Role of Mitogen Activated Protein Kinase Signaling in Parkinson’s Disease

{kind=link}
{kind=link}
Abstract
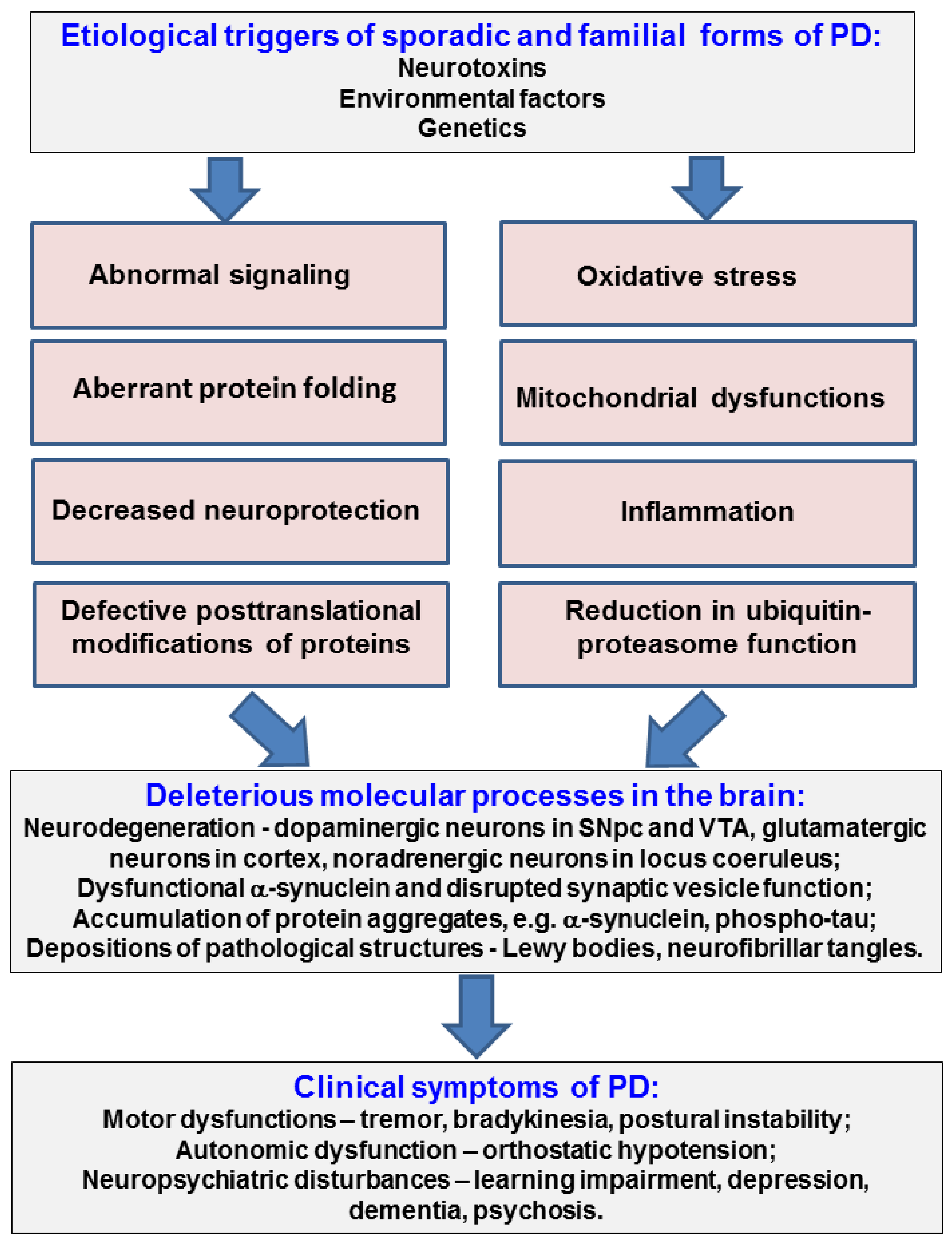
:1. Introduction
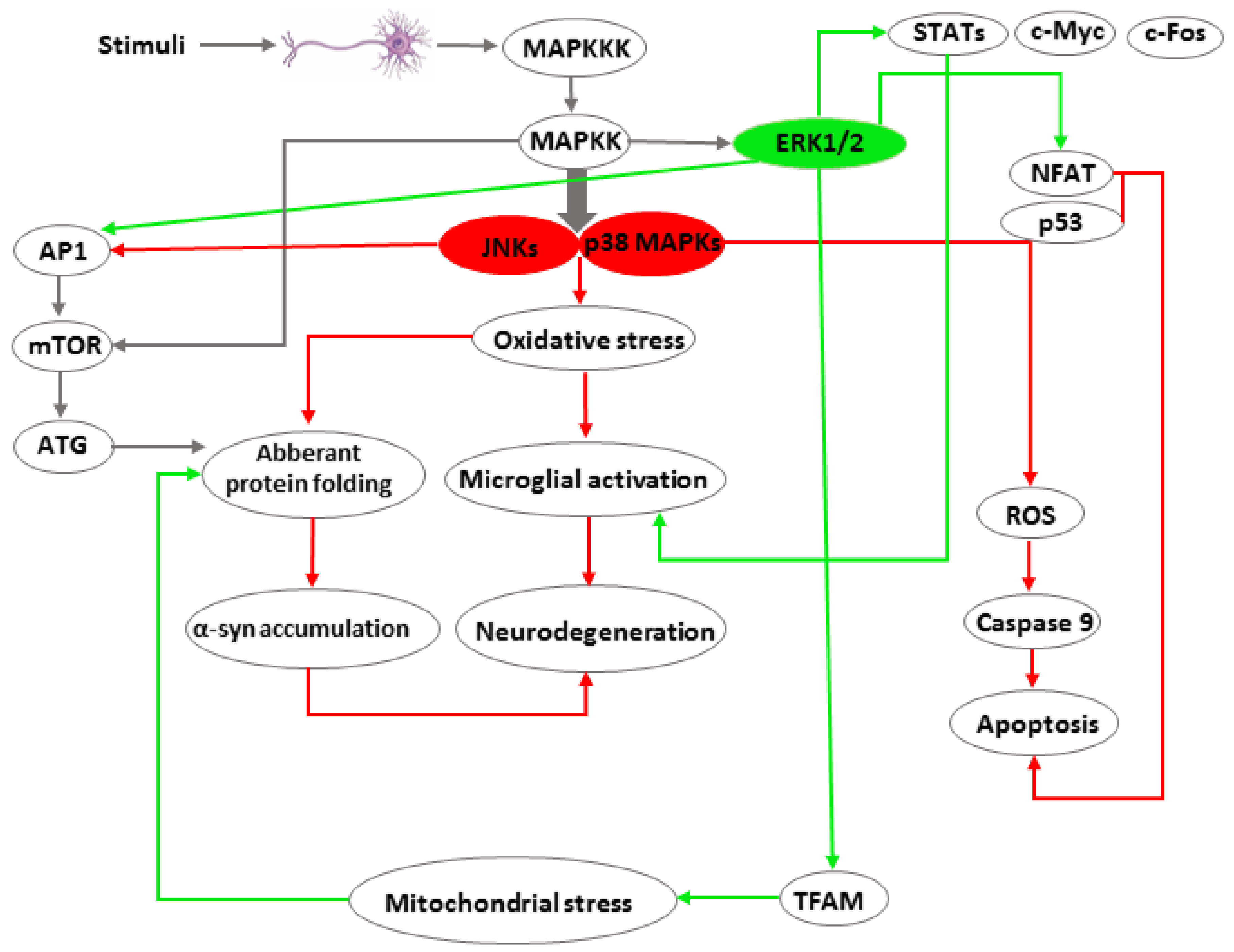
1.1. MAPK Signalling
1.2. JNK Signaling
1.3. ERK1/2 Signaling
1.4. p38 MAPK Signaling
2. Conclusions
Author Contributions
Acknowledgements
Conflicts of Interest
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (agonist of the AMPA receptor) |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| AKT | protein kinase B |
| ATG13 | autophagy-related protein 13 |
| bFGF | basic fibroblast growth factor |
| Cdc42 | cell division control protein 42 homolog |
| CNS | central nervous system |
| CSF-1 | colony stimulating factor 1 |
| ERK1/2 | extracellular regulated kinase 1 and 2 |
| EGFR | epidermal growth factor receptor |
| GPCR | G protein-coupled receptors |
| HMGB1 | high mobility group protein 1 |
| IL-1β | interleukin 1β |
| JIP1 | JNK interacting protein 1 |
| JNK | c-Jun N-terminal kinase |
| LBs | Lewy bodies |
| L-DOPA | L-3,4-dihydroxyphenylalanine |
| LRRK2 | leucine-rich repeat kinase 2 |
| LTD | long-term depression |
| MAP kinase | mitogen activated protein kinase |
| MEK3 and 6 | mitogen-activated protein kinase kinase 3 and 6 |
| MEKK1–3 | mitogen-activated protein kinase kinase kinase 1 to 3 |
| MLK2/3 | mixed lineage kinases 2/3 |
| MN9D | cell line used as a model of dopaminergic neurons |
| MNK | MAPK interacting protein kinase |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MSK | mitogen and stress activated protein kinase |
| NGF | neurite growth factor |
| NMDA | N-Methyl-d-aspartic acid |
| NO | nitric oxide |
| 6-OHDA | 6-hydroxydopamine |
| PD | Parkinson’s disease |
| LTP | long-term potentiation |
| TH | tyrosine hydroxylase |
| Rac1 | GTPase, member of the Rho family |
| ROS | reactive oxygen species |
| RTKs | receptor tyrosine kinases |
| RSK | ribosomal S6 kinase |
| SN | substantia nigra |
| STAT | signal transducer and activator of transcription |
| TAK1 | transforming growth factor beta-activated kinase 1 |
| TAO1/2 | thousand and one amino acid kinases 1/2 |
| TFAM | mitochondrial transcription factor A |
| TNF-α | tumor necrosis factor α |
| Tpl2 | tumor progression locus 2 kinase |
| UNC51-like kinase-1 | serine/threonine kinase involved in the autophagic cascade |
| NFAT | nuclear factor of activated T-cells |
| PI3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
References
- Nussbaum, R.L.; Ellis, C.E. Alzheimer’s disease and Parkinson’s disease. J. N. Engl. Med. 2003, 348, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Penney, E.B.; Mccabe, B.D. Parkinson’s Disease: Insights from Invertebrates. In Parkinson’s Disease Molecular and Therapeutic Insights from Model. Systems; Nass, R., Przedborski, S., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2008; pp. 323–333. ISBN 978-0123740281. [Google Scholar]
- Lindenbach, D.; Dupre, K.B.; Eskow Jaunarajs, K.L.; Ostock, C.Y.; Goldenberg, A.A.; Bishop, C. Effects of 5-HT1A receptor stimulation on striatal and cortical M1 pERK induction by L-DOPA and a D1 receptor agonist in a rat model of Parkinson’s disease. Brain Res. 2013, 1537, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Domingo-Sàbat, M.; Ariza, A. Molecular pathology of Lewy body diseases. Int. J. Mol. Sci. 2009, 10, 724–745. [Google Scholar] [CrossRef] [PubMed]
