Effects of Sunitinib and Other Kinase Inhibitors on Cells Harboring a PDGFRB Mutation Associated with Infantile Myofibromatosis

 , ,
, ,
Abstract
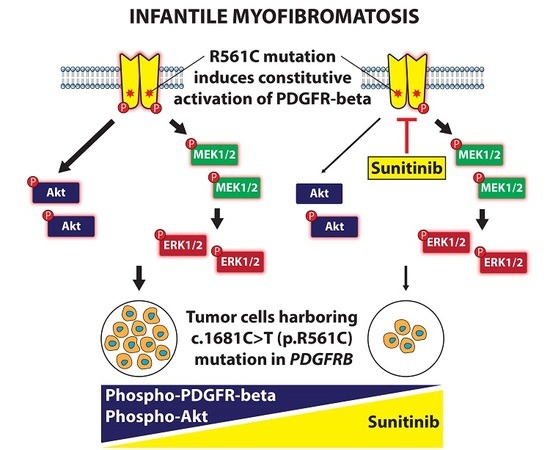
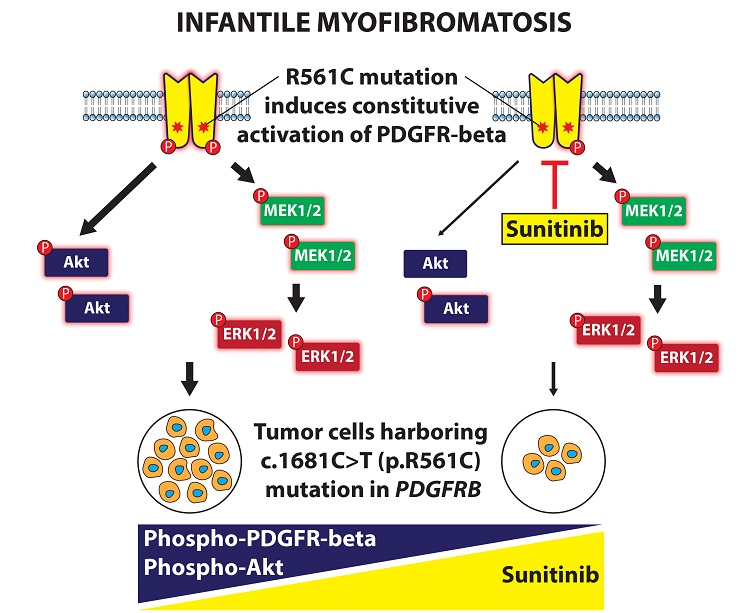
:
1. Introduction
2. Results
2.1. Germline Mutations in PDGFRB Were Identified in Both Children, and the Same Mutation in PDGFRB Was Confirmed in NSTS-47 Cells
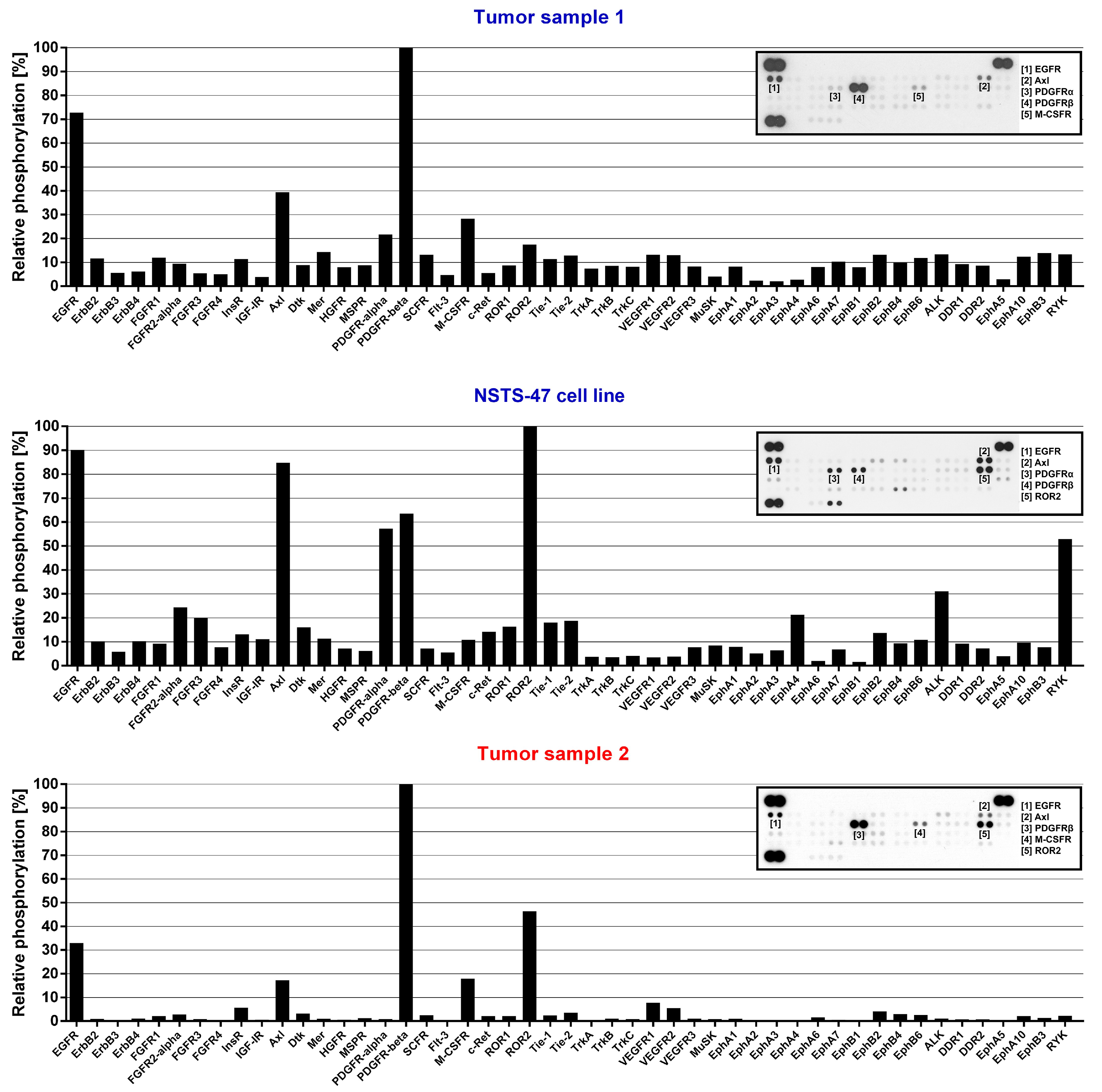
2.2. PDGFR-Beta, EGFR and ERK1/2 Kinases Are Highly Phosphorylated in Cells Harboring c.1681C>T (p.R561C) Mutation in PDGFRB
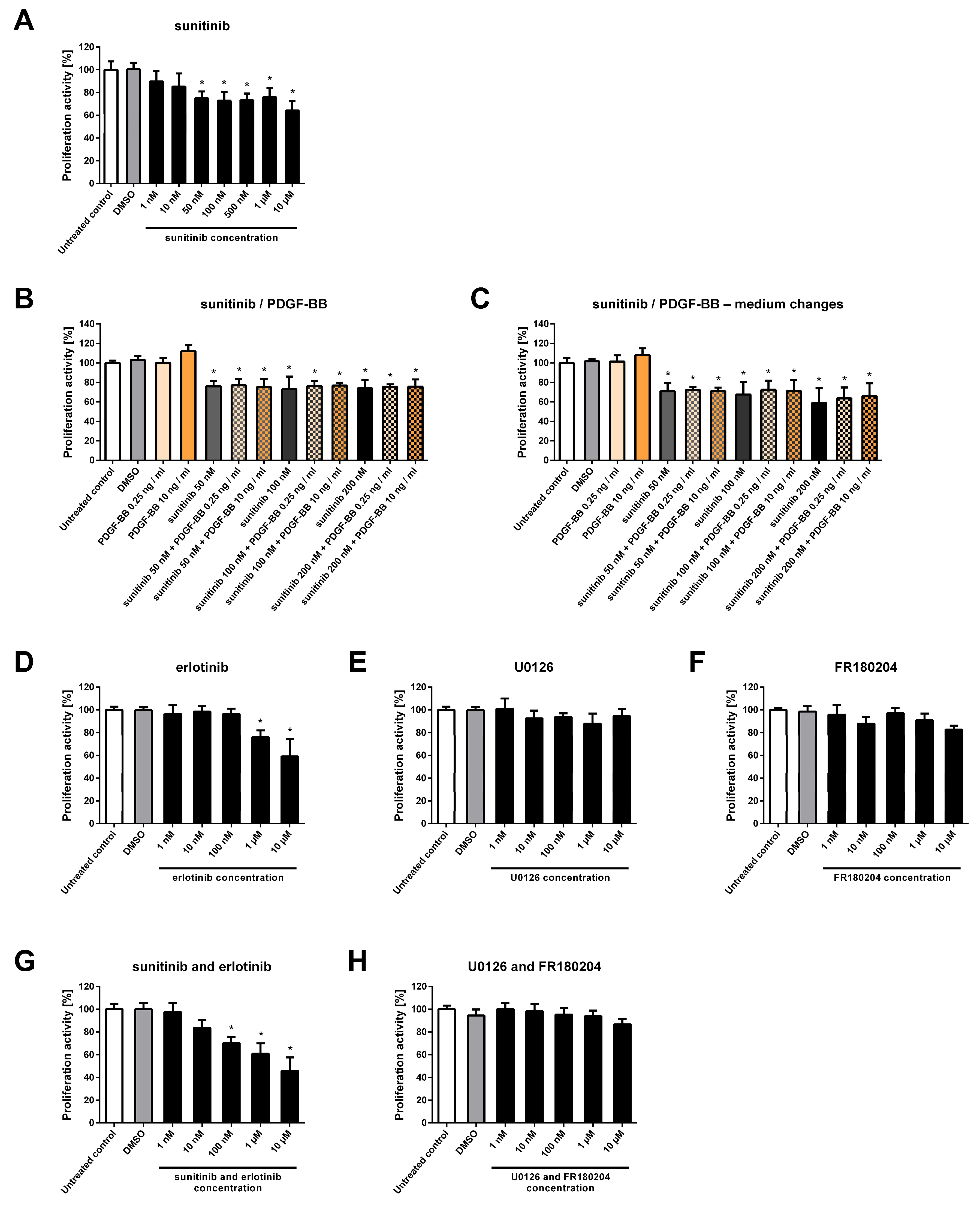
2.3. NSTS-47 Cells Are Sensitive to Sunitinib and Erlotinib
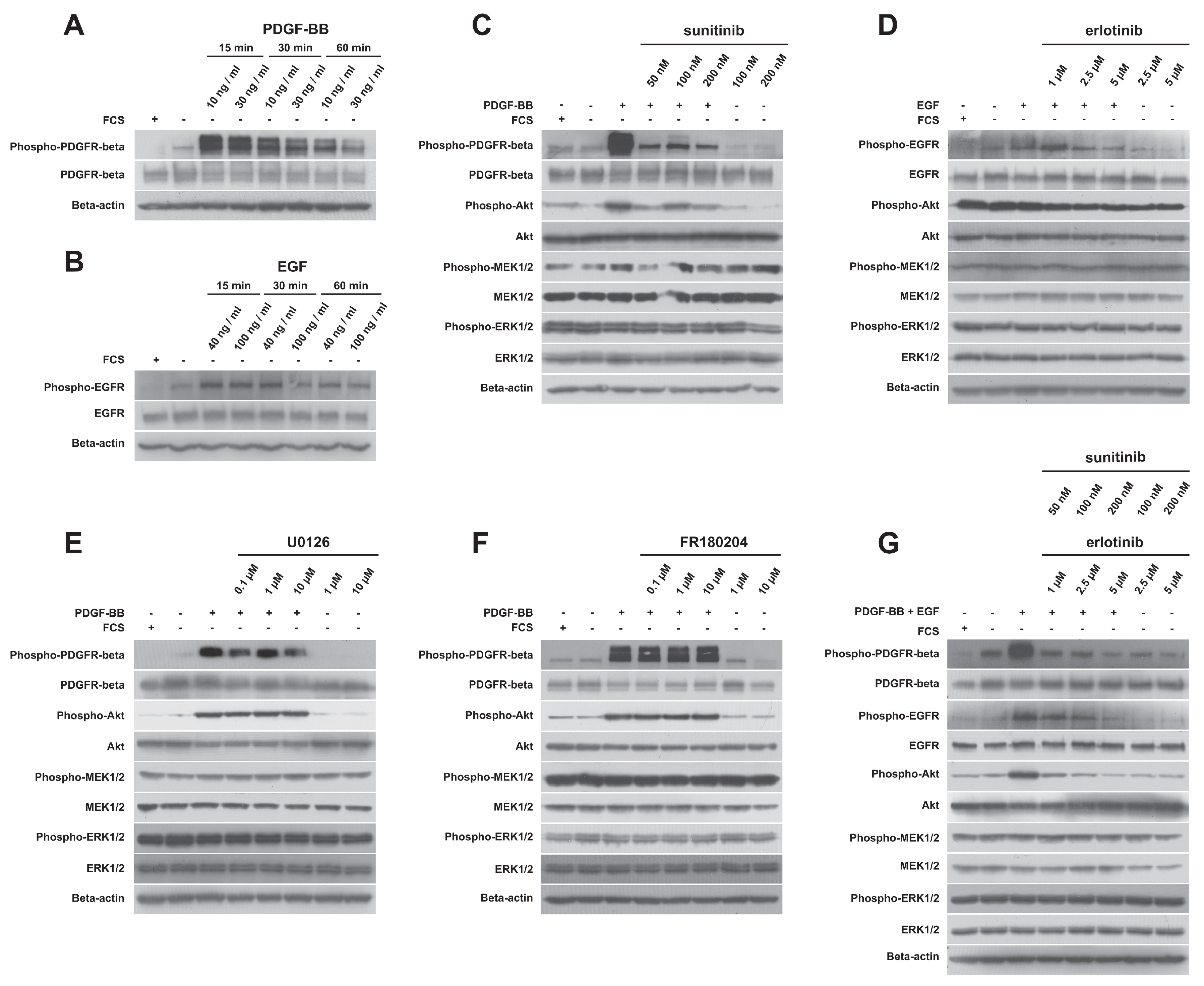
2.4. PDGFR-Beta and EGFR Exhibited Ligand-Dependent Tyrosine Phosphorylation
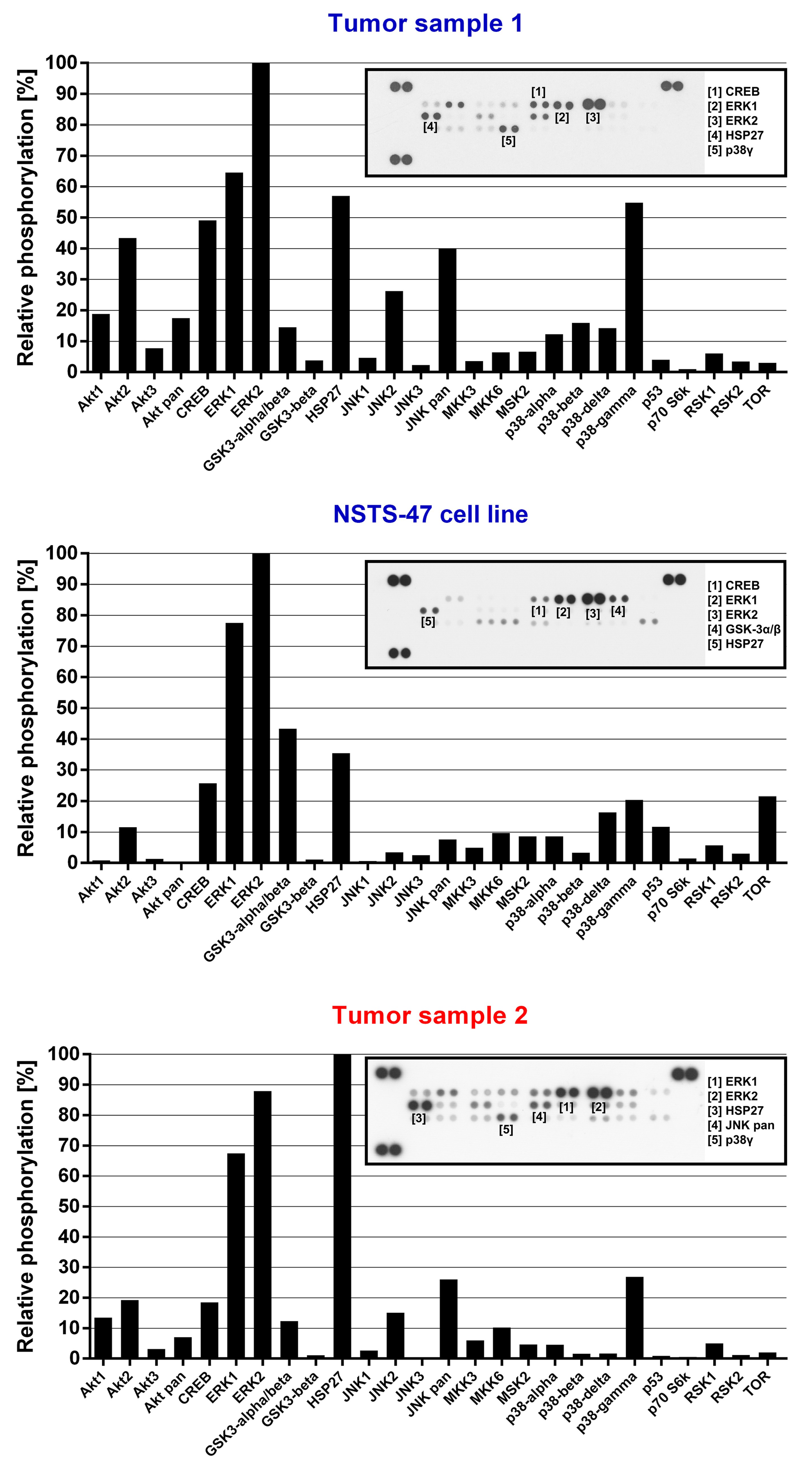
2.5. Detailed Analysis of Signaling Pathways Revealed Constitutive Phosphorylation of MEK1/2 and ERK1/2 Proteins
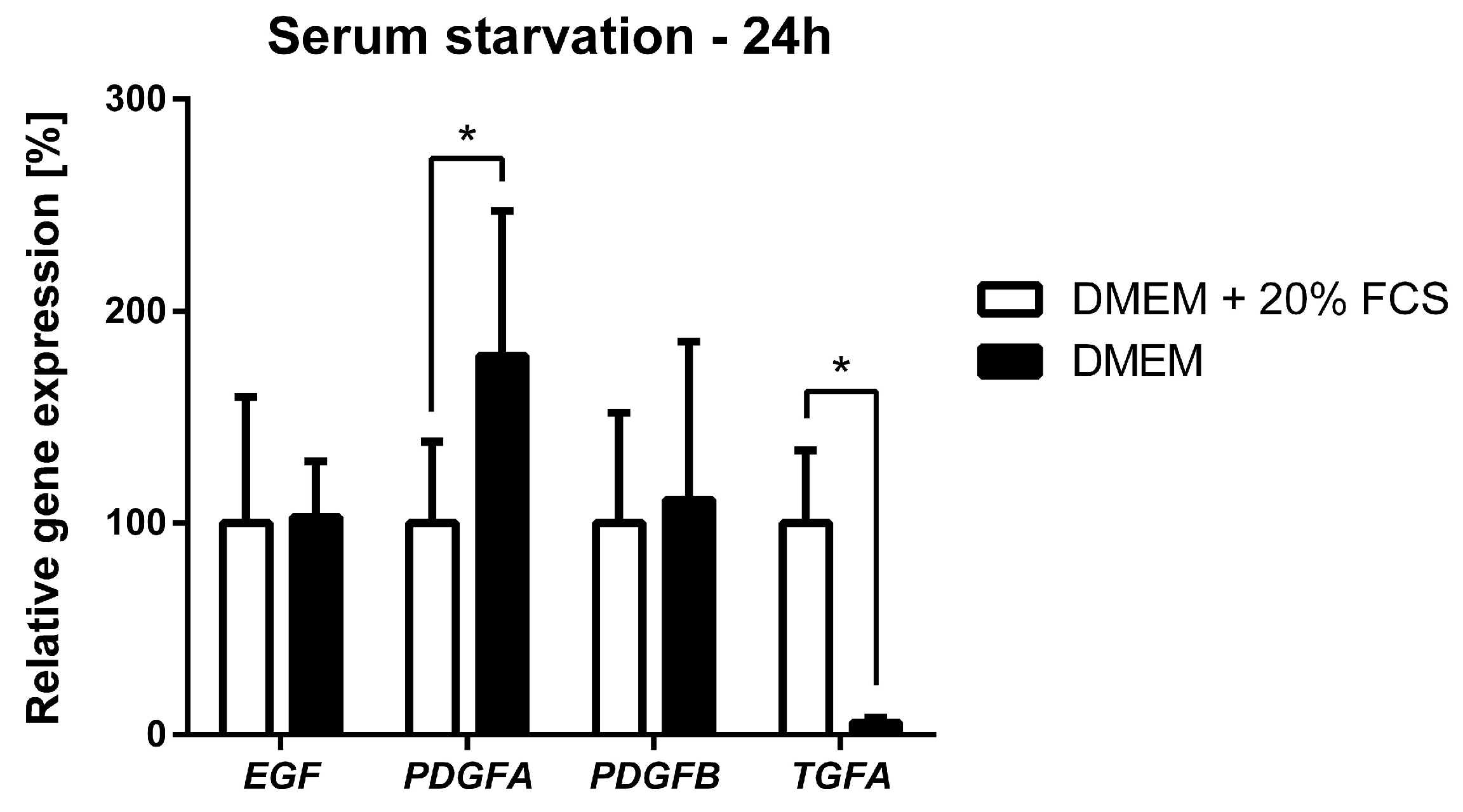
2.6. Serum Starvation of NSTS-47 Cells Induces an Increase in PDGFA Expression
3. Discussion
4. Materials and Methods
4.1. Tumor Samples
4.2. Cell Line and Cell Culture
4.3. Genetic Analyses
4.4. Chemicals
4.5. Phospho-RTK and Phospho-MAPK Array Analysis
4.6. MTT Assay
4.7. Western Blotting and Immunodetection
4.8. RT-qPCR
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | dimethyl sulfoxide |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular signal-regulated kinase |
