3K, AKT and CREB in hippocampus ↓ levels of amyloid plaques in the hippocampus | |||
| Treatment: Injection saline/DMSO (control) and IGF-2 at 250 ng | ↑ Memory consolidation (Morris Water Maze) via PI3K/AKT pathway | ||
| ↓ Memory decline | |||
| Animal: APP.PS1/CHGFP and Wild Type (control) mice | ↓ Aβ plaque numbers | [109] | |
| ↑ p75NGFR compared to vehicle both in APP.PS1/CHGFP and WT | |||
| ↑ ChAT in APP.PS1/CHGFP and WT hippocampus | |||
| Treatment: Infusion of vehicle or 50 ng/h hIGF-2 for 7 days | ↑ BMP-9 level in APP.PS1/CHGFP and WT hippocampus (basal level is higher in APP.PS1/CHGFP) | ||
| ↓ ALK1 expression in WT but not in APP.PS1/CHGFP hippocampus | |||
| ↓ FGF-2 level in APP.PS1/CHGFP hippocampus | |||
| ↑ Hippocampal neurogenesis (DCX) in APP.PS1/CHGFP and WT hippocampus | |||
| FGF | |||
| FGF-2 | Animal: APP.PS1 mice (AD model) and WT Tg2576 mice (control). | In vivo: FGF-2 in APP.PS1 mice Reverse learning deficit memory ↓ Aβ hippocampal deposition ↑ Neurogenesis in subgranular zone of dental gyrus In vitro: FGF-2 ↑ Dose-dependent Aβ phagocytosis in microglia ↓ Production of Aβ in neural stem cells ↑ Neuronal differentiation of neural stem cells | [110] |
| Treatment: hippocampal injection of hybrid virus expressing FGF-2 or GFP (control) at 1 × 1010 per brain at 4 months (presymptomatic) and 7–8 months (postsymptomatic) of age | |||
| Cells: Primary microglia culture and neural stem cells (mouse embryonic brain day 14) | |||
| Treatment: Microglia: FGF-2 (0.1 or 1 ng/mL) + 10 µg fibrillar Aβ1–42 for 1 h | |||
| Neural stem cell: hybrid virus + 1 µM Aβ1–42 oligomer for 7 days | |||
| Cells: Primary astrocyte culture weight FGF-2 (HMW, 23 kDa) at 10 ng/mL | LMW and HMW FGF-2: | [111] | |
| Induce ERK and AKT pathway activation | |||
| Protective effect against cytotoxicity induced by Aβ (20 μM) or oxidative stress | |||
| Treatment: Medium with or without purified low molecular weight FGF-2 (LMW, 17 kDa) or high molecular | ↑ Bcl-XL transcripts | ||
| LMW FGF-2: | |||
| ↑ Proliferation by upregulation of c-Myc, Cyclin D1, and Cyclin E through PI3K/AKT pathway | |||
| FGF-21 | Animal: male APP.PS1 transgenic mice (6 months old) | In vivo: | [112] |
| Subcutaneous injection: | |||
| ↑ Learning abilities after 5 days (Morris water maze) | |||
| ↓ Brain Aβ burden | |||
| Treatment: Subcutaneously injection with 5 mg/kg/day twice a day for 1 month or intracerebroventricular with a mini pump 0.4 µg/day for 14 days | ↓ Tau phosphorylation positive area | ||
| Intracerebroventricular injection: | |||
| Rescue neurodegeneration through the FGF-21/FGFR1 signaling pathway (Morris water Maze) | |||
| In vitro: | |||
| Cells: Rat (PC12) pheochromocytoma cells and rat astrocyte (C6) line in coculture treated with FGF-21 at different concentrations between 0.07 and 8 µM | ↑ Cell viability against Aβ25-35 toxicity (higher effect in the presence of astrocytes) | ||
| ↓ Tau hyperphosphorylation | |||
| ↓ ROS levels | |||
| Rescues the lactate system deficiency induced by Aβ25–35 | |||
| Superfamily | Peptide Sequence | Experimental Conditions | Effect on CNS Cells In Vitro or In Vivo | Refs |
|---|---|---|---|---|
| Neurotrophin | ||||
| NGF | Dimeric dipeptide: | Cells: mouse hippocampal HT-22 immortalized neurons and primary culture of embryonic rat hippocampal neurons (18 days old embryos) | Strong neuroprotective properties at 10−8 M | [230] |
