ERK5 Phosphorylates Kv4.2 and Inhibits Inactivation of the A-Type Current in PC12 Cells

 and
and 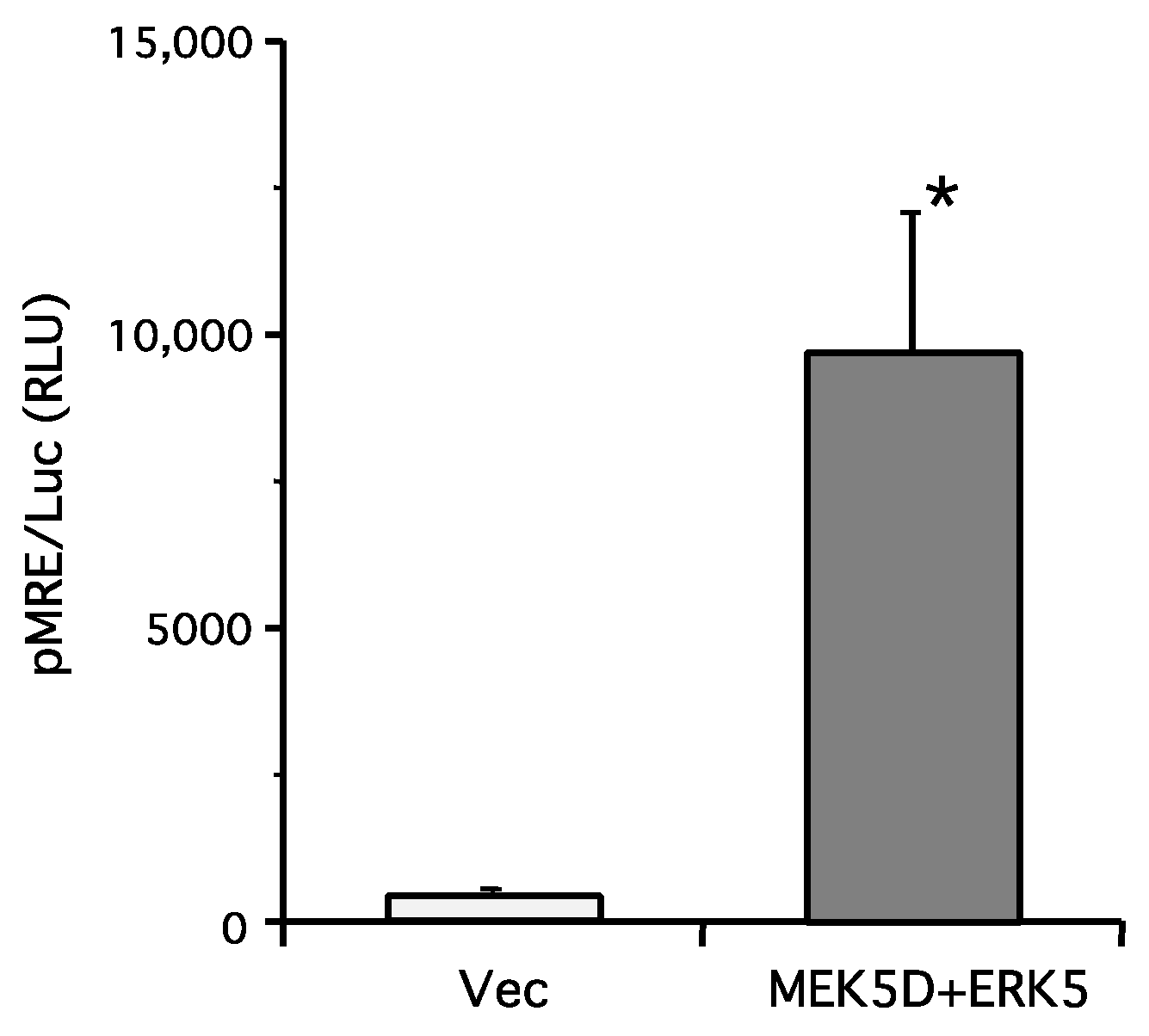{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
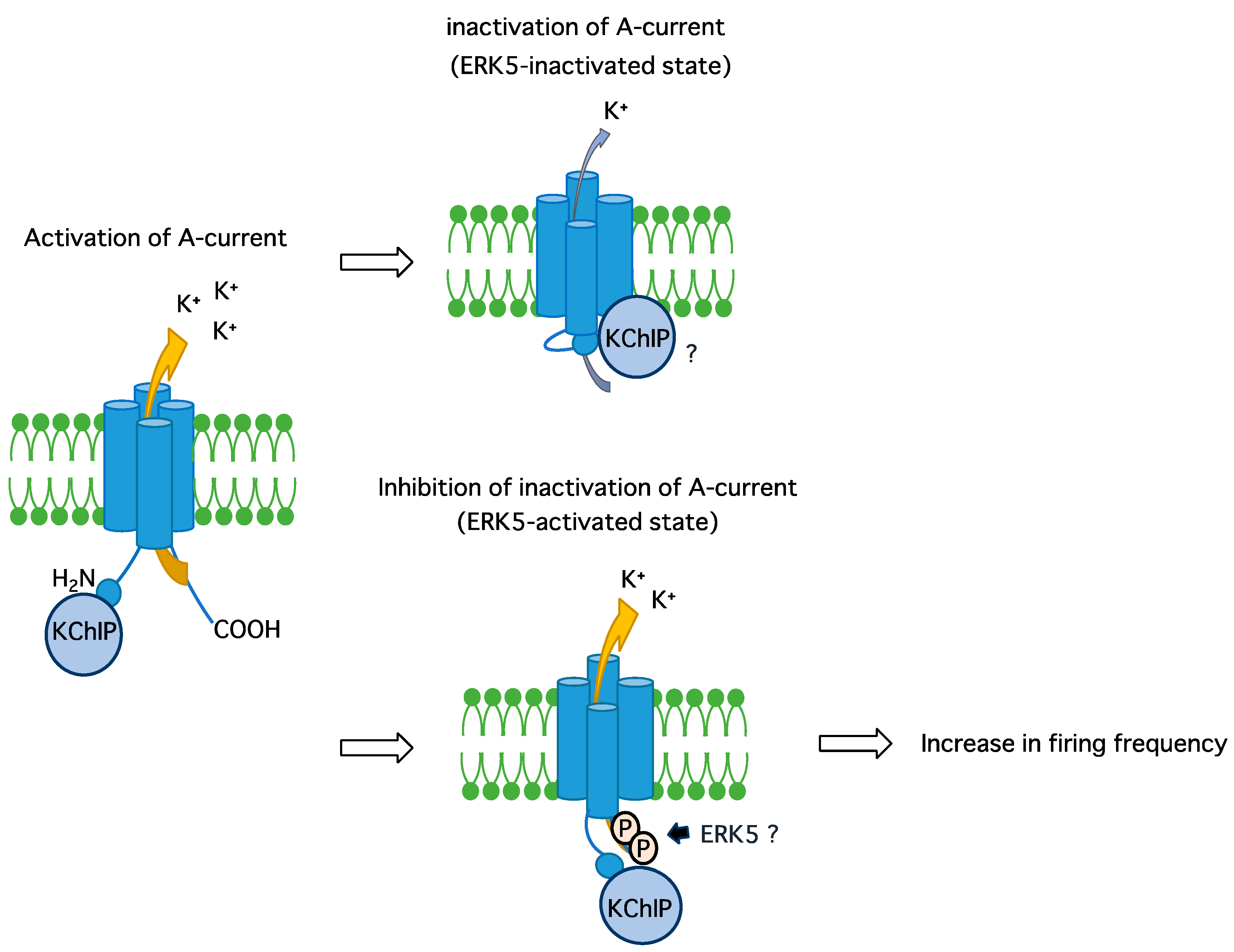
Abstract
:1. Introduction
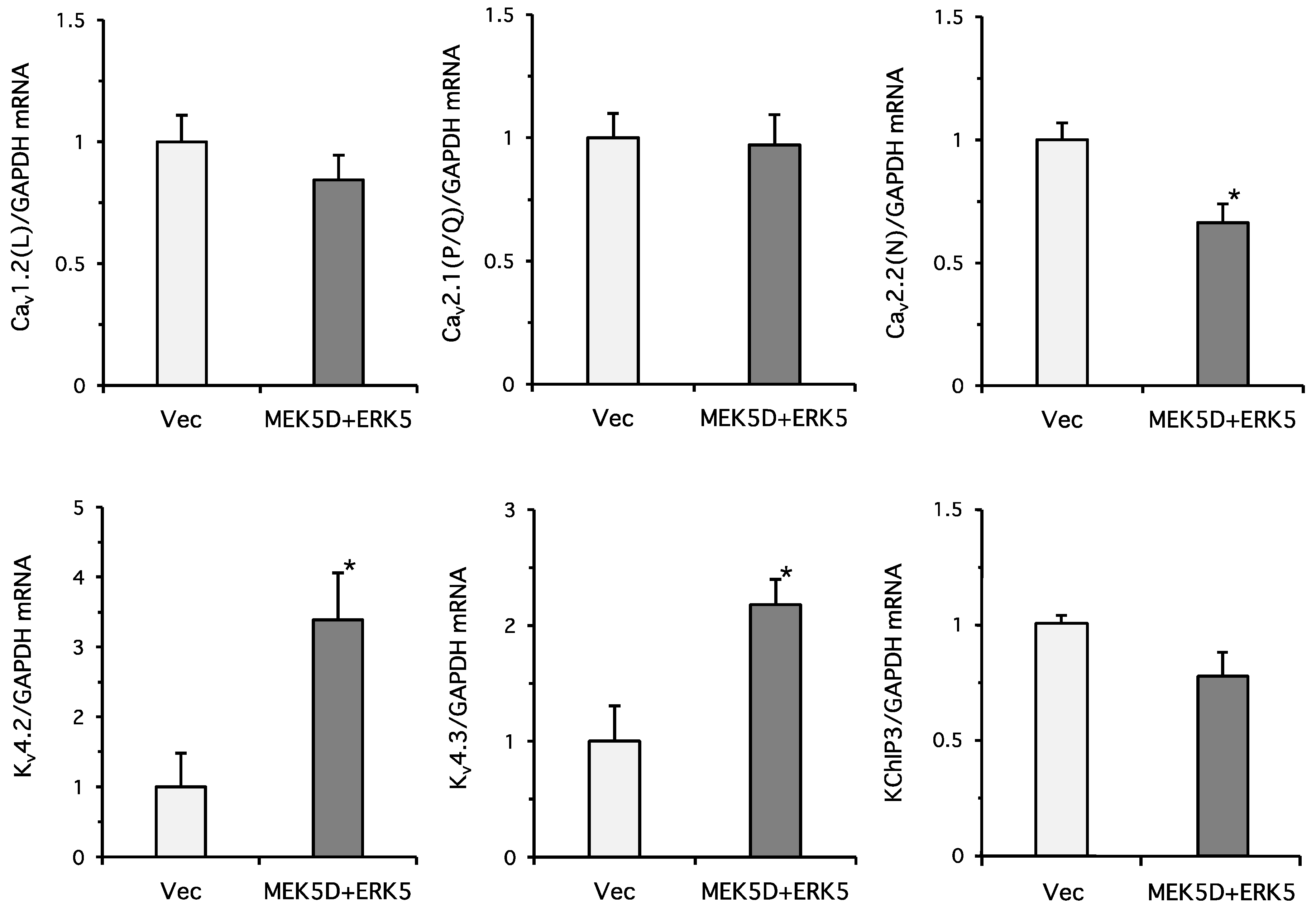
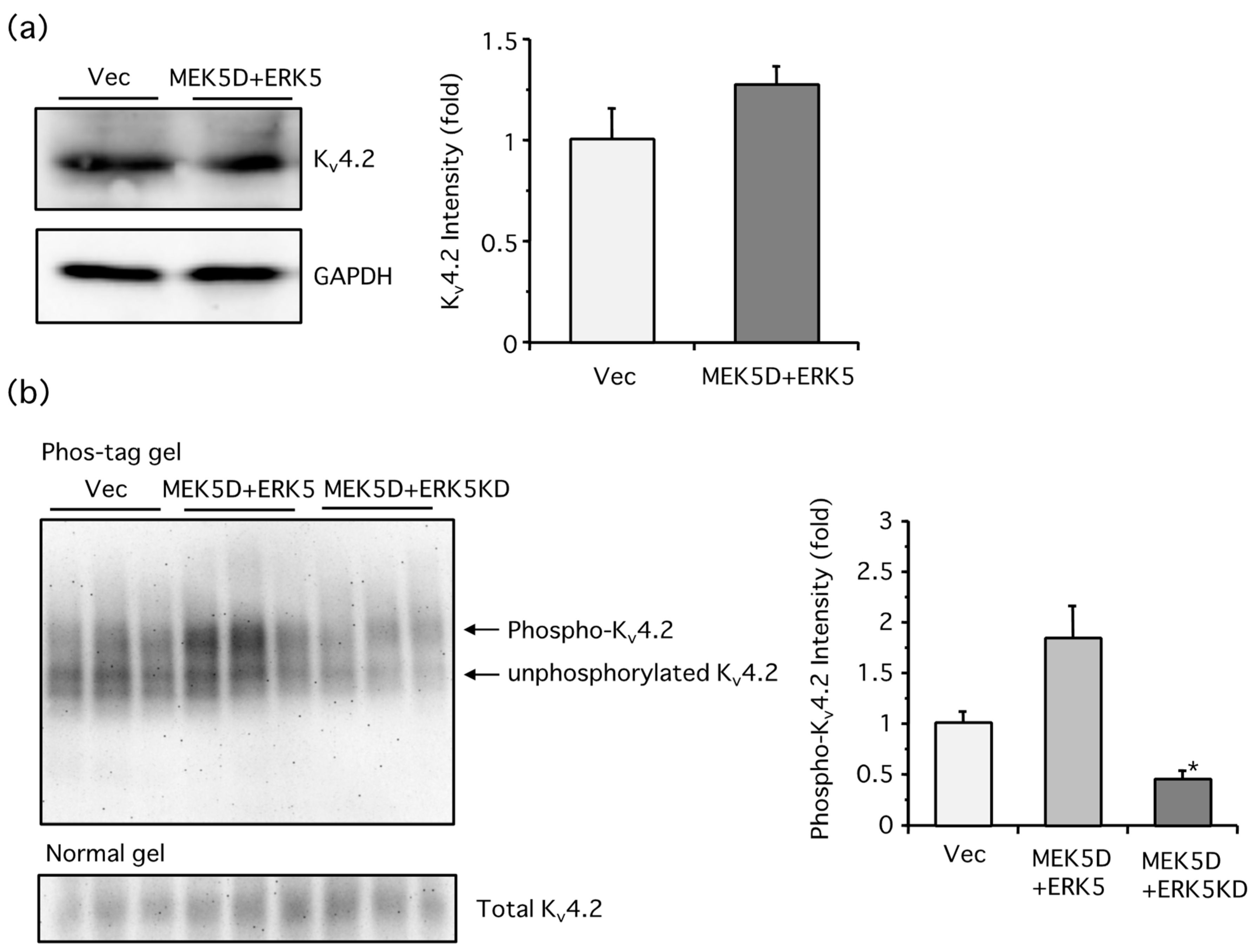
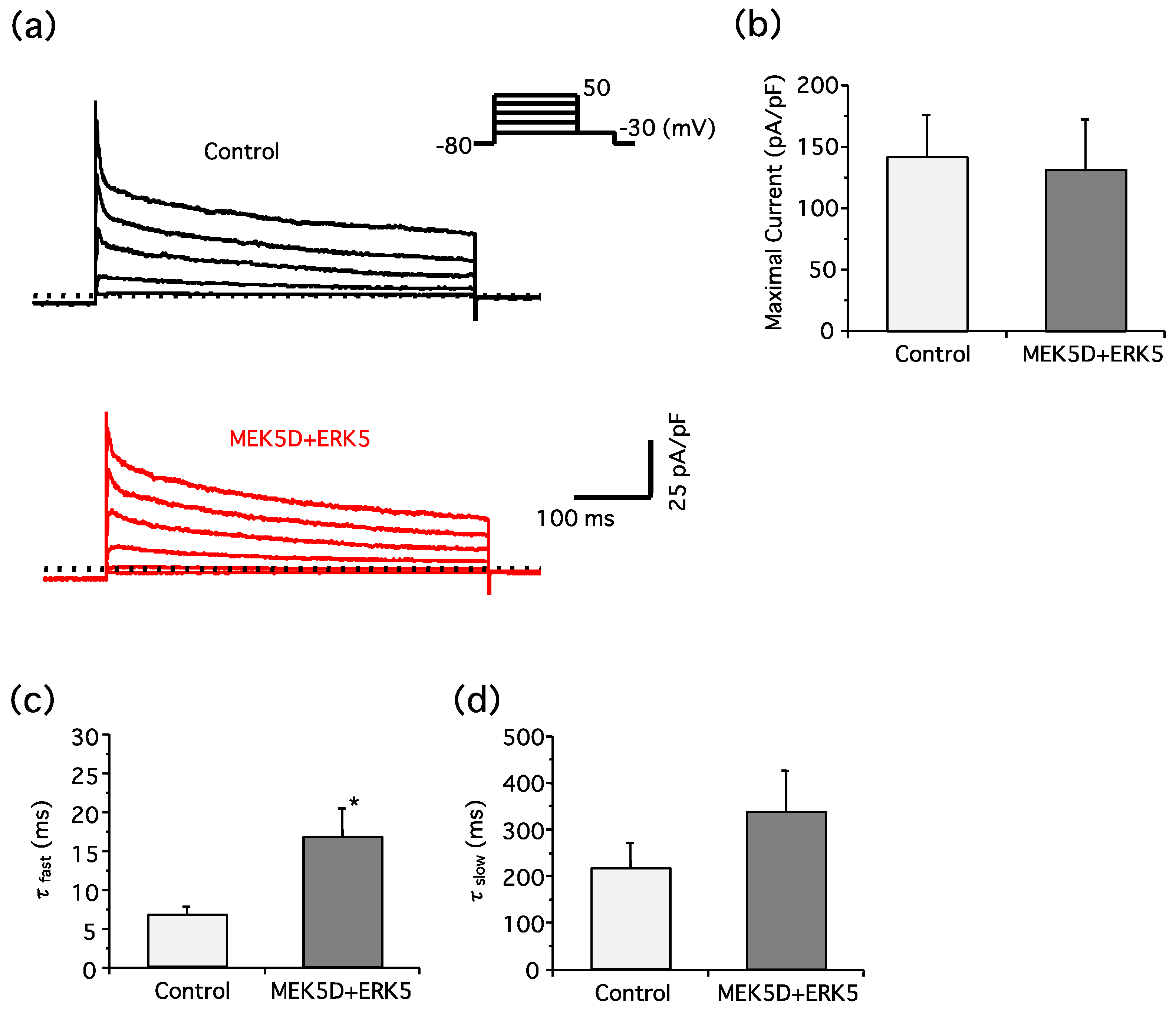
2. Results
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MAPK | Mitogen-activated protein kinase |
| ERK | Extracellular signal-regulated kinase |
| MEK | MAPK/ERK kinase |
| MEF | Myocyte-enhancer factor |
| MRE HEK293 cells | MEF2 response elementhuman embryonic kidney 293 cells |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| TRP | Transient receptor potential |
| NFAT | Nuclear factor of activated T cells |
| KChIPCA | K+ channel-interacting proteincornu ammonis |
| PACAP | Pituitary adenylate cyclase-activating polypeptide |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| ECL | Enhanced chemiluminescence |
| EGFP | Enhanced green fluorescent protein |
| DMEM | Dulbecco’s modified Eagle’s medium |
| TBST | Tris-buffered saline containing 0.1% tween-20 |
References
