Identifying Therapeutic Targets for Spinocerebellar Ataxia Type 3/Machado–Joseph Disease through Integration of Pathological Biomarkers and Therapeutic Strategies

Abstract
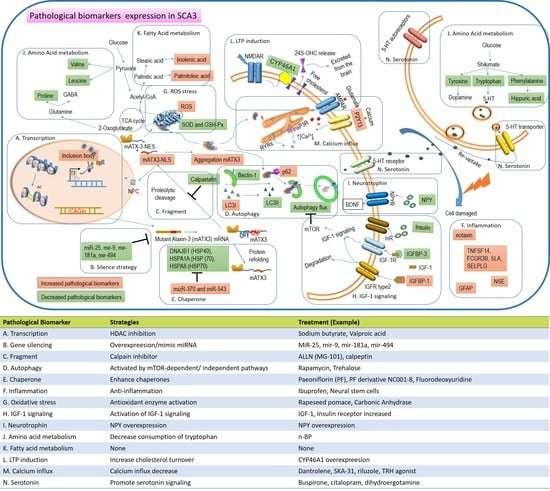
:
1. Introduction
2. Content
2.1. Update on Clinical Trials for SCA3/MJD Treatment
2.1.1. Neurotransmitter Modulators
2.1.2. Growth Factors
2.1.3. HDAC Inhibitors
2.1.4. Autophagy Enhancers
2.1.5. Stem Cells
2.2. Experimental Therapeutic Strategies for SCA3/MJD
2.2.1. RNAi Silencing of Ataxin-3 Expression
2.2.2. Reduced Cleavage Protein Formation
2.2.3. Inhibition of Ataxin-3 Fragment Nuclear Entry
2.3. Decreasing Ataxin-3 Aggregation
2.3.1. Phosphorylation/Dephosphorylation of Ataxin-3
2.3.2. SUMOylation Process of Ataxin-3
2.3.3. Autophagy
2.3.4. Proteosome System
2.3.5. Chaperones
2.4. Reducing Inflammation and Oxidative Stress
2.4.1. Inhibition of Inflammation
2.4.2. Mitigation of Oxidative Stress
2.5. Rescue of Cellular Dysfunction
2.5.1. Growth and Neurotrophic Factors
2.5.2. Metabolism
2.5.3. Enzymes
2.5.4. Transcription Regulation
2.6. Neuronal Homeostasis
2.6.1. Glutamate Receptor Signaling
2.6.2. Ion Channel Homeostasis
2.6.3. Adenosinergic System
2.6.4. Serotonergic System
3. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Li, T.; Martins, S.; Peng, Y.; Wang, P.; Hou, X.; Chen, Z.; Wang, C.; Tang, Z.; Qiu, R.; Chen, C.; et al. Is the High Frequency of Machado-Joseph Disease in China Due to New Mutational Origins? Front. Genet. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Soong, B.-W.; Lu, Y.-C.; Choo, K.-B.; Lee, H.-Y. Frequency Analysis of Autosomal Dominant Cerebellar Ataxias in Taiwanese Patients and Clinical and Molecular Characterization of Spinocerebellar Ataxia Type 6. Arch. Neurol. 2001, 58, 1105–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettencourt, C.; Lima, M. Machado-Joseph Disease: From first descriptions to new perspectives. Orphanet J. Rare Dis. 2011, 6, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieling, C.; Prestes, P.R.; Saraiva-Pereira, M.L.; Jardim, L.B. Survival estimates for patients with Machado-Joseph disease (SCA3). Clin. Genet. 2007, 72, 543–545. [Google Scholar] [CrossRef] [PubMed]
- Matos, C.A.; de Almeida, L.P.; Nóbrega, C. Machado-Joseph disease/spinocerebellar ataxia type 3: Lessons from disease pathogenesis and clues into therapy. J. Neurochem. 2019, 148, 8–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.H. Machado-joseph disease/spinocerebellar ataxia 3 responsive to buspirone. Mov. Disord. 1997, 12, 613–614. [Google Scholar] [CrossRef] [PubMed]
- Takei, A.; Honma, S.; Kawashima, A.; Yabe, I.; Fukazawa, T.; Hamada, K.; Hamada, T.; Tashiro, K. Beneficial effects of tandospirone on ataxia of a patient with Machado-Joseph disease. Psychiatry Clin. Neurosci. 2002, 56, 181–185. [Google Scholar] [CrossRef]
- Takei, A.; Fukazawa, T.; Hamada, T.; Sohma, H.; Yabe, I.; Sasaki, H.; Tashiro, K. Effects of tandospirone on “5-HT1A receptor-associated symptoms” in patients with Machado-Josephe disease: An open-label study. Clin. Neuropharmacol. 2004, 27, 9–13. [Google Scholar] [CrossRef]
- Arpa, J.; Sanz-Gallego, I.; Medina-Báez, J.; Portela, L.V.C.; Jardim, L.B.; Torres-Aleman, I.; Saute, J.A.M. Subcutaneous insulin-like growth factor-1 treatment in spinocerebellar ataxias: An open label clinical trial. Mov. Disord. 2011, 26, 358–359. [Google Scholar] [CrossRef]
- Zesiewicz, T.A.; Greenstein, P.E.; Sullivan, K.L.; Wecker, L.; Miller, A.; Jahan, I.; Chen, R.; Perlman, S.L. A randomized trial of varenicline (Chantix) for the treatment of spinocerebellar ataxia type 3. Neurology 2012, 78, 545–550. [Google Scholar] [CrossRef]
- Lei, L.F.; Yang, G.P.; Wang, J.L.; Chuang, D.M.; Song, W.H.; Tang, B.S.; Jiang, H. Safety and efficacy of valproic acid treatment in SCA3/MJD patients. Parkinsonism Relat. Disord. 2016, 26, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Saute, J.A.; de Castilhos, R.M.; Monte, T.L.; Schumacher-Schuh, A.F.; Donis, K.C.; D’Avila, R.; Souza, G.N.; Russo, A.D.; Furtado, G.V.; Gheno, T.C.; et al. A randomized, phase 2 clinical trial of lithium carbonate in Machado-Joseph disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Coarelli, G.; Marcotulli, C.; Leonardi, L.; Piccolo, F.; Spadaro, M.; Frontali, M.; Ferraldeschi, M.; Vulpiani, M.C.; Ponzelli, F.; et al. Riluzole in patients with hereditary cerebellar ataxia: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015, 14, 985–991. [Google Scholar] [CrossRef]
- Tsai, Y.A.; Liu, R.S.; Lirng, J.F.; Yang, B.H.; Chang, C.H.; Wang, Y.C.; Wu, Y.S.; Ho, J.H.; Lee, O.K.; Soong, B.W. Treatment of Spinocerebellar Ataxia With Mesenchymal Stem Cells: A Phase I/IIa Clinical Study. Cell Transplant. 2017, 26, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, B.S.; Prashanth, L.K.; Shah, B.B.; Marras, C.; Lang, A.E. A randomized trial of varenicline (chantix) for the treatment of spinocerebellar ataxia type 3. Neurology 2012, 79, 2218. [Google Scholar] [CrossRef] [Green Version]
- Teixeira-Castro, A.; Jalles, A.; Esteves, S.; Kang, S.; da Silva Santos, L.; Silva-Fernandes, A.; Neto, M.F.; Brielmann, R.M.; Bessa, C.; Duarte-Silva, S.; et al. Serotonergic signalling suppresses ataxin 3 aggregation and neurotoxicity in animal models of Machado-Joseph disease. Brain J. Neurol. 2015, 138, 3221–3237. [Google Scholar] [CrossRef] [Green Version]