- Barr, R.K.; Bogoyevitch, M.A. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int. J. Biochem. Cell Biol. 2001, 33, 1047–1063. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Seger, R. The ERK signaling cascade—Views from different subcellular compartments. Biofactors 2009, 35, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2016, 6, 321. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.; Kawasaki, H.; Nishina, H. Diverse Roles of JNK and MKK Pathways in the Brain. J. Signal Transduct. 2012, 2012, 459265. [Google Scholar] [CrossRef] [PubMed]
- Kuan, C.Y.; Yang, D.D.; Samanta Roy, D.R.; Davis, R.J.; Rakic, P.; Flavell, R.A. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 1999, 22, 667–676. [Google Scholar] [CrossRef]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Raber, J.; Yang, D.; Su, B.; Mucke, L. Dynamic regulation of c-Jun N-terminal kinase activity in mouse brain by environmental stimuli. Proc. Natl. Acad. Sci. USA 1997, 94, 12655–12660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Ma, C.; Mao, Z.; Li, M. JNK inhibition as a potential strategy in treating Parkinson’s disease. Drug News Perspect. 2004, 10, 646. [Google Scholar] [CrossRef]
- Kulich, S.M.; Chu, C.T. Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: Implications for Parkinson’s disease. J. Neurochem. 2001, 77, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Strudwick, N.; Suwara, M.; Sutcliffe, L.K.; Mihai, A.D.; Ali, A.A.; Watson, J.N.; Schröder, M. An initial phase of JNK activation inhibits cell death early in the endoplasmic reticulum stress response. J. Cell Sci. 2016, 129, 2317–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Qian, J.; Zhang, Y.; Ma, J.; Wang, G.; Xiao, Q.; Chen, S.; Ding, J. Small peptide inhibitor of JNKs protects against MPTP-induced nigral dopaminergic injury via inhibiting the JNK-signaling pathway. Lab. Investig. 2010, 90, 156–167. [Google Scholar] [CrossRef]
- Zhang, S.; Gui, X.H.; Huang, L.P.; Deng, M.Z.; Fang, R.M.; Ke, X.H.; He, Y.P.; Li, L.; Fang, Y.Q. Neuroprotective Effects of β-Asarone Against 6-Hydroxy Dopamine-Induced Parkinsonism via JNK/Bcl-2/Beclin-1 Pathway. Mol. Neurobiol. 2016, 53, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Badshah, H.; Ali, T.; Rehman, S.-U.; Amin, F.-U.; Ullah, F.; Kim, T.H.; Kim, M.O. Protective Effect of Lupeol Against Lipopolysaccharide-Induced Neuroinflammation via the p38/c-Jun N-Terminal Kinase Pathway in the Adult Mouse Brain. J. Neuroimmune Pharmacol. 2016, 11, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zhang, F.; Hao, L.; Wu, J.; Jia, J.; Liu, J.Y.; Zheng, L.T.; Zhen, X. 1-O-tigloyl-1-O-deacetyl-nimbolinin B inhibits LPS-stimulated inflammatory responses by suppressing NF-κB and JNK activation in microglia cells. J. Pharmacol. Sci. 2014, 125, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Brecht, S.; Kirchhof, R.; Chromik, A.; Willesen, M.; Nicolaus, T.; Raivich, G.; Wessig, J.; Waetzig, V.; Goetz, M.; Claussen, M.; et al. Specific pathophysiological functions of JNK isoforms in the brain. Eur. J. Neurosci. 2005, 21, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Chun, H.J.; Lee, K.M.; Jung, Y.S.; Lee, J. Silibinin suppresses astroglial activation in a mouse model of acute Parkinson’s disease by modulating the ERK and JNK signaling pathways. Brain Res. 2015, 1627, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Xiao, Q.; Sheng, C.Y.; Hong, Z.; Yang, H.Q.; Wang, G.; Ding, J.Q.; Chen, S.D. Blockade of the translocation and activation of c-Jun N-terminal kinase 3 (JNK3) attenuates dopaminergic neuronal damage in mouse model of Parkinson’s disease. Neurochem. Int. 2009, 54, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Crocker, C.E.; Khan, S.; Cameron, M.D.; Robertson, H.A.; Robertson, G.S.; Lograsso, P. JNK Inhibition Protects Dopamine Neurons and Provides Behavioral Improvement in a Rat 6-hydroxydopamine Model of Parkinson’s Disease. ACS Chem. Neurosci. 