- Coulombe, P.; Meloche, S. Atypical mitogen-activated protein kinase: Structure, regulation and functions. Biochim. Biophys. Acta 2007, 1773, 1376–1387. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.A.; Burow, M.E.; Beckman, B.S. MEK5/ERK5 pathway: The first fifteen years. Biochim. Biophys. Acta 2012, 1825, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Tournier, C. Regulation of cellular functions by the ERK5 signalling pathway. Cell. Signal. 2006, 18, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Tatake, R.J.; O’Neill, M.M.; Kennedy, C.A.; Wayne, A.L.; Jakes, S.; Wu, D.; Kugler, S.Z., Jr.; Kashem, M.A.; Kaplita, P.; Snow, R.J. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem. Biophys. Res. Commun. 2008, 377, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Obara, Y.; Yamauchi, A.; Takehara, S.; Nemoto, W.; Takahashi, M.; Stork, P.J.; Nakahata, N. ERK5 Activity Is Required for Nerve Growth Factor-induced Neurite Outgrowth and Stabilization of Tyrosine Hydroxylase in PC12 Cells. J. Biol. Chem. 2009, 284, 23564–23573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Deng, X.; Lu, B.; Cameron, M.; Fearns, C.; Patricelli, M.P.; Yates, J.R., 3rd; Gray, N.S.; Lee, J.D. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell. 2010, 18, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Hoang, V.T.; Yan, T.J.; Cavanaugh, J.E.; Flaherty, P.T.; Beckman, B.S.; Burow, M.E. MEK5-ERK5 signaling in cancer: Implications for targeted therapy. Cancer Lett. 2017, 392, 51. [Google Scholar] [CrossRef] [PubMed]