- Watanave, M.; Matsuzaki, Y.; Nakajima, Y.; Ozawa, A.; Yamada, M.; Hirai, H. Contribution of Thyrotropin-Releasing Hormone to Cerebellar Long-Term Depression and Motor Learning. Front. Cell. Neurosci. 2018, 12, 490. [Google Scholar] [CrossRef]
- Nishizawa, M.; Onodera, O.; Hirakawa, A.; Shimizu, Y.; Yamada, M. Effect of rovatirelin in patients with cerebellar ataxia: Two randomised double-blind placebo-controlled phase 3 trials. J. Neurol. Neurosurg. Psychiatry 2020. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.M. Taltirelin (Tanabe Seiyaku). Idrugs Investig. Drugs J. 2001, 4, 1389–1400. [Google Scholar]
- Tan, S.; Wang, R.-H.; Niu, H.-X.; Shi, C.-H.; Mao, C.-Y.; Zhang, R.; Song, B.; Sun, S.-L.; Liu, X.-J.; Hou, H.-M.; et al. Nerve growth factor for the treatment of spinocerebellar ataxia type 3: An open-label study. Chin. Med. J. 2015, 128, 291–294. [Google Scholar] [CrossRef]
- Shi, Y.; Huang, F.; Tang, B.; Li, J.; Wang, J.; Shen, L.; Xia, K.; Jiang, H. MicroRNA profiling in the serums of SCA3/MJD patients. Int. J. Neurosci. 2014, 124, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhang, L.; Long, Z.; Chen, Z.; Hou, X.; Wang, C.; Peng, H.; Wang, J.; Li, J.; Duan, R.; et al. miR-25 alleviates polyQ-mediated cytotoxicity by silencing ATXN3. FEBS Lett. 2014, 588, 4791–4798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona, V.; Cunha-Santos, J.; Onofre, I.; Simões, A.T.; Vijayakumar, U.; Davidson, B.L.; de Almeida, L.P. Unravelling Endogenous MicroRNA System Dysfunction as a New Pathophysiological Mechanism in Machado-Joseph Disease. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1038–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Gong, X.; Zhang, L.; Li, T.; Yuan, H.; Xie, Y.; Peng, Y.; Qiu, R.; Xia, K.; Tang, B.; et al. Identification of a potential exosomal biomarker in spinocerebellar ataxia Type 3/Machado-Joseph disease. Epigenomics 2019, 11, 1037–1056. [Google Scholar] [CrossRef] [Green Version]
- Simões, A.T.; Gonçalves, N.; Koeppen, A.; Déglon, N.; Kügler, S.; Duarte, C.B.; Pereira de Almeida, L. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado–Joseph disease. Brain J. Neurol. 2012, 135, 2428–2439. [Google Scholar] [CrossRef]
- Watchon, M.; Yuan, K.C.; Mackovski, N.; Svahn, A.J.; Cole, N.J.; Goldsbury, C.; Rinkwitz, S.; Becker, T.S.; Nicholson, G.A.; Laird, A.S. Calpain Inhibition Is Protective in Machado-Joseph Disease Zebrafish Due to Induction of Autophagy. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 7782–7794. [Google Scholar] [CrossRef] [Green Version]
- Haacke, A.; Hartl, F.U.; Breuer, P. Calpain inhibition is sufficient to suppress aggregation of polyglutamine-expanded ataxin-3. J. Biol. Chem. 2007, 282, 18851–18856. [Google Scholar] [CrossRef] [Green Version]
- Onofre, I.; Mendonça, N.; Lopes, S.; Nobre, R.; de Melo, J.B.; Carreira, I.M.; Januário, C.; Gonçalves, A.F.; de Almeida, L.P. Fibroblasts of Machado Joseph Disease patients reveal autophagy impairment. Sci. Rep. 2016, 6, 28220. [Google Scholar] [CrossRef]
- Nascimento-Ferreira, I.; Santos-Ferreira, T.; Sousa-Ferreira, L.; Auregan, G.; Onofre, I.; Alves, S.; Dufour, N.; Colomer Gould, V.F.; Koeppen, A.; Déglon, N.; et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado–Joseph disease. Brain J. Neurol. 2011, 134, 1400–1415. [Google Scholar] [CrossRef]
- Ou, Z.; Luo, M.; Niu, X.; Chen, Y.; Xie, Y.; He, W.; Song, B.; Xian, Y.; Fan, D.; OuYang, S.; et al. Autophagy Promoted the Degradation of Mutant ATXN3 in Neurally Differentiated Spinocerebellar Ataxia-3 Human Induced Pluripotent Stem Cells. BioMed Res. Int. 2016, 2016, 6701793. [Google Scholar] [CrossRef] [Green Version]
- Marcelo, A.; Brito, F.; Carmo-Silva, S.; Matos, C.A.; Alves-Cruzeiro, J.; Vasconcelos-Ferreira, A.; Koppenol, R.; Mendonça, L.; de Almeida, L.P.; Nóbrega, C. Cordycepin activates autophagy through AMPK phosphorylation to reduce abnormalities in Machado-Joseph disease models. Hum. Mol. Genet. 2019, 28, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Santos, J.; Duarte-Neves, J.; Carmona, V.; Guarente, L.; Pereira de Almeida, L.; Cavadas, C. Caloric restriction blocks neuropathology and motor deficits in Machado-Joseph disease mouse models through SIRT1 pathway. Nat. Commun. 2016, 7, 11445. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, M.P.; Rujano, M.A.; Van Waarde, M.A.; Vis, E.; Brunt, E.R.; Kampinga, H.H. Levels of DNAJB family members (HSP40) correlate with disease onset in patients with spinocerebellar ataxia type 3. Eur. J. Neurosci. 2010, 32, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Evert, B.O.; Nalavade, R.; Jungverdorben, J.; Matthes, F.; Weber, S.; Rajput, A.; Bonn, S.; Brüstle, O.; Peitz, M.; Krauß, S. Upregulation of miR-370 and miR-543 is associated with reduced expression of heat shock protein 40 in spinocerebellar ataxia type 3. PLoS ONE 2018, 13, e0201794. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Experimental and Clinical Strategies for Treating Spinocerebellar Ataxia Type 3. Neuroscience 2018, 371, 138–154. [Google Scholar] [CrossRef] [PubMed]
- Raposo, M.; Bettencourt, C.; Maciel, P.; Gao, F.; Ramos, A.; Kazachkova, N.; Vasconcelos, J.; Kay, T.; Rodrigues, A.J.; Bettencourt, B.; et al. Novel candidate blood-based transcriptional biomarkers of Machado-Joseph disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 968–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendonça, L.S.; Nóbrega, C.; Tavino, S.; Brinkhaus, M.; Matos, C.; Tomé, S.; Moreira, R.; Henriques, D.; Kaspar, B.K.; Pereira de Almeida, L. Ibuprofen enhances synaptic function and neural progenitors proliferation markers and improves neuropathology and motor coordination in Machado-Joseph disease models. Hum. Mol. Genet. 2019, 28, 3691–3703. [Google Scholar] [CrossRef]