| FCS | fetal calf serum |
| IM | infantile myofibromatosis |
| MAPK | mitogen-activated protein kinase |
| MEK | MAPK/ERK kinase |
| MTT | 3-(4-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PDGFR | platelet-derived growth factor receptor |
| PKIs | protein kinase inhibitors |
| RTKs | receptor tyrosine kinases |
| TGFA | transforming growth factor alpha |
| WES | whole exome sequencing |
References
- Martignetti, J.A.; Tian, L.; Li, D.; Ramirez, M.C.; Camacho-Vanegas, O.; Camacho, S.C.; Guo, Y.; Zand, D.J.; Bernstein, A.M.; Masur, S.K.; et al. Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. Am. J. Hum. Genet. 2013, 92, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Levine, E.; Fréneaux, P.; Schleiermacher, G.; Brisse, H.; Pannier, S.; Teissier, N.; Mesples, B.; Orbach, D. Risk-adapted therapy for infantile myofibromatosis in children. Pediatr. Blood Cancer 2012, 59, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Wang, K.C.; Lee, J.Y.; Phi, J.H.; Park, S.H.; Cheon, J.E.; Jang, Y.E.; Kim, S.K. Congenital solitary infantile myofibromatosis involving the spinal cord. J. Neurosurg. Pediatr. 2013, 11, 82–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, V.; Kumar, B.P.; Kumar, K.A.; Mohan, A.P. Myofibroma-a rare entity with unique clinical presentation. J. Maxillofac. Oral Surg. 2015, 14, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Hausbrandt, P.A.; Leithner, A.; Beham, A.; Bodo, K.; Raith, J.; Windhager, R. A rare case of