- Simoes, A.E.; Rodrigues, C.M.; Borralho, P.M. The MEK5/ERK5 signalling pathway in cancer: A promising novel therapeutic target. Drug Discov. Today 2016, 21, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Obara, Y.; Nagasawa, R.; Nemoto, W.; Pellegrino, M.J.; Takahashi, M.; Habecker, B.A.; Stork, P.J.; Ichiyanagi, O.; Ito, H.; Tomita, Y.; et al. ERK5 induces ankrd1 for catecholamine biosynthesis and homeostasis in adrenal medullary cells. Cell. Signal. 2016, 28, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Obara, Y.; Nakahata, N. The signaling pathway leading to extracellular signal-regulated kinase 5 (ERK5) activation via G-proteins and ERK5-dependent neurotrophic effects. Mol. Pharmacol. 2010, 77, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Obara, Y.; Labudda, K.; Dillon, T.J.; Stork, P.J. PKA phosphorylation of Src mediates Rap1 activation in NGF and cAMP signaling in PC12 cells. J. Cell. Sci. 2004, 117, 6085–6094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- York, R.D.; Yao, H.; Dillon, T.; Ellig, C.L.; Eckert, S.P.; McCleskey, E.W.; Stork, P.J. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 1998, 392, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cundiff, P.; Abel, G.; Wang, Y.; Faigle, R.; Sakagami, H.; Xu, M.; Xia, Z. Extracellular signal-regulated kinase (ERK) 5 is necessary and sufficient to specify cortical neuronal fate. Proc. Natl. Acad. Sci. USA 2006, 103, 9697–9702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.W.; Chan, G.C.; Kuo, C.T.; Storm, D.R.; Xia, Z. Inhibition of adult neurogenesis by inducible and targeted deletion of ERK5 mitogen-activated protein kinase specifically in adult neurogenic regions impairs contextual fear extinction and remote fear memory. J. Neurosci. 2012, 32, 6444–6455. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.W.; Zou, J.; Wang, W.; Sakagami, H.; Garelick, M.G.; Abel, G.; Kuo, C.T.; Storm, D.R.; Xia, Z. Inducible and conditional deletion of extracellular signal-regulated kinase 5 disrupts adult hippocampal neurogenesis. J. Biol. Chem. 2012, 287, 23306–23317. [Google Scholar] [CrossRef] [PubMed]
- Finegan, K.G.; Wang, X.; Lee, E.J.; Robinson, A.C.; Tournier, C. Regulation of neuronal survival by the extracellular signal-regulated protein kinase 5. Cell. Death Differ. 2009, 16, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, F.L.; Heerssen, H.M.; Bhattacharyya, A.; Klesse, L.; Lin, M.Z.; Segal, R.A. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat. Neurosci. 