- De Assis, A.M.; Saute, J.A.M.; Longoni, A.; Haas, C.B.; Torrez, V.R.; Brochier, A.W.; Souza, G.N.; Furtado, G.V.; Gheno, T.C.; Russo, A.; et al. Peripheral Oxidative Stress Biomarkers in Spinocerebellar Ataxia Type 3/Machado-Joseph Disease. Front. Neurol. 2017, 8, 485. [Google Scholar] [CrossRef] [Green Version]
- Pohl, F.; Teixeira-Castro, A.; Costa, M.D.; Lindsay, V.; Fiúza-Fernandes, J.; Goua, M.; Bermano, G.; Russell, W.; Maciel, P.; Kong Thoo Lin, P. GST-4-Dependent Suppression of Neurodegeneration in C. elegans Models of Parkinson’s and Machado-Joseph Disease by Rapeseed Pomace Extract Supplementation. Front. Neurosci. 2019, 13, 1091. [Google Scholar] [CrossRef]
- Hsieh, M.; Hsieh, B.Y.; Ma, C.-Y.; Li, Y.-T.; Liu, C.-S.; Lo, C.-M. Protective roles of carbonic anhydrase 8 in Machado-Joseph Disease. J. Neurosci. Res. 2019, 97, 1278–1297. [Google Scholar] [CrossRef]
- Tort, A.B.L.; Portela, L.V.C.; Rockenbach, I.C.; Monte, T.L.; Pereira, M.L.; Souza, D.O.; Rieder, C.R.M.; Jardim, L.B. S100B and NSE serum concentrations in Machado Joseph disease. Clin. Chim Acta 2005, 351, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lei, L.; Shi, Y.; Wang, J.; Jiang, H.; Shen, L.; Tang, B. Serum concentrations of NSE and S100B in spinocerebellar ataxia type 3/Machado-Joseph disease. J. Cent. South Univ. Med. Sci. 2011, 36, 504–510. [Google Scholar] [CrossRef]
- Mendonça, L.S.; Nóbrega, C.; Hirai, H.; Kaspar, B.K.; Pereira de Almeida, L. Transplantation of cerebellar neural stem cells improves motor coordination and neuropathology in Machado-Joseph disease mice. Brain J. Neurol. 2015, 138, 320–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saute, J.A.M.; da Silva, A.C.F.; Muller, A.P.; Hansel, G.; de Mello, A.S.; Maeda, F.; Vedolin, L.; Saraiva-Pereira, M.L.; Souza, D.O.; Arpa, J.; et al. Serum insulin-like system alterations in patients with spinocerebellar ataxia type 3. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 731–735. [Google Scholar] [CrossRef] [Green Version]
- Raj, K.; Sarkar, S. Tissue-Specific Upregulation of Drosophila Insulin Receptor (InR) Mitigates Poly(Q)-Mediated Neurotoxicity by Restoration of Cellular Transcription Machinery. Mol. Neurobiol. 2019, 56, 1310–1329. [Google Scholar] [CrossRef]
- Duarte-Neves, J.; Gonçalves, N.; Cunha-Santos, J.; Simões, A.T.; den Dunnen, W.F.A.; Hirai, H.; Kügler, S.; Cavadas, C.; Pereira de Almeida, L. Neuropeptide Y mitigates neuropathology and motor deficits in mouse models of Machado-Joseph disease. Hum. Mol. Genet. 2015, 24, 5451–5463. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-H.; Shi, C.-H.; Zhou, L.-N.; Li, Y.-S.; Yang, J.; Liu, Y.-T.; Mao, C.-Y.; Luo, H.-Y.; Xu, G.-W.; Xu, Y.-M. Metabolic Profiling Reveals Biochemical Pathways and Potential Biomarkers of Spinocerebellar Ataxia 3. Front. Mol. Neurosci. 2019, 12, 159. [Google Scholar] [CrossRef]
- Rajamani, K.; Liu, J.-W.; Wu, C.-H.; Chiang, I.T.; You, D.-H.; Lin, S.-Y.; Hsieh, D.-K.; Lin, S.-Z.; Harn, H.-J.; Chiou, T.-W. n-Butylidenephthalide exhibits protection against neurotoxicity through regulation of tryptophan 2, 3 dioxygenase in spinocerebellar ataxia type 3. Neuropharmacology 2017, 117, 434–446. [Google Scholar] [CrossRef]
- Nóbrega, C.; Mendonça, L.; Marcelo, A.; Lamazière, A.; Tomé, S.; Despres, G.; Matos, C.A.; Mechmet, F.; Langui, D.; den Dunnen, W.; et al. Restoring brain cholesterol turnover improves autophagy and has therapeutic potential in mouse models of spinocerebellar ataxia. Acta Neuropathol. 2019, 138, 837–858. [Google Scholar] [CrossRef]
- Chen, X.; Tang, T.-S.; Tu, H.; Nelson, O.; Pook, M.; Hammer, R.; Nukina, N.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 12713–12724. [Google Scholar] [CrossRef] [Green Version]
- Shakkottai, V.G.; do Carmo Costa, M.; Dell’Orco, J.M.; Sankaranarayanan, A.; Wulff, H.; Paulson, H.L. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 13002–13014. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Schmidt, T.; Golla, M.; Lehmann, L.; Weber, J.J.; Hübener-Schmid, J.; Riess, O. In vivo assessment of riluzole as a potential therapeutic drug for spinocerebellar ataxia type 3. J. Neurochem. 2016, 138, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.-Y.; Yang, C.-C.; Soong, B.-W.; Yu, C.-Y.; Chen, S.-H.; Huang, H.-P.; Kuo, H.-C. Modeling spinocerebellar ataxias 2 and 3 with iPSCs reveals a role for glutamate in disease pathology. Sci. Rep. 2019, 9, 1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buijsen, R.A.M.; Toonen, L.J.A.; Gardiner, S.L.; van Roon-Mom, W.M.C. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurother. J. Am. Soc. Exp. Neurother. 2019, 16, 263–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, L.R.; Keller, L.; Bushart, D.D.; Delatorre, R.G.; Li, D.; McLoughlin, H.S.; do Carmo Costa, M.; Shakkottai, V.G.; Smith, G.D.; Paulson, H.L. Antisense oligonucleotide therapy rescues aggresome formation in a novel spinocerebellar ataxia type 3 human embryonic stem cell line. Stem Cell Res. 2019, 39, 101504. [Google Scholar] [CrossRef]