infantile myofibromatosis and review of literature. J. Pediatr. Orthop. B 2010, 19, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Weaver, M.S.; Navid, F.; Huppmann, A.; Meany, H.; Angiolillo, A. Vincristine and Dactinomycin in Infantile Myofibromatosis with a Review of Treatment Options. J. Pediatr. Hematol. Oncol. 2015, 37, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.H.; Gayden, T.; Campeau, P.M.; LeDuc, C.A.; Russo, D.; Nguyen, V.H.; Guo, J.; Qi, M.; Guan, Y.; Albrecht, S.; et al. A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am. J. Hum. Genet. 2013, 92, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Mudry, P.; Slaby, O.; Neradil, J.; Soukalova, J.; Melicharkova, K.; Rohleder, O.; Jezova, M.; Seehofnerova, A.; Michu, E.; Veselska, R.; et al. Case report: Rapid and durable response to PDGFR targeted therapy in a child with refractory multiple infantile myofibromatosis and a heterozygous germline mutation of the PDGFRB gene. BMC Cancer 2017, 17, 119. [Google Scholar] [CrossRef] [PubMed]
- Gatibelza, M.E.; Vazquez, B.R.; Bereni, N.; Denis, D.; Bardot, J.; Degardin, N. Isolated infantile myofibromatosis of the upper eyelid: Uncommon localization and long-term results after surgical management. J. Pediatr. Surg. 2012, 47, 1457–1459. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.; Hanna, B.; Graf, N.; Fu, H.; Mylene, V.; Campeau, P.M.; Ronan, A. The spectrum of infantile myofibromatosis includes both non-penetrance and adult recurrence. Eur. J. Med. Genet. 