2001, 4, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.; Muzzio, I.A.; Bourtchouladze, R.; Van-Strien, N.; Lapidus, K.; Yin, D.; Winder, D.G.; Adams, J.P.; Sweatt, J.D.; Kandel, E.R. Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron 2003, 39, 309–325. [Google Scholar] [CrossRef]
- Gupte, R.P.; Kadunganattil, S.; Shepherd, A.J.; Merrill, R.; Planer, W.; Bruchas, M.R.; Strack, S.; Mohapatra, D.P. Convergent phosphomodulation of the major neuronal dendritic potassium channel Kv4.2 by pituitary adenylate cyclase-activating polypeptide. Neuropharmacology 2016, 101, 291–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.J.; Carrasquillo, Y.; Karim, F.; Jung, W.E.; Nerbonne, J.M.; Schwarz, T.L.; Gereau, R.W.T. The kv4.2 potassium channel subunit is required for pain plasticity. Neuron 2006, 50, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.J.; Zhao, Q.R.; Liu, D.D.; Chow, C.W.; Mei, Y.A. Neuritin Up-regulates Kv4.2 alpha-Subunit of Potassium Channel Expression and Affects Neuronal Excitability by Regulating the Calcium-Calcineurin-NFATc4 Signaling Pathway. J. Biol. Chem. 2016, 291, 17369–17381. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kravchenko, V.V.; Tapping, R.I.; Han, J.; Ulevitch, R.J.; Lee, J.D. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. Embo. J. 1997, 16, 7054–7066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, T.; Obara, Y.; Yamauchi, A.; Couvillon, A.D.; Mason, J.J.; Ishii, K.; Nakahata, N. Phosphorylation of ERK5 on Thr732 is associated with ERK5 nuclear localization and ERK5-dependent transcription. PLoS ONE 2015, 10, e0117914. [Google Scholar] [CrossRef] [PubMed]
- Schrader, L.A.; Birnbaum, S.G.; Nadin, B.M.; Ren, Y.; Bui, D.; Anderson, A.E.; Sweatt, J.D. ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am. J. Physiol. Cell. Physiol 2006, 290, C852-861. [Google Scholar] [CrossRef] [PubMed]
- Obara, Y.; Ishii, K. Transcriptome Analysis Reveals That Midnolin Regulates mRNA Expression Levels of Multiple Parkinson’s Disease Causative Genes. Biol. Pharm. Bull. 2018, 41, 20–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.P.; Anderson, A.E.; Varga, A.W.; Dineley, K.T.; Cook, R.G.; Pfaffinger, P.J.; Sweatt, J.D. The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J. Neurochem. 2000, 75, 2277–2287. [Google Scholar] [CrossRef] [PubMed]
- Lugo, J.N.; Barnwell, L.F.; Ren, Y.; Lee, W.L.; Johnston, L.D.; Kim, R.; Hrachovy, R.A.; Sweatt, J.D.; Anderson, A.E. Altered phosphorylation and localization of the A-type channel, Kv4.2 in status epilepticus. J. Neurochem. 