- Alves, S.; Nascimento-Ferreira, I.; Dufour, N.; Hassig, R.; Auregan, G.; Nóbrega, C.; Brouillet, E.; Hantraye, P.; Pedroso de Lima, M.C.; Déglon, N.; et al. Silencing ataxin-3 mitigates degeneration in a rat model of Machado–Joseph disease: No role for wild-type ataxin-3? Hum. Mol. Genet. 2010, 19, 2380–2394. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.D.C.; Luna-Cancalon, K.; Fischer, S.; Ashraf, N.S.; Ouyang, M.; Dharia, R.M.; Martin-Fishman, L.; Yang, Y.; Shakkottai, V.G.; Davidson, B.L.; et al. Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1898–1908. [Google Scholar] [CrossRef] [Green Version]
- Nóbrega, C.; Codêsso, J.M.; Mendonça, L.; Pereira de Almeida, L. RNA Interference Therapy for Machado-Joseph Disease: Long-Term Safety Profile of Lentiviral Vectors Encoding Short Hairpin RNAs Targeting Mutant Ataxin-3. Hum. Gene Ther. 2019, 30, 841–854. [Google Scholar] [CrossRef]
- Conceição, M.; Mendonça, L.; Nóbrega, C.; Gomes, C.; Costa, P.; Hirai, H.; Moreira, J.N.; Lima, M.C.; Manjunath, N.; Pereira de Almeida, L. Intravenous administration of brain-targeted stable nucleic acid lipid particles alleviates Machado-Joseph disease neurological phenotype. Biomaterials 2016, 82, 124–137. [Google Scholar] [CrossRef]
- Bonneau, E.; Neveu, B.; Kostantin, E.; Tsongalis, G.J.; De Guire, V. How close are miRNAs from clinical practice? A perspective on the diagnostic and therapeutic market. EJIFCC 2019, 30, 114–127. [Google Scholar]
- Evers, M.M.; Toonen, L.J.A.; van Roon-Mom, W.M.C. Ataxin-3 protein and RNA toxicity in spinocerebellar ataxia type 3: Current insights and emerging therapeutic strategies. Mol. Neurobiol. 2014, 49, 1513–1531. [Google Scholar] [CrossRef] [Green Version]
- Goti, D.; Katzen, S.M.; Mez, J.; Kurtis, N.; Kiluk, J.; Ben-Haïem, L.; Jenkins, N.A.; Copeland, N.G.; Kakizuka, A.; Sharp, A.H.; et al. A Mutant Ataxin-3 Putative-Cleavage Fragment in Brains of Machado-Joseph Disease Patients and Transgenic Mice Is Cytotoxic above a Critical Concentration. J. Neurosci. 2004, 24, 10266. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.J.; Haas, E.; Maringer, Y.; Hauser, S.; Casadei, N.L.P.; Chishti, A.H.; Riess, O.; Hübener-Schmid, J. Calpain-1 ablation partially rescues disease-associated hallmarks in models of Machado-Joseph disease. Hum. Mol. Genet. 2020, 29, 892–906. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, T.; Lacomblez, L.; Abitbol, J.L.; Ludolph, A.; Mora, J.S.; Robberecht, W.; Shaw, P.J.; Pruss, R.M.; Cuvier, V.; Meininger, V.; et al. A phase II-III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur. J. Neurol. 2014, 21, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Clemens, L.E.; Weber, J.J.; Wlodkowski, T.T.; Yu-Taeger, L.; Michaud, M.; Calaminus, C.; Eckert, S.H.; Gaca, J.; Weiss, A.; Magg, J.C.D.; et al. Olesoxime suppresses calpain activation and mutant huntingtin fragmentation in the BACHD rat. Brain J. Neurol. 2015, 138, 3632–3653. [Google Scholar] [CrossRef] [Green Version]
- Bichelmeier, U.; Schmidt, T.; Hübener, J.; Boy, J.; Rüttiger, L.; Häbig, K.; Poths, S.; Bonin, M.; Knipper, M.; Schmidt, W.J.; et al. Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: In vivo evidence. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 7418–7428. [Google Scholar] [CrossRef] [Green Version]
- Sowa, A.S.; Martin, E.; Martins, I.M.; Schmidt, J.; Depping, R.; Weber, J.J.; Rother, F.; Hartmann, E.; Bader, M.; Riess, O.; et al. Karyopherin α-3 is a key protein in the pathogenesis of spinocerebellar ataxia type 3 controlling the nuclear localization of ataxin-3. Proc. Natl. Acad. Sci. USA 2018, 115, E2624–E2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, L.; Xu, K.; Chen, Z.; Tang, B.; Jiang, H. Roles of Post-translational Modifications in Spinocerebellar Ataxias. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Matos, C.A.; Nóbrega, C.; Louros, S.R.; Almeida, B.; Ferreiro, E.; Valero, J.; Pereira de Almeida, L.; Macedo-Ribeiro, S.; Carvalho, A.L. Ataxin-3 phosphorylation decreases neuronal defects in spinocerebellar ataxia type 3 models. J. Cell Biol. 2016, 212, 465–480. [Google Scholar] [CrossRef]
- Mueller, T.; Breuer, P.; Schmitt, I.; Walter, J.; Evert, B.O.; Wüllner, U. CK2-dependent phosphorylation determines cellular localization and stability of ataxin-3. Hum. Mol. Genet. 2009, 18, 3334–3343. [Google Scholar] [CrossRef] [Green Version]
- Pastori, V.; Sangalli, E.; Coccetti, P.; Pozzi, C.; Nonnis, S.; Tedeschi, G.; Fusi, P. CK2 and GSK3 phosphorylation on S29 controls wild-type ATXN3 nuclear uptake. Biochim. Biophys. Acta 2010, 1802, 583–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.-F.; Liao, S.-S.; Luo, Y.-Y.; Tang, J.-G.; Wang, J.-L.; Lei, L.-F.; Chi, J.-W.; Du, J.; Jiang, H.; Xia, K.; et al. SUMO-1 modification on K166 of polyQ-expanded ataxin-3 strengthens its stability and increases its cytotoxicity. PLoS ONE 2013, 8, e54214. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.P.; Lee, D.H. Autophagy mediates SUMO-induced degradation of a polyglutamine protein ataxin-3. Anim. Cells Syst. 2017, 21, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Almeida, B.; Abreu, I.A.; Matos, C.A.; Fraga, J.S.; Fernandes, S.; Macedo, M.G.; Gutiérrez-Gallego, R.; Pereira, P.J.B.; Carvalho, A.L.; Macedo-Ribeiro, S. SUMOylation of the brain-predominant Ataxin-3 isoform modulates its interaction with p97. Biochim. Biophys. Acta 2015, 1852, 1950–1959. [Google Scholar] [CrossRef] [Green Version]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Bové, J.; Martínez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Herzog, L.K.; Kevei, É.; Marchante, R.; Böttcher, C.; Bindesbøll, C.; Lystad, A.H.; Pfeiffer, A.; Gierisch, M.E.; Salomons, F.A.; Simonsen, A.; et al. The Machado-Joseph disease deubiquitylase ataxin-3 interacts with LC3C/GABARAP and promotes autophagy. Aging Cell 2020, 19, e13051. [Google Scholar] [CrossRef]