2017, 60, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Lepelletier, C.; Al-Sarraj, Y.; Bodemer, C.; Shaath, H.; Fraitag, S.; Kambouris, M.; Hamel-Teillac, D.; Shanti, H.E.; Hadj-Rabia, S. Heterozygous PDGFRB Mutation in a Three-generation Family with Autosomal Dominant Infantile Myofibromatosis. Acta Derm. Venereol. 2017, 97, 858–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H. Targeting the PDGF signaling pathway in tumor treatment. Cell. Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol. Med. 2013, 19, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoch, R.V.; Soriano, P. Roles of PDGF in animal development. Development 2003, 130, 4769–4784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol 2017, 12, 245–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Hansson, E.M.; Tikka, S.; Lanner, F.; Sahlgren, C.; Farnebo, F.; Baumann, M.; Kalimo, H.; Lendahl, U. Notch signaling regulates platelet-derived growth factor receptor-beta expression in vascular smooth muscle cells. Circ. Res. 2008, 102, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Linhares, N.D.; Freire, M.C.; Cardenas, R.G.; Pena, H.B.; Bahia, M.; Pena, S.D. Exome sequencing identifies a novel homozygous variant in NDRG4 in a family with infantile myofibromatosis. Eur. J. Med. Genet. 2014, 57, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Linhares, N.D.; Freire, M.C.; Cardenas, R.G.; Bahia, M.; Puzenat, E.; Aubin, F.; Pena, S.D. Modulation of expressivity in PDGFRB-related infantile myofibromatosis: A role for PTPRG? Genet. Mol. Res. 2014, 13, 6287–6292. [Google Scholar] [CrossRef] [PubMed]