| GK-2 β-turn loop L4 | Treatment: peptide (10−5–10−10 M) added 24 h before adding H2O2 (1.5 mM) for 30 min or glutamate (5 mM) for 24 h | |||
 | Incubation time: 4 h and 24 h | |||
| Dimeric dipeptides | Cells: mouse hippocampal HT-22 immortalized neurons and rat pheochromocytoma PC12 | Both GK-2 (10−8 M) and GK-6 (10−6 M): | [231] | |
| GK-2 and | ↑ Phosphorylation of TrkA | |||
| GK-6 β-turn loop L1 | Treatment: peptide (10−5–10−10 M) added 24 h before oxidative stress | GK-6 (10−6 M): | ||
 | Exhibits slight neuroprotective properties | |||
| ↑ Differentiation (↑ neurite outgrowth in PC-12 at 7 days) | ||||
| NGF N terminus | Cells: Rat PC12 pheochromocytoma cells | ↑ Internalization of TrkA and p75NTR receptors | [232] | |
| Linear peptide NGF(1-14) | ||||
| SSSHPIFHRGEFSV-NH2 | Treatment: peptide at 50 µM or NGF (50 ng/mL) | ↑ Proliferation of PC12 cells at 48 h | ||
| Dimeric peptide d-NGF(1-15) | ||||
 | Incubation time: 10 min to 72 h | ↑ Differentiation (↑neurite total length at 72 h) | ||
| Cyclic peptide | Cells: p75NTR- and TrkA-NIH-3T3 cells and E17 fetal rat cortical neurons | No effect on NGF (0.5 nM) binding to TrkA, supporting its specificity for p75NTR | [233] | |
| ↓ Dose-dependent Aβ1–40 (0.5 nM) binding to p75NTR in rat cortical neurons | ||||
 | Treatment: Cyclic peptide (0–300 nM) for 30 min or 24 h and then Aβ1–40 (0.5 nM, 25 nM or 20 µM) for 30 min | ↓ Aβ1–40 (20 µM) signaling through p75NTR: ↓ c-jun mRNA and ↓ phosphorylation of cJUN | ||
| protects at 250 nM E17 neurons or 3T3 from Aβ1–40 (20 µM) -induced toxicity | ||||
| Dimeric dipeptide: | Animal: Mongrel male rats with bilaterally injection of Streptozotocin 3 mg/kg into their cerebral ventricles | GK-2 treatment can counteract the cognitive deficit in AD model (spatial memory impairment in Morris water maze) | [234] | |
| GK-2 | Treatment: GK-2 (0.5 mg/kg) or memantine (10 mg/kg) 4 h after the surgery and then once a day for 2 weeks | Effect similar to memantine | ||
| Dimeric dipeptide: | Animal: Ischemic stroke animal model; male Wistar rats (8–9 weeks) with intravascular thread occlusion of the middle cerebral artery | ↑ Hippocampal and striatum neurogenesis in rat cerebral ischemia | [235] | |
| GK-2 | Treatment: GK-2 (1 mg/kg, intraperitoneal); 6 or 24 h after surgery, once a day for 6 days | ↓ Volume of the ischemic injury (60% when injected 6 h after surgery) | ||
| Cyclic complex peptides derived from loops L1 and L4 with or without NGF N terminus | Cells: PC12 cells (clone 615) stably overexpressing TrkA, dorsal root ganglia (DRG) from 8-day-old chick embryos and cerebellar granule neurons from 8-day-old Sprague Dawley rat pups | In vitro: | [236] | |
| Both NL1L4 and L1L4 (3 µM) have neurotrophic properties | ||||
| NL1L4 | Treatment: NL1L4 (3, 6 and 10 µM), L1L4 (50, 100 nM, 3, 6 and 10 µM), and NGF (0.192 nM, control) | ↑ DRG differentiation within 2 days like NGF | ||
 | L1L4 dose-dependent ↑ PC12 differentiation at 3 days (EC50 1 µM) | |||
| Incubation time: 10 min (TrkA activation); 2 weeks (DRG) and 3 days (PC12) | ↑ TrkA phosphorylation (pTrkA) in PC12 cells at 10 min (NL1L4 and L1L4 (3 µM): 57 and 80% of pTrkA level obtained using NGF, respectively) | |||
| No effect on TrkB phosphorylation in cerebellar granule neurons | ||||
| L1L4 | Animal: CCI model (adult male Sprague Dawley rats) treated by L1L4 (37.5 µg/µL) through intrathecal lumbar spinal catheter (1 µL/h for 7 days) | In vivo: | ||