- Yiu, E.M.; Tai, G.; Peverill, R.E.; Lee, K.J.; Croft, K.D.; Mori, T.A.; Scheiber-Mojdehkar, B.; Sturm, B.; Praschberger, M.; Vogel, A.P.; et al. An open-label trial in Friedreich ataxia suggests clinical benefit with high-dose resveratrol, without effect on frataxin levels. J. Neurol. 2015, 262, 1344–1353. [Google Scholar] [CrossRef]
- Chai, Y.; Koppenhafer, S.L.; Shoesmith, S.J.; Perez, M.K.; Paulson, H.L. Evidence for Proteasome Involvement in Polyglutamine Disease: Localization to Nuclear Inclusions in SCA3/MJD and Suppression of Polyglutamine Aggregation in vitro. Hum. Mol. Genet. 1999, 8, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.C.; Chang, C.-N.; Chen, W.-L.; Lin, T.-H.; Chao, C.-Y.; Lin, C.-H.; Lin, H.-Y.; Cheng, M.-L.; Chiang, M.-C.; Lin, J.-Y.; et al. Targeting Ubiquitin Proteasome Pathway with Traditional Chinese Medicine for Treatment of Spinocerebellar Ataxia Type 3. Am. J. Chin. Med. 2019, 47, 63–95. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.-T.; Fen, F.; Ding, M.-P. Effects of puerarin with aspirin on the markers of damaged vascular endothelial cells in patients with acute cerebral infarction. J. Chin. Mater. Med. 2008, 33, 2827–2829. [Google Scholar]
- Chen, L.; Bi, X.-Y.; Zhu, L.-X.; Qiu, Y.-Q.; Ding, S.-J.; Deng, B.-Q. Flavonoids of puerarin versus tanshinone II A for ischemic stroke: A randomized controlled trial. Chin. J. Integr. Med. 2011, 9, 1215–1220. [Google Scholar] [CrossRef]
- Hyrskyluoto, A.; Bruelle, C.; Lundh, S.H.; Do, H.T.; Kivinen, J.; Rappou, E.; Reijonen, S.; Waltimo, T.; Petersén, Å.; Lindholm, D.; et al. Ubiquitin-specific protease-14 reduces cellular aggregates and protects against mutant huntingtin-induced cell degeneration: Involvement of the proteasome and ER stress-activated kinase IRE1α. Hum. Mol. Genet. 2014, 23, 5928–5939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, D.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Qi, H.; Jiang, D.; Wang, R.; Chen, A.; Yan, Z.; Xiao, J. The new use of an ancient remedy: A double-blinded randomized study on the treatment of rheumatoid arthritis. Am. J. Chin. Med. 2013, 41, 263–280. [Google Scholar] [CrossRef]
- Sadakane, C.; Watanabe, J.; Fukutake, M.; Nisimura, H.; Maemura, K.; Kase, Y.; Kono, T. Pharmacokinetic Profiles of Active Components After Oral Administration of a Kampo Medicine, Shakuyakukanzoto, to Healthy Adult Japanese Volunteers. J. Pharm. Sci. 2015, 104, 3952–3959. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Carvalho, G.; Saute, J.A.M.; Haas, C.B.; Torrez, V.R.; Brochier, A.W.; Souza, G.N.; Furtado, G.V.; Gheno, T.; Russo, A.; Monte, T.L.; et al. Cytokines in Machado Joseph Disease/Spinocerebellar Ataxia 3. Cerebellum 2016, 15, 518–525. [Google Scholar] [CrossRef]
- Babiloni, C.; Frisoni, G.B.; Del Percio, C.; Zanetti, O.; Bonomini, C.; Cassetta, E.; Pasqualetti, P.; Miniussi, C.; De Rosas, M.; Valenzano, A.; et al. Ibuprofen treatment modifies cortical sources of EEG rhythms in mild Alzheimer’s disease. Clin. Neurophysiol. 2009, 120, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Scali, C.; Prosperi, C.; Bracco, L.; Piccini, C.; Baronti, R.; Ginestroni, A.; Sorbi, S.; Pepeu, G.; Casamenti, F. Neutrophils CD11b and fibroblasts PGE(2) are elevated in Alzheimer’s disease. Neurobiol. Aging 2002, 23, 523–530. [Google Scholar] [CrossRef]
- Rosenbloom, A.L. Recombinant human insulin-like growth factor I (rhIGF-I) and rhIGF-I/rhIGF-binding-protein-3: New growth treatment options? J. Pediatr. 2007, 150, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Purohit, S.; Sharma, S.; Bai, S.; Zhi, W.; Ponny, S.R.; Hopkins, D.; Steed, L.; Bode, B.; Anderson, S.W.; et al. IGF-Binding Proteins in Type-1 Diabetes Are More Severely Altered in the Presence of Complications. Front. Endocrinol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, A.J.; Codogno, P. Autophagy: A Sweet Process in Diabetes. Cell Metab. 2008, 8, 275–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, K.; Busby, W.H., Jr.; Clarke, J.B.; Clemmons, D.R. Tissue transglutaminase facilitates the polymerization of insulin-like growth factor-binding protein-1 (IGFBP-1) and leads to loss of IGFBP-1′s ability to inhibit insulin-like growth factor-I-stimulated protein synthesis. J. Biol. Chem. 2001, 276, 8740–8745. [Google Scholar] [CrossRef] [Green Version]
- Floyd, S.; Favre, C.; Lasorsa, F.M.; Leahy, M.; Trigiante, G.; Stroebel, P.; Marx, A.; Loughran, G.; O’Callaghan, K.; Marobbio, C.M.T.; et al. The insulin-like growth factor-I-mTOR signaling pathway induces the mitochondrial pyrimidine nucleotide carrier to promote cell growth. Mol. Biol. Cell 2007, 18, 3545–3555. [Google Scholar] [CrossRef] [Green Version]
- Bitto, A.; Lerner, C.; Torres, C.; Roell, M.; Malaguti, M.; Perez, V.; Lorenzini, A.; Hrelia, S.; Ikeno, Y.; Matzko, M.E.; et al. Long-term IGF-I exposure decreases autophagy and cell viability. PLoS ONE 2010, 5, e12592. [Google Scholar] [CrossRef] [Green Version]
- Soong, B.W.; Liu, R.S. Regional decrease in brain glucose metabolism in asymptomatic gene carriers of Machado-Joseph disease: A preliminary report. Chin. Med. J. 1998, 61, 121–126. [Google Scholar]