- Mirenda, M.; Toffali, L.; Montresor, A.; Scardoni, G.; Sorio, C.; Laudanna, C. Protein tyrosine phosphatase receptor type γ is a JAK phosphatase and negatively regulates leukocyte integrin activation. J. Immunol. 2015, 194, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
- Arts, F.A.; Chand, D.; Pecquet, C.; Velghe, A.; Constantinescu, S.; Hallberg, B.; Demoulin, J.B. PDGFRB mutants found in patients with familial infantile myofibromatosis or overgrowth syndrome are oncogenic and sensitive to imatinib. Oncogene 2016, 35, 3239–3248. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Demetri, G.; Sargent, W.; Raymond, E. Molecular basis for sunitinib efficacy and future clinical development. Nat. Rev. Drug Discov. 2007, 6, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Koker, M.; Weir, B.A.; Heynck, S.; Rabinovsky, R.; Zander, T.; Seeger, J.M.; Weiss, J.; Fischer, F.; Frommolt, P.; et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009, 69, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.L.; Worlikar, S.; MacKerell, A.D.; Shapiro, P.; Fletcher, S. Small-molecule inhibitors of the ERK signaling pathway: Towards novel anticancer therapeutics. ChemMedChem 2011, 6, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Abouantoun, T.J.; Castellino, R.C.; MacDonald, T.J. Sunitinib induces PTEN expression and inhibits PDGFR signaling and migration of medulloblastoma cells. J. Neurooncol. 