2011, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, M.; Du, Y.; Zhang, W.; Bai, M.; Zhang, Z.; Li, Z.; Miao, J. Inhibition of c-Jun N-terminal kinase activation reverses Alzheimer disease phenotypes in APPswe/PS1dE9 mice. Ann. Neurol. 2015, 77, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Ryu, M.J.; Han, J.; Jang, Y.; Kim, J.; Lee, M.J.; Ryu, I.; Ju, X.; Oh, E.; Chung, W.; et al. Activation of the HMGB1-RAGE axis upregulates TH expression in dopaminergic neurons via JNK phosphorylation. Biochem. Biophys. Res. Commun. 2017, 493, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Spigolon, G.; Cavaccini, A.; Trusel, M.; Tonini, R.; Fisone, G. cJun N-terminal kinase (JNK) mediates cortico-striatal signaling in a model of Parkinson’s disease. Neurobiol. Dis. 2018, 110, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Bellone, C. Modulation of the glutamatergic transmission by Dopamine: A focus on Parkinson, Huntington and Addiction diseases. Front. Cell Neurosci. 2015, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Nisticò, R.; Florenzano, F.; Mango, D.; Ferraina, C.; Grilli, M.; Di Prisco, S.; Nobili, A.; Saccucci, S.; D’Amelio, M.; Morbin, M.; et al. Presynaptic c-Jun N-terminal Kinase 2 regulates NMDA receptor-dependent glutamate release. Sci. Rep. 2015, 12, 9035. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Galletti, F.; Saggese, E.; Ghiglieri, V.; Picconi, B. Neuronal networks and synaptic plasticity in Parkinson’s disease: Beyond motor deficits. Parkinsonism Relat. Disord. 2007, 13 (Suppl. 3), 259–262. [Google Scholar] [CrossRef]
- Kim, M.J.; Futai, K.; Jo, J.; Hayashi, Y.; Cho, K.; Sheng, M. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron 2007, 56, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.M.; Lin, D.T.; Nuriya, M.; Huganir, R.L. Rapid and bi-directional regulation of AMPA receptor phosphorylation and trafficking by JNK. EMBO J. 2008, 27, 361–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, D.T.; Kawasaki, M.; Walcoff, M.; Hisamoto, N.; Matsumoto, K.; Jin, Y. UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron 2001, 32, 787–800. [Google Scholar] [CrossRef]
- Daire, V.; Giustiniani, J.; Leroy-Gori, I.; Quesnoit, M.; Drevensek, S.; Dimitrov, A.; Perez, F.; Poüs, C. Kinesin-1 regulates microtubule dynamics via a c-Jun N-terminal kinase-dependent mechanism. J. Biol. Chem. 2009, 284, 31992–32001. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Zhang, B.; Lee, V.M.; Trojanowski, J.Q. Axonal transport defects: A common theme in neurodegenerative diseases. Acta Neuropathol. 2005, 109, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown, R.H., Jr.; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, X.; Wu, J.; Li, J.; Zhang, L.; Xiong, T.; Tang, J.; Qu, Y.; Mu, D. Involvement of the JNK/FOXO3a/Bim Pathway in Neuronal Apoptosis after Hypoxic-Ischemic Brain Damage in Neonatal Rats. PLoS ONE 2015, 10, e0132998. [Google Scholar] [CrossRef] [PubMed]
- Kanamoto, T.; Mota, M.; Takeda, K.; Rubin, L.L.; Miyazono, K.; Ichijo, H.; Bazenet, C.E. Role of apoptosis signal-regulating kinase in regulation of the c-Jun N-terminal kinase pathway and apoptosis in sympathetic neurons. Mol. Cell. Biol. 2000, 20, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.K.; Chen, J.; Zhang, Y.; Clemens, D.; Follenzi, A.; Zern, M.A. Role of MAPK phosphatase-1 in sustained activation of JNK during ethanol-induced apoptosis in hepatocyte-like VL-17A cells. J. Biol. Chem. 2007, 282, 31900–31908. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, C.; Ballerio, R.; Gaillard, P.; Giachetti, C.; Carboni, S.; Vitte, P.A.; Gotteland, J.P.; Cirillo, R. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. Br. J. Pharmacol. 2004, 142, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, E.W.; Mo, S.J.; Rhee, D.K.; Pyo, S. Inhibition of ICAM-1 expression by garlic component, allicin, in gamma-irradiated human vascular endothelial cells via downregulation of the JNK signaling pathway. Int. Immunopharmacol. 2006, 6, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Shklover, J.; Mishnaevski, K.; Levy-Adam, F.; Kurant, E. JNK pathway activation is able to synchronize neuronal death and glial phagocytosis in Drosophila. Cell. Death. Dis. 2015, 6, e1649. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Andersen, J.K. The role of c-Jun N-terminal kinase (JNK) in Parkinson’s disease. IUBMB Life 2003, 55, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Hsu, L.J.; Rockenstein, E.; Takenouchi, T.; Mallory, M.; Masliah, E. Alpha-Synuclein protects against oxidative stress via inactivation of the c-Jun N-terminal kinase stress-signaling pathway in neuronal cells. J. Biol. Chem. 2002, 277, 11465–11472. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Liu, S.; Wang, C.; Wang, F.; Song, Y.; Yan, N.; Xi, S.; Liu, Z.; Sun, G. JNK and NADPH oxidase involved in fluoride-induced oxidative stress in BV-2 microglia cells. Mediat. Inflamm. 2013, 2013, 895975. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Xie, Q.; Tibbetts, R.S. Opposing roles of p38 and JNK in a Drosophila model of TDP-43 proteinopathy reveal oxidative stress and innate immunity as pathogenic components of neurodegeneration. Hum. Mol. Genet. 2015, 24, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.C.; Bohmann, D.; Jasper, H. JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev. Cell 2003, 5, 811–816. [Google Scholar] [CrossRef]
- Szebenyi, G.; Morfini, G.A.; Babcock, A.; Gould, M.; Selkoe, K.; Stenoien, D.L.; Young, M.; Faber, P.W.; MacDonald, M.E.; McPhaul, M.J.; et al. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 2003, 40, 41–52. [Google Scholar] [CrossRef]
- Ren, H.; Fu, K.; Mu, C.; Li, B.; Wang, D.; Wang, G. DJ-1, a cancer and Parkinson’s disease associated protein, regulates autophagy through JNK pathway in cancer cells. Cancer Lett. 2010, 297, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Bandoski, C.; Bartlett, J.D. Fluoride induces oxidative damage and SIRT1/autophagy through ROS-mediated JNK signaling. Free Radic. Biol. Med. 2015, 89, 369–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi-Niki, K.; Kato-Ose, I.; Murata, H.; Maita, H.; Iguchi-Ariga, S.M.; Ariga, H. Epidermal Growth Factor-dependent Activation of the Extracellular Signal-regulated Kinase Pathway by DJ-1 Protein through Its Direct Binding to c-Raf Protein. J. Biol. Chem. 2015, 29, 17838–17847. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Doronin, S.; Malbon, C.C. Insulin activation of mitogen-activated protein kinases Erk1,2 is amplified via beta-adrenergic receptor expression and requires the integrity of the Tyr350 of the receptor. J. Biol. Chem. 2000, 275, 36086–36093. [Google Scholar] [CrossRef] [PubMed]
- Naor, Z.; Benard, O.; Seger, R. Activation of MAPK cascades by G-protein-coupled receptors: The case of gonadotropin-releasing hormone receptor. Trends Endocrinol. Metab. 2000, 11, 91–99. [Google Scholar] [CrossRef]