2008, 106, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Katsura, H.; Obata, K.; Mizushima, T.; Sakurai, J.; Kobayashi, K.; Yamanaka, H.; Dai, Y.; Fukuoka, T.; Sakagami, M.; Noguchi, K. Activation of extracellular signal-regulated protein kinases 5 in primary afferent neurons contributes to heat and cold hyperalgesia after inflammation. J. Neurochem. 2007, 102, 1614–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Takimoto, K. GATA and FOG2 transcription factors differentially regulate the promoter for Kv4.2 K(+) channel gene in cardiac myocytes and PC12 cells. Cardiovasc. Res. 2003, 60, 278–287. [Google Scholar] [CrossRef]
- Gong, N.; Bodi, I.; Zobel, C.; Schwartz, A.; Molkentin, J.D.; Backx, P.H. Calcineurin increases cardiac transient outward K+ currents via transcriptional up-regulation of Kv4.2 channel subunits. J. Biol. Chem. 2006, 281, 38498–38506. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.T.; Xiong, Q.; Graef, I.A.; Crabtree, G.R.; Chow, C.W. Recruitment of the extracellular signal-regulated kinase/ribosomal S6 kinase signaling pathway to the NFATc4 transcription activation complex. Mol. Cell. Biol. 2005, 25, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Rush, M.E.; Rinzel, J. The potassium A-current, low firing rates and rebound excitation in Hodgkin-Huxley models. Bull. Math. Biol. 1995, 57, 899–929. [Google Scholar] [CrossRef] [PubMed]