2011, 101, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, C.; Daryani, V.M.; Billups, C.A.; Boyett, J.M.; Leary, S.; Tanos, R.; Goldsmith, K.C.; Stewart, C.F.; Blaney, S.M.; Gajjar, A. Phase II evaluation of sunitinib in the treatment of recurrent or refractory high-grade glioma or ependymoma in children: A children’s Oncology Group Study ACNS1021. Cancer Med. 2016, 5, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Jakacki, R.I.; Hamilton, M.; Gilbertson, R.J.; Blaney, S.M.; Tersak, J.; Krailo, M.D.; Ingle, A.M.; Voss, S.D.; Dancey, J.E.; Adamson, P.C. Pediatric phase I and pharmacokinetic study of erlotinib followed by the combination of erlotinib and temozolomide: A Children’s Oncology Group Phase I Consortium Study. J. Clin. Oncol. 2008, 26, 4921–4927. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veselska, R.; Kuglik, P.; Cejpek, P.; Svachova, H.; Neradil, J.; Loja, T.; Relichova, J. Nestin expression in the cell lines derived from glioblastoma multiforme. BMC Cancer 2006, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Skoda, J.; Neradil, J.; Zitterbart, K.; Sterba, J.; Veselska, R. EGFR signaling in the HGG-02 glioblastoma cell line with an unusual loss of EGFR gene copy. Oncol. Rep. 2014, 31, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gender | Age | PDGFRB Mutation |