- Chiri, S.; Bogliolo, S.; Ehrenfeld, J.; Ciapa, B. Activation of extracellular signal-regulated kinase ERK after hypo-osmotic stress in renal epithelial A6 cells. Biochim. Biophys. Acta 2004, 1664, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. Recept Signal. Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.J.; John, S. Signal transducer and activator of transcription (STAT) signalling and T-cell lymphomas. Immunology 2005, 114, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Chen, Q.; Wu, Y.; Liu, F.; Chen, X.; Zhang, Y. Unphosphorylated STAT1 represses apoptosis in macrophages during Mycobacterium tuberculosis infection. J. Cell. Sci. 2017, 130, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Selcher, J.C.; Nekrasova, T.; Paylor, R.; Landreth, G.E.; Sweatt, J.D. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn. Mem. 2001, 8, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Scholl, F.A.; Dumesic, P.A.; Barragan, D.I.; Harada, K.; Bissonauth, V.; Charron, J.; Khavari, P.A. Mek1/2 MAPK kinases are essential for Mammalian development, homeostasis, and Raf-induced hyperplasia. Dev. Cell 2007, 12, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Fiore, R.S.; Bayer, V.E.; Pelech, S.L.; Posada, J.; Cooper, J.A.; Baraban, J.M. p42 mitogen-activated protein kinase in brain: Prominent localization in neuronal cell bodies and dendrites. Neuroscience 1993, 55, 463–472. [Google Scholar] [CrossRef]
- Di Cristo, G.; Berardi, N.; Cancedda, L.; Pizzorusso, T.; Putignano, E.; Ratto, G.M.; Maffei, L. Requirement of ERK activation for visual cortical plasticity. Science 2001, 292, 2337–2340. [Google Scholar] [CrossRef] [PubMed]
- Fieblinger, T.; Sebastianutto, I.; Alcacer, C.; Bimpisidis, Z.; Maslava, N.; Sandberg, S.; Engblom, D.; Cenci, M.A. Mechanisms of dopamine D1 receptor-mediated ERK1/2 activation in the parkinsonian striatum and their modulation by metabotropic glutamate receptor type 5. J. Neurosci. 2014, 34, 4728–4740. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.E.; Dash, P.K. Extracellular signal-regulated kinase activity in the entorhinal cortex is necessary for long-term spatial memory. Learn. Mem. 2002, 9, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Yufune, S.; Satoh, Y.; Takamatsu, I.; Ohta, H.; Kobayashi, Y.; Takaenoki, Y.; Pagès, G.; Pouysségur, J.; Endo, S.; Kazama, T. Transient Blockade of ERK Phosphorylation in the Critical Period Causes Autistic Phenotypes as an Adult in Mice. Sci. Rep. 2015, 5, 10252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, E.C.; Slack, R.S. Emerging role for ERK as a key regulator of neuronal apoptosis. Sci. STKE 2004, 251, PE45. [Google Scholar] [CrossRef] [PubMed]
- Bhat, N.R.; Zhang, P. Hydrogen peroxide activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: Role of extracellular signal-regulated kinase in hydrogen peroxide-induced cell death. J. Neurochem. 1999, 72, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Canals, S.; Casarejos, M.J.; de Bernardo, S.; Solano, R.M.; Mena, M.A. Selective and persistent activation of extracellular signal-regulated protein kinase by nitric oxide in glial cells induces neuronal degeneration in glutathione-depleted midbrain cultures. Mol. Cell. Neurosci. 2003, 24, 1012–1026. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.; Kou, C.; Lu, C.; Xu, J.; Xie, J.; Lu, J.; Liu, Y.; Wang, Z.; Wang, D. Involvement of the ERK pathway in the protective effects of glycyrrhizic acid against the MPP+-induced apoptosis of dopaminergic neuronal cells. Int. J. Mol. Med. 2014, 34, 742–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Rusnak, M.; Lombroso, P.J.; Sidhu, A. Dopamine promotes striatal neuronal apoptotic death via ERK signaling cascades. Eur. J. Neurosci. 2009, 29, 287–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, A.H. Mitochondrial complex I deficiency in Parkinson’s disease. Adv. Neurol. 1993, 60, 288–291. [Google Scholar] [PubMed]