- Soong, B.W.; Liu, R.S. Positron emission tomography in asymptomatic gene carriers of Machado-Joseph disease. J. Neurol. Neurosurg. Psychiatry 1998, 64, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.L.; Chen, H.L.; Duan, J.A.; Liu, J.W. Research progress of structures and pharmacological activities of phthalides from Angelica sinensis. China J. Chin. Mater. Med. 2016, 41, 167–176. [Google Scholar] [CrossRef]
- Hsueh, K.W.; Chiou, T.W.; Chiang, S.F.; Yamashita, T.; Abe, K.; Borlongan, C.V.; Sanberg, P.R.; Huang, A.Y.; Lin, S.Z.; Harn, H.J. Autophagic down-regulation in motor neurons remarkably prolongs the survival of ALS mice. Neuropharmacology 2016, 108, 152–160. [Google Scholar] [CrossRef]
- Kanai, M.; Funakoshi, H.; Takahashi, H.; Hayakawa, T.; Mizuno, S.; Matsumoto, K.; Nakamura, T. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol. Brain 2009, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Kacher, R.; Lamazière, A.; Heck, N.; Kappes, V.; Mounier, C.; Despres, G.; Dembitskaya, Y.; Perrin, E.; Christaller, W.; Sasidharan Nair, S.; et al. CYP46A1 gene therapy deciphers the role of brain cholesterol metabolism in Huntington’s disease. Brain J. Neurol. 2019, 142, 2432–2450. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.M.; Lam, M.; Mast, N.; Moon, J.; Li, Y.; Maxfield, E.; Pikuleva, I.A. CYP46A1 Activation by Efavirenz Leads to Behavioral Improvement without Significant Changes in Amyloid Plaque Load in the Brain of 5XFAD Mice. Neurother. J. Am. Soc. Exp. Neurother. 2019, 16, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Mitroi, D.N.; Pereyra-Gómez, G.; Soto-Huelin, B.; Senovilla, F.; Kobayashi, T.; Esteban, J.A.; Ledesma, M.D. NPC1 enables cholesterol mobilization during long-term potentiation that can be restored in Niemann–Pick disease type C by CYP46A1 activation. EMBO Rep. 2019, 20, e48143. [Google Scholar] [CrossRef]
- Araujo, J.; Breuer, P.; Dieringer, S.; Krauss, S.; Dorn, S.; Zimmermann, K.; Pfeifer, A.; Klockgether, T.; Wuellner, U.; Evert, B.O. FOXO4-dependent upregulation of superoxide dismutase-2 in response to oxidative stress is impaired in spinocerebellar ataxia type 3. Hum. Mol. Genet. 2011, 20, 2928–2941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doss-Pepe, E.W.; Stenroos, E.S.; Johnson, W.G.; Madura, K. Ataxin-3 interactions with rad23 and valosin-containing protein and its associations with ubiquitin chains and the proteasome are consistent with a role in ubiquitin-mediated proteolysis. Mol. Cell Biol. 2003, 23, 6469–6483. [Google Scholar] [CrossRef] [Green Version]
- Shimohata, T.; Nakajima, T.; Yamada, M.; Uchida, C.; Onodera, O.; Naruse, S.; Kimura, T.; Koide, R.; Nozaki, K.; Sano, Y.; et al. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat. Genet. 2000, 26, 29–36. [Google Scholar] [CrossRef]
- Evert, B.O.; Araujo, J.; Vieira-Saecker, A.M.; de Vos, R.A.I.; Harendza, S.; Klockgether, T.; Wüllner, U. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 11474–11486. [Google Scholar] [CrossRef]
- Konno, A.; Shuvaev, A.N.; Miyake, N.; Miyake, K.; Iizuka, A.; Matsuura, S.; Huda, F.; Nakamura, K.; Yanagi, S.; Shimada, T.; et al. Mutant ataxin-3 with an abnormally expanded polyglutamine chain disrupts dendritic development and metabotropic glutamate receptor signaling in mouse cerebellar Purkinje cells. Cerebellum 2014, 13, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.R.; Heck, N.; Lohof, A.M.; Rochefort, C.; Morel, M.-P.; Wehrlé, R.; Doulazmi, M.; Marty, S.; Cannaya, V.; Avci, H.X.; et al. Mature Purkinje cells require the retinoic acid-related orphan receptor-α (RORα) to maintain climbing fiber mono-innervation and other adult characteristics. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 9546–9562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanave, M.; Hoshino, C.; Konno, A.; Fukuzaki, Y.; Matsuzaki, Y.; Ishitani, T.; Hirai, H. Pharmacological enhancement of retinoid-related orphan receptor α function mitigates spinocerebellar ataxia type 3 pathology. Neurobiol. Dis. 2019, 121, 263–273. [Google Scholar] [CrossRef]
- Jeub, M.; Herbst, M.; Spauschus, A.; Fleischer, H.; Klockgether, T.; Wuellner, U.; Evert, B.O. Potassium channel dysfunction and depolarized resting membrane potential in a cell model of SCA3. Exp. Neurol. 2006, 201, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Carrasquero, L.M.G.; Delicado, E.G.; Bustillo, D.; Gutiérrez-Martín, Y.; Artalejo, A.R.; Miras-Portugal, M.T. P2 × 7 and P2Y13 purinergic receptors mediate intracellular calcium responses to BzATP in rat cerebellar astrocytes. J. Neurochem. 2009, 110, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-F.; Chern, Y. Adenosine Receptors and Huntington’s Disease. Int Rev. Neurobio. 2014, 119, 195–232. [Google Scholar] [CrossRef]
- Chou, A.-H.; Chen, Y.-L.; Chiu, C.-C.; Yuan, S.-J.; Weng, Y.-H.; Yeh, T.-H.; Lin, Y.-L.; Fang, J.-M.; Wang, H.-L. T1-11 and JMF1907 ameliorate polyglutamine-expanded ataxin-3-induced neurodegeneration, transcriptional dysregulation and ataxic symptom in the SCA3 transgenic mouse. Neuropharmacology 2015, 99, 308–317. [Google Scholar] [CrossRef]
- Gonçalves, N.; Simões, A.T.; Cunha, R.A.; de Almeida, L.P. Caffeine and adenosine A(2A) receptor inactivation decrease striatal neuropathology in a lentiviral-based model of Machado-Joseph disease. Ann. Neurol. 2013, 73, 655–666. [Google Scholar] [CrossRef]