|---|---|---|
| Male | 3.5 months | c.1681C>T (p.R561C) |
| Female | 8 years | c.1681C>T (p.R561C) |
| Primary Antibodies | ||||
| Antigen | Manufacturer | Catalog No. | Dilution | |
| Beta-actin | Sigma-Aldrich | A5441 | 1:20,000 | |
| Akt (pan) | Cell Signaling Technology | 4691 | 1:1000 | |
| Phospho-Akt (Ser473) | Cell Signaling Technology | 4060 | 1:2000 | |
| ERK1/2 | Cell Signaling Technology | 4695 | 1:1000 | |
| Phospho-ERK1/2 (Thr202/Tyr204) | Cell Signaling Technology | 4370 | 1:2000 | |
| MEK1/2 | Cell Signaling Technology | 9122 | 1:1000 | |
| Phospho-MEK1/2 (Ser217/221) | Cell Signaling Technology | 9121 | 1:1000 | |
| EGFR | Cell Signaling Technology | 2646 | 1:1000 | |
| Phospho-EGFR (Tyr1068) | Cell Signaling Technology | 2236 | 1:1000 | |
| PDGFR-beta | Cell Signaling Technology | 3169 | 1:1000 | |
| Phospho-PDGFR-beta (Tyr751) | Cell Signaling Technology | 4549 | 1:1000 | |
| Secondary antibodies | ||||
| Specificity | Conjugate | Manufacturer | Catalog No. | Dilution |
| Anti-Mouse IgG | horseradish peroxidase | Cell Signaling Technology | 7076 | 1:2000–1:20,000 |
| Anti-Rabbit IgG | horseradish peroxidase | Cell Signaling Technology | 7074 | 1:2000 |
| Gene | Gene Symbol | Primer Sequence |
|---|---|---|
| Epidermal growth factor | EGF | F: 5′-AGGATTGACACAGAAGGAACCAA-3′ R: 5′-ACATACTCTCTCTTGCCTTGACC-3′ |
| Heat shock protein 90 alpha family class B member 1 | HSP90AB1 | F: 5′-CGCATGAAGGAGACACAGAA-3′ R: 5′-TCCCATCAAATTCCTTGAGC-3′ |