- Zhu, J.H.; Guo, F.; Shelburne, J.; Watkins, S.; Chu, C.T. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003, 13, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Monick, M.M.; Powers, L.S.; Barrett, C.W.; Hinde, S.; Ashare, A.; Groskreutz, D.J.; Nyunoya, T.; Coleman, M.; Spitz, D.R.; Hunninghake, G.W. Constitutive ERK MAPK activity regulates macrophage ATP production and mitochondrial integrity. J. Immunol. 2008, 180, 7485–7496. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.Z.; Zhu, J.; Dagda, R.K.; Uechi, G.; Cherra, S.J.; Gusdon, A.M.; Balasubramani, M.; Chu, C.T. ERK-mediated phosphorylation of TFAM downregulates mitochondrial transcription: Implications for Parkinson’s disease. Mitochondrion 2014, 17, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, J.; Chen, S.; Wang, Y.; Cao, L.; Zhang, Y.; Kang, W.; Li, H.; Gui, Y.; Chen, S.; et al. DJ-1 modulates the expression of Cu/Zn-superoxide dismutase-1 through the Erk1/2-Elk1 pathway in neuroprotection. Ann. Neurol. 2011, 70, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Cai, P.; Ye, J.; Zhu, J.; Liu, D.; Chen, D.; Wei, X.; Johnson, N.R.; Wang, Z.; Zhang, H.; Cao, G.; et al. Inhibition of Endoplasmic Reticulum Stress is Involved in the Neuroprotective Effect of bFGF in the 6-OHDA-Induced Parkinson’s Disease Model. Aging Dis. 2016, 7, 336–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Bai, L.; Zhang, S.; Zhou, X.; Li, Y.; Bai, J. Trx-1 ameliorates learning and memory deficits in MPTP-induced Parkinson’s disease model in mice. Free Radic. Biol. Med. 2018, 124, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Shin, K.S.; Zhao, T.T.; Park, H.; Lee, K.E.; Lee, M.K. L-DOPA modulates cell viability through the ERK-c-Jun system in PC12 and dopaminergic neuronal cells. Neuropharmacology 2016, 101, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Qin, L.; Huang, F.; Wang, X.; Yang, L.; Shi, H.; Wu, H.; Zhang, B.; Chen, Z.; Wu, X. Amentoflavone protects dopaminergic neurons in MPTP-induced Parkinson’s disease model mice through PI3K/Akt and ERK signaling pathways. Toxicol. Appl. Pharmacol. 2017, 319, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Liu, J.; Ju, C.; Chen, G.; Xu, S.; Zeng, Y.; Yan, X.; Wang, W.; Liu, D.; Fu, S. Licochalcone A Prevents the Loss of Dopaminergic Neurons by Inhibiting Microglial Activation in Lipopolysaccharide (LPS)-Induced Parkinson’s Disease Models. Int. J. Mol. Sci. 2017, 18, 2043. [Google Scholar] [CrossRef] [PubMed]
- Kamakura, S.; Moriguchi, T.; Nishida, E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J. Biol. Chem. 1999, 274, 26563–26571. [Google Scholar] [CrossRef] [PubMed]
- Mody, N.; Leitch, J.; Armstrong, C.; Dixon, J.; Cohen, P. Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett. 2001, 502, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Cavanaugh, J.E.; Jaumotte, L.D.; Lakoski, J.M.; Zigmond, M.J. Neuroprotective Role of ERK1/2 and ERK5 in a Dopaminergic Cell Line Under Basal Conditions and in Response to Oxidative Stress. J. Neurosci. Res. 2006, 84, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Gram, H. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J. Biol. Chem. 1997, 272, 30122–30128. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, M.; Svensson, L.; Roach, M.; Hambor, J.; McNeish, J.; Gabel, C.A. Deficiency of the stress kinase p38alpha results in embryonic lethality: Characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J. Exp. Med. 2000, 191, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Beardmore, V.A.; Hinton, H.J.; Eftychi, C.; Apostolaki, M.; Armaka, M.; Darragh, J.; McIlrath, J.; Carr, J.M.; Armit, L.J.; Clacher, C.; et al. Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol. Cell. Biol. 2005, 25, 10454–10464. [Google Scholar] [CrossRef] [PubMed]