- Yohrling Iv, G.J.; Jiang, G.C.T.; DeJohn, M.M.; Robertson, D.J.; Vrana, K.E.; Cha, J.-H.J. Inhibition of tryptophan hydroxylase activity and decreased 5-HT1A receptor binding in a mouse model of Huntington’s disease. J. Neurochem. 2002, 82, 1416–1423. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.; Liu, J.-W.; Harn, H.-J.; Hsueh, K.-W.; Rajamani, K.; Deng, Y.-C.; Chia, C.-M.; Shyu, W.-C.; Lin, S.-Z.; Chiou, T.-W. Human Olfactory Ensheathing Cell Transplantation Improves Motor Function in a Mouse Model of Type 3 Spinocerebellar Ataxia. Cell Transplant. 2017, 26, 1611–1621. [Google Scholar] [CrossRef]
- Koch, P.; Breuer, P.; Peitz, M.; Jungverdorben, J.; Kesavan, J.; Poppe, D.; Doerr, J.; Ladewig, J.; Mertens, J.; Tüting, T.; et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 2011, 480, 543–546. [Google Scholar] [CrossRef]
- Liu, J.; Tang, T.-S.; Tu, H.; Nelson, O.; Herndon, E.; Huynh, D.P.; Pulst, S.M.; Bezprozvanny, I. Deranged Calcium Signaling and Neurodegeneration in Spinocerebellar Ataxia Type 2. J. Neurosci. 2009, 29, 9148. [Google Scholar] [CrossRef]
- Shimobayashi, E.; Josef, P.K. Calcium Signaling, PKC Gamma, IP3R1 and CAR8 Link Spinocerebellar Ataxias and Purkinje Cell Dendritic Development. Curr. Neuropharmacol. 2018, 16, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Schorge, S.; van de Leemput, J.; Singleton, A.; Houlden, H.; Hardy, J. Human ataxias: A genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci. 2010, 33, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, E.A.; Rafacho, A. Implications of Palmitoleic Acid (Palmitoleate) On Glucose Homeostasis, Insulin Resistance and Diabetes. Curr. Drug Targets 2017, 18, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Tricò, D.; Mengozzi, A.; Nesti, L.; Hatunic, M.; Gabriel Sanchez, R.; Konrad, T.; Lalić, K.; Lalić, N.M.; Mari, A.; Natali, A.; et al. Circulating palmitoleic acid is an independent determinant of insulin sensitivity, beta cell function and glucose tolerance in non-diabetic individuals: A longitudinal analysis. Diabetologia 2020, 63, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, M.F.; Mutch, D.M.; Leri, F. The Relationship between Fatty Acids and Different Depression-Related Brain Regions, and Their Potential Role as Biomarkers of Response to Antidepressants. Nutrients 2017, 9, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Indication | Drug Name | Mechanisms | Status | Outcome | NCT no. | Year |
|---|---|---|---|---|---|---|
| Cerebellar ataxia | Buspirone | 5-HT1A serotonin agonist | Case-study/completed | Improved gait ataxia | - | 1994 [6] |
| SCA3 | Tandospirone | 5-HT1A serotonin agonist | Case-study/completed | Leg pain, insomnia, anorexia, and depression remarkably alleviated | - | 1994 [7] |
| SCA3 | Tandospirone | 5-HT1A serotonin agonist | Double-blind study/completed | 1. ARS reduction in 7/10 patients. 2. SDS reduction in 3/6 patients. 3. Insomnia and leg pain alleviation in 5/7 patients. | - | 2001 [8] |
| SCA3 and SCA7 | Insulin-like growth factor-1 (IGF-1) | Neuromodulatory functions | Open label/completed | SARA improved after 8 months and worsened after 20 months. | - | 2007 [9] |
| SCA3 | Varenicline | agonist at α4β2 neuronal nicotinic acetylcholine receptors | Phase 2/completed | 1. Side effect of nausea. 2. Improved axial symptoms and rapid alternating movements. | NCT00992771 | 9 Oct 2009 [10] |
| SCA3 | Sodium phenylbutyrate | HDAC inhibitors | Withdrawn # | - | NCT01096095 | 30 Mar 2010 |
| SCA3 | VPA | HDAC inhibitors | Phase 1/completed | SARA score (−2.05) greater in the VPA group than in the placebo (−0.75) groups | ChiCTR-TRC10000754 | 6 Jan 2010 [11] |
| SCA3 | Lithium carbonate | Interfere with ion transport processes | Phase 2/Phase 3/completed | No effect on progression (NESSCA) | NCT01096082 | 30 Mar 2010 [12] |
| Cerebellar Ataxia | Riluzole | Glutamate release inhibitor | Phase 2/Phase 3/completed | 1. 50% patient with decrease SARA score. 2. No severe adverse events were recorded | NCT01104649 | 15 Apr 2010 [13] |
| SCA3 | NGF | Neuroprotection | Open label/Completed | Total SARA score decreased significantly | - | Nov 2011 |
| Cerebellar ataxia | Allogeneic adult Ad-MSC | Neuroprotection | Phase 1/Phase 2/completed | 1. No adverse events 2. Increased brain glucose metabolism | NCT01649687 | 25 Jul 2012 [14] |
| SCA3 | Cabaletta (trehalose) | Chemical chaperone | Phase 2, completed | Stable on the SARA scale. | NCT02147886 | 28 May 2014 |
| SCA1, 2, 3, and 6 | Dalfampridine | Potassium channel blocker | Completed | No difference in change of T25FW and SARA score | NCT01811706 | 12 Jan 2015 |
| Cerebellar Ataxia | Stemchymal® | Neuroprotection | Unknown, Phase 2 | NCT02540655 | 4 Sep 2015 | |
| SCA1, 2, 3, and 6 | hUC-MSC | - | Phase 2, unknown | - | NCT03378414 | 19 Dec 2017 |
| SCA1, 2, 3, 6, 7, 8, and 10 | Troriluzole | Glutamate release inhibitor | Phase 3 | - | NCT03701399 | 10 Oct 2018 |
| SCA1, 2, 3, 6, and MSA-C | BHV-4157 (pro-drug of riluzole) | Glutamate release inhibitor | Phase 3, active, not recruiting | - | NCT03408080 | 23 Jan 2018 |
| Ataxia, Cerebellar | Nilotinib (Bcr-Abl TKI) | Autophagy enhancer | Phase 2, active, not recruiting | - | NCT03932669 | 1 May 2019 |
| Spinocerebellar Degeneration | C-Trelin OD Tab (analogue of TRH) | Inhibition of activation of glutamate | Recruiting, Phase 4 | - | NCT04107740 | 27 Sep 2019 |
| Mechanism | Biomarkers | Function | Expressions Level in Subject | Treatment | Therapy Results |
|---|---|---|---|---|---|
| RNAi-mediated knockdown of ataxin-3 | |||||