| Platelet derived growth factor subunit A | PDGFA | F: 5′-TCCGTAGGGAGTGAGGATTCTTT-3′ R: 5′-GGCTTCTTCCTGACGTATTCCA-3′ |
| Platelet derived growth factor subunit B | PDGFB | F: 5′-GATCCGCTCCTTTGATGATCTCC-3′ R: 5′-ATCTCGATCTTTCTCACCTGGAC-3′ |
| Transforming growth factor alpha | TGFA | F: 5′-TGCCACTCAGAAACAGTGGTC-3′ R: 5′-AGTCCGTCTCTTTGCAGTTCTT-3′ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sramek, M.; Neradil, J.; Macigova, P.; Mudry, P.; Polaskova, K.; Slaby, O.; Noskova, H.; Sterba, J.; Veselska, R. Effects of Sunitinib and Other Kinase Inhibitors on Cells Harboring a PDGFRB Mutation Associated with Infantile Myofibromatosis. Int. J. Mol. Sci. 2018, 19, 2599. https://doi.org/10.3390/ijms19092599
Sramek M, Neradil J, Macigova P, Mudry P, Polaskova K, Slaby O, Noskova H, Sterba J, Veselska R. Effects of Sunitinib and Other Kinase Inhibitors on Cells Harboring a PDGFRB Mutation Associated with Infantile Myofibromatosis. International Journal of Molecular Sciences. 2018; 19(9):2599. https://doi.org/10.3390/ijms19092599
Chicago/Turabian StyleSramek, Martin, Jakub Neradil, Petra Macigova, Peter Mudry, Kristyna Polaskova, Ondrej Slaby, Hana Noskova, Jaroslav Sterba, and Renata Veselska. 2018. "Effects of Sunitinib and Other Kinase Inhibitors on Cells Harboring a PDGFRB Mutation Associated with Infantile Myofibromatosis" International Journal of Molecular Sciences 19, no. 9: 2599. https://doi.org/10.3390/ijms19092599