- Obara, Y.; Imai, T.; Sato, H.; Takeda, Y.; Kato, T.; Ishii, K. Midnolin is a novel regulator of parkin expression and is associated with Parkinson’s Disease. Sci. Rep. 2017, 7, 5885. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Kawamura, K.; Nakamura, Y.; Ono, K. Pathological impact of hyperpolarization-activated chloride current peculiar to rat pulmonary vein cardiomyocytes. J. Mol. Cell. Cardiol. 2014, 66, 53–62. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kashino, Y.; Obara, Y.; Okamoto, Y.; Saneyoshi, T.; Hayashi, Y.; Ishii, K. ERK5 Phosphorylates Kv4.2 and Inhibits Inactivation of the A-Type Current in PC12 Cells. Int. J. Mol. Sci. 2018, 19, 2008. https://doi.org/10.3390/ijms19072008
Kashino Y, Obara Y, Okamoto Y, Saneyoshi T, Hayashi Y, Ishii K. ERK5 Phosphorylates Kv4.2 and Inhibits Inactivation of the A-Type Current in PC12 Cells. International Journal of Molecular Sciences. 2018; 19(7):2008. https://doi.org/10.3390/ijms19072008
Chicago/Turabian StyleKashino, Yurina, Yutaro Obara, Yosuke Okamoto, Takeo Saneyoshi, Yasunori Hayashi, and Kuniaki Ishii. 2018. "ERK5 Phosphorylates Kv4.2 and Inhibits Inactivation of the A-Type Current in PC12 Cells" International Journal of Molecular Sciences 19, no. 7: 2008. https://doi.org/10.3390/ijms19072008