- Schonhoff, C.M.; Park, S.W.; Webster, C.R.L.; Anwer, M.S. p38 MAPK α and β isoforms differentially regulate plasma membrane localization of MRP2. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G999–G1005. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.K.; Jha, N.K.; Kar, R.; Ambasta, R.K.; Kumar, P. p38 MAPK and PI3K/AKT Signalling Cascades in Parkinson’s Disease. Int. J. Mol. Cell Med. 2015, 4, 67–86. [Google Scholar] [PubMed]
- Harper, S.J.; LoGrasso, P. Signalling for survival and death in neurones: The role of stress-activated kinases, JNK and p38. Cell Signal. 2001, 13, 299–310. [Google Scholar] [CrossRef]
- Mielke, K.; Herdegen, T. JNK and p38 stresskinases—Degenerative effectors of signal-transduction-cascades in the nervous system. Prog. Neurobiol. 2000, 61, 45–60. [Google Scholar] [CrossRef]
- De Chiara, G.; Marcocci, M.E.; Torcia, M.; Lucibello, M.; Rosini, P.; Bonini, P.; Higashimoto, Y.; Damonte, G.; Armirotti, A.; Amodei, S.; et al. Bcl-2 Phosphorylation by p38 MAPK: Identification of target sites and biologic consequences. J. Biol. Chem. 2006, 281, 21353–21361. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Chang, S.H.; Becker, E.B.; Bonni, A.; Xia, Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J. Biol. Chem. 2006, 281, 25215–25222. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.S.; Eom, D.S.; Han, B.S.; Kim, W.K.; Han, B.H.; Choi, E.J.; Oh, T.H.; Markelonis, G.J.; Cho, J.W.; Oh, Y.J. Phosphorylation of p38 MAPK induced by oxidative stress is linked to activation of both caspase-8- and -9-mediated apoptotic pathways in dopaminergic neurons. J. Biol. Chem. 2004, 279, 20451–20460. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Zhang, X.; Meng, X.; Lu, L.; Mai, D.; Qu, S. Simvastatin Inhibits Activation of NADPH Oxidase/p38 MAPK Pathway and Enhances Expression of Antioxidant Protein in Parkinson Disease Models. Front. Mol. Neurosci. 2018, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Hu, Y.; Liu, L.; Cai, J.; Peng, C.; Li, Q. Gastrodin protects against MPP(+)-induced oxidative stress by up regulates heme oxygenase-1 expression through p38 MAPK/Nrf2 pathway in human dopaminergic cells. Neurochem. Int. 2014, 75, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Liedhegner, E.A.; Steller, K.M.; Mieyal, J.J. Levodopa activates apoptosis signaling kinase 1 (ASK1) and promotes apoptosis in a neuronal model: Implications for the treatment of Parkinson’s disease. Chem. Res. Toxicol. 2011, 24, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, M.; Shen, X. Critical role of ASK1 in the 6-hydroxydopamine-induced apoptosis in human neuroblastoma SH-SY5Y cells. J. Neurochem. 2006, 97, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, D.; Hu, Q.; Wang, G. Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS ONE 2013, 8, e72046. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.K.; Han, B.S.; Choi, W.S.; Youdim, M.B.; Oh, Y.J. Translocation and oligomerization of Bax is regulated independently by activation of p38 MAPK and caspase-2 during MN9D dopaminergic neurodegeneration. Apoptosis 2011, 16, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.L.; Lee, T.R.; Hung, S.Y.; Kang, K.H.; Lee, M.J.; Fu, W.M. Impairment of oxidative stress-induced heme oxygenase-1 expression by the defect of Parkinson-related gene of PINK1. J. Neurochem. 2011, 17, 643–653. [Google Scholar] [CrossRef]