 | ↓ Neuropathic pain in CCI model (restores mechanical and thermal sensitivity) | |||
| BDNF | B-3 (Ac-SKKR-CONH2) | Cells: NIH 3T3 cells transfected with TrkB receptor; | ↑ TrkB phosphorylation at TrkB at Tyr 706 at 1 h | [237] |
| mouse E18 primary | No cytotoxic effect on cells at 5 days | |||
| hippocampal neurons | ↑ Neuronal differentiation (↑ b-III-tubulin, anti-neurofilament-M, and NeuN) in E18 hippocampal neurons at 5 days | |||
| B-5 (Ac-IKRG-CONH2) | Treatment: peptides (2 nM to 10 µM) | ↑ BDNF synthesis induced by B-3 (0.1 and 1 µM) and B-5 (0.1 µM) in primary E18 hippocampal cells at 5 days | ||
| Incubation time: 1 h; 2 and 5 days | TrkB synthesis induced by B-3 and B-5 (1 µM) in NIH-3T3 at 5 days | |||
| GSB-106 | Animal: male C57Bl/6 mice (chronic social defeat stress (CSDS)) | ↑ Locomotion in CSDS mice | [238] | |
 | Treatment: GSB-106 0.1 mg/kg once a day, for 21 days | Restores decreased synaptophysin level in hippocampus of CSDS mice | ||
| BMP | ||||
| BMP-9 | pBMP-9 | Cells: SH-SY5Y cells | ↑ Neuronal differentiation (↑ neurite outgrowth; ↑ MAP-2, NSE. NeuN at 5 days). | [239] |
| Ac-CGGKVGKACCVPTKLSPISVLYK-NH2 | Treatment: peptides or BMP-9 (control) at 0.1 or 1 nM with or without retinoic acid (RA) in serum-free medium | SpBMP-9 ↑ differentiation in cholinergic phenotype. (↑ acetylcholine, VAChT, ChAT) compared to BMP-9 or pBMP-9 | ||
| SpBMP-9 | ||||
| Ac-CGGKVGKASSVPTKLSPISVLYK-NH2 | Incubation time: 1, 3, and 5 days. | Adding RA ↑ peptide-induced differentiation | ||
| SpBMP-9 | Cells: SH-SY5Y cells | SpBMP-9 plus NGF or bFGF | [240] | |
| and NSpBMP-9 (negative peptide) | Treatment: peptides at 0.1 nM with or without NGF (100 ng/mL) or bFGF (FGF-2; 20 ng/mL) in serum-free medium | ↑ Neuronal differentiation (↑ neurite outgrowth, ↑ NSE expression) compared to growth factor alone | ||
| Ac-CGGKVGKAGGVPTKLSPIGGLYK-NH2 | ↑ Neuronal differentiation in cholinergic phenotype. (↑ VAChT vesicles located in the neurites) compared with growth factor alone | |||
| Incubation time: 5 days. | NSpBMP-9 has no effect | |||
| BMP-2 | GBMP1a (H-PFPLADHLNSTNHAIVQTLVNS-NH2) | Cells: primary human glioblastoma cells (glioma stem cells subpopulation) | ↑ Astroglial differentiation (↑ GFAP protein expression; ↑ S100) | [241] |
| Treatment: 60 ng/mL GBMP1a | ||||
| Incubation time: 5 days | ↓ Cell proliferation | |||
| FGF | ||||
| FGF-2 | FK-18 FFFERLESNNYNTYSRK | Cells: SH-SY5Y cell | ↓ Glutamate-induced apoptosis via Akt activation | [242] |
| Treatment: FK18 at 10 µg/mL or bFGF at 100 ng/mL 2 h before, at the same time, or 30 min after stimulation with glutamate (4–10 mM) | ↑ Bcl-2/Bax ratio | |||
| ↓ Cleaved caspase-3 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gascon, S.; Jann, J.; Langlois-Blais, C.; Plourde, M.; Lavoie, C.; Faucheux, N. Peptides Derived from Growth Factors to Treat Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 6071. https://doi.org/10.3390/ijms22116071
Gascon S, Jann J, Langlois-Blais C, Plourde M, Lavoie C, Faucheux N. Peptides Derived from Growth Factors to Treat Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(11):6071. https://doi.org/10.3390/ijms22116071
Chicago/Turabian StyleGascon, Suzanne, Jessica Jann, Chloé Langlois-Blais, Mélanie Plourde, Christine Lavoie, and Nathalie Faucheux. 2021. "Peptides Derived from Growth Factors to Treat Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 11: 6071. https://doi.org/10.3390/ijms22116071