| Target to ataxin-3 | MiR-25 | Bind to ATXN3 3′-UTR | Underexpressed in SCA3 patients [21] | MiR-25 mimics | Suppressed 3′UTR of ATXN3 mRNA [22] |
| Target to ataxin-3 | Mir-9 | Bind to ATXN3 3′-UTR | Underexpressed in SCA3 patients (CSF-derived exosome and neurons) [23,24]. | miRNA overexpression | Suppressed 3′UTR of ATXN3 mRNA [23] |
| Target to ataxin-3 | Mir-181a | Bind to ATXN3 3′-UTR | Underexpressed in SCA3 patients (CSF-derived exosome and neurons) [23,24]. | miRNA overexpression | Suppressed 3′UTR of ATXN3 [23] |
| Target to ataxin-3 | Mir-494 | Bind to ATXN3 3′-UTR | Underexpressed in SCA3 patients (neurons) [23]. | miRNA overexpression | Suppressed 3′UTR of ATXN3 [23] |
| Reduced cleavage protein formation | |||||
| Calpain inhibitor | Calpastatin | Calpain inhibitor | Underexpressed in SCA3 patients [25]. | ALLN (MG-101) or calpeptin | Reduced full-length and small fragment ataxin-3 via Calpeptin [26]. Reduced small fragment ataxin-3 via ALLN [27]. |
| Decreasing ataxin-3 aggregation | |||||
| Autophagy | Beclin-1 | Autophagy initiator | Underexpressed in symptomatic SCA3 patients [28,29] | Beclin-1 overexpression | mTOR-dependent pathways activation [28,29] |
| Autophagy | Ratio of LC3II/LC3I | Autophagosome | Underexpressed in SCA3 patient’s fibroblasts [28] | Rapamycin or cordycepin | mTOR-dependent pathways activation [28,29,30,31] |
| Autophagy | P62 | Deliver ubiquitinated proteins | Higher in SCA3 patient’s fibroblasts [28] | Rapamycin or cordycepin | mTOR-dependent pathways activation [28,29,30,31] |
| Autophagy | Sirtuin-1 | NAD +-dependent deacetylase | Underexpressed in SCA3 patient’s fibroblasts [32] | Caloric restriction or resveratrol | Rescuing SIRT1 levels, motor incoordination, imbalance [32] |
| Chaperon | DNAJB1 | Protein refolding machine | Significantly Underexpressed in SCA3 with early-onset patients [33]. Underexpressed in SCA3 patient-derived iPSC lines [34] | DNAJB1 overexpression [33] | Largely reduced ATX3Q82 aggregation in HEK cell [33] |
| Chaperon | HSPA1A | Protein refolding machine | Underexpressed in SCA3 patient’s fibroblast [33] | Paeoniflorin (PF), PF derivative NC001-8, or Fluorodeoxyuridine | Enhancing the expression of HSF-1 and HSP70 chaperones [35] |
| Chaperon | HSPA8 | Protein refolding machine | Underexpressed in SCA3 patient’s fibroblast [33] | Paeoniflorin (PF), PF derivative NC001-8, or Fluorodeoxyuridine | Enhancing the expression of HSF-1 and HSP70 chaperones [35] |
| Reducing inflammation and oxidative stress | |||||
| Inflammatory factors | TNFSF14 | Neurodegenerative | Higher in SCA3 patients with duration ≤9 years [36] | Ibuprofen [37] | Reduced Il1b, TNFa mRNA and IKB-α protein phosphorylation levels [37] |
| Oxidative Stress | SOD | Antioxidant enzyme activities | Underexpressed in symptomatic SCA3 [38] | RSP [39], or CA8 overexpression [40] | Induction of GST-4iva RSP [39]. Rescued abnormal Ca2+ release via CA8overexpression [40]. |
| Oxidative Stress | GSH-Px | Antioxidant enzyme activities | Underexpressed in symptomatic SCA3 [38] | RSP [39], or CA8 overexpression [40] | Induction of GST-4iva RSP [39]. Rescued abnormal Ca2+ release via CA8overexpression [40]. |
| Neural degeneration | NSE | Peripheral marker of neuronal disruption | Higher in SCA3 [41,42] | Neural stem cells injection [43] | Decreased pro-inflammatory mediators IL1B and TNFA [43] |
| Rescue of cellular dysfunction. | |||||
| Growth factors | Insulin | Growth factors | Underexpressed in SCA3 [44] | IGF-1 | Significantly decreased in SARA scores [9] |
| Growth factors | IGF-1/IGFBP-3 | Free IGF-1 | Higher in SCA3 [44] | Insulin receptor Upregulation [45] | Increased autophagy-mediated to rescue phenotype [45] |
| Neurotrophic | Neuropeptide Y | Neuroprotective molecule | Underexpressed in SCA3 [46] | NPY overexpression [46] | Increased BDNF levels [46] |
| Metabolism | Tryptophan | Amino acid metabolism | Underexpressed in SCA3 [47] | n-BP [48] | Decreased TDO2 expression [48] |
| Enzyme | CYP46A1 | brain cholesterol turnover Activation | Underexpressed in SCA3 [49] | CYP46A1 overexpression [49] | Decreased aggregation ataxin-3 protein and increased Purkinje cell number [49] |
| Ion-channel homostatasis | P2RY13 | Increase of intracellular calcium | Higher in SCA3 patients [36] | Dantrolene [50], SKA-31 [51], or riluzole [52] | Inhibited calcium release from ER via dantrolene [50]. Activated Kv3.1 channels via SKA-31 [51]. Prevented calcium influx increase in the cells via riluzole [53]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-S.; Hong, Z.-X.; Lin, S.-Z.; Harn, H.-J. Identifying Therapeutic Targets for Spinocerebellar Ataxia Type 3/Machado–Joseph Disease through Integration of Pathological Biomarkers and Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 3063. https://doi.org/10.3390/ijms21093063
Chen Y-S, Hong Z-X, Lin S-Z, Harn H-J. Identifying Therapeutic Targets for Spinocerebellar Ataxia Type 3/Machado–Joseph Disease through Integration of Pathological Biomarkers and Therapeutic Strategies. International Journal of Molecular Sciences. 2020; 21(9):3063. https://doi.org/10.3390/ijms21093063
Chicago/Turabian StyleChen, Yu-Shuan, Zhen-Xiang Hong, Shinn-Zong Lin, and Horng-Jyh Harn. 2020. "Identifying Therapeutic Targets for Spinocerebellar Ataxia Type 3/Machado–Joseph Disease through Integration of Pathological Biomarkers and Therapeutic Strategies" International Journal of Molecular Sciences 21, no. 9: 3063. https://doi.org/10.3390/ijms21093063