- Hwang, C.J.; Kim, Y.E.; Son, D.J.; Park, M.H.; Choi, D.Y.; Park, P.H.; Hellström, M.; Han, S.B.; Oh, K.W.; Park, E.K.; et al. Parkin deficiency exacerbate ethanol-induced dopaminergic neurodegeneration by P38 pathway dependent inhibition of autophagy and mitochondrial function. Redox Biol. 2017, 11, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ren, Y.; Gui, C.; Zhao, M.; Wu, X.; Mao, K.; Li, W.; Zou, F. Phosphorylation of Parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T α-synuclein model of Parkinson’s disease. Cell Death Dis. 2018, 9, 700. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, Z.; Gu, J.H.; Ge, J.B.; Liang, Z.Q.; Qin, Z.H. p38(MAPK)/p53-Mediated Bax induction contributes to neurons degeneration in rotenone-induced cellular and rat models of Parkinson’s disease. Neurochem. Int. 2013, 63, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Mo, J.S.; Kim, M.Y.; Ann, E.J.; Ahn, J.S.; Jo, E.H.; Lee, H.J.; Lee, Y.C.; Seol, W.; Yarmoluk, S.M.; et al. LRRK2 functions as a scaffolding kinase of ASK1-mediated neuronal cell death. Biochim. Biophys. Acta 2017, 1864, 2356–2368. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Chen, H.; Ma, J.; He, Q.; Huang, Q.; Liu, Q.; Li, M.; Yuan, Z. c-Abl-p38α signaling plays an important role in MPTP-induced neuronal death. Cell Death Differ. 2016, 23, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, S.; Saeed, U.; Mishra, M.; Valli, R.K.; Joshi, S.D.; Meka, D.P.; Seth, P.; Ravindranath, V. Selective activation of p38 mitogen-activated protein kinase in dopaminergic neurons of substantia nigra leads to nuclear translocation of p53 in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. J. Neurosci. 2008, 28, 12500–12509. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.S.; Kim, H.; Lee, H.J.; Sapkota, K.; Park, S.E.; Kim, S.; Kim, S.J. Celastrol from ‘Thunder God Vine’ protects SH-SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson’s disease. Neurochem. Res. 2014, 39, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, R.M.; Ahmed, M.A.E.; El-Sahar, A.E.; El Sayed, N.S. Montelukast attenuates rotenone-induced microglial activation/p38 MAPK expression in rats: Possible role of its antioxidant, anti-inflammatory and antiapoptotic effects. Toxicol. Appl. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Timmer, M.; Cesnulevicius, K.; Hitti, E.; Kotlyarov, A.; Gaestel, M. MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing neuroinflammation--relevance in a mouse model of Parkinson’s disease. J. Neurochem. 2008, 105, 2039–2052. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; She, H.; Zhang, T.; Xu, H.; Cheng, L.; Yepes, M.; Zhao, Y.; Mao, Z. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J. Cell. Biol. 2018, 217, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, Y.; Zhu, X. MicroRNA-181a Regulates Apoptosis and Autophagy Process in Parkinson’s Disease by Inhibiting p38 Mitogen-Activated Protein Kinase (MAPK)/c-Jun N-Terminal Kinases (JNK) Signaling Pathways. Med. Sci. Monit. 2017, 23, 1597–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bohush, A.; Niewiadomska, G.; Filipek, A. Role of Mitogen Activated Protein Kinase Signaling in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 2973. https://doi.org/10.3390/ijms19102973
Bohush A, Niewiadomska G, Filipek A. Role of Mitogen Activated Protein Kinase Signaling in Parkinson’s Disease. International Journal of Molecular Sciences. 2018; 19(10):2973. https://doi.org/10.3390/ijms19102973
Chicago/Turabian StyleBohush, Anastasiia, Grazyna Niewiadomska, and Anna Filipek. 2018. "Role of Mitogen Activated Protein Kinase Signaling in Parkinson’s Disease" International Journal of Molecular Sciences 19, no. 10: 2973. https://doi.org/10.3390/ijms19102973