Biochemical Characterization and Structural Modeling of Fused Glucose-6-Phosphate Dehydrogenase-Phosphogluconolactonase from Giardia lamblia

 , , , , , , , and
, , , , , , , and
Abstract
:
1. Introduction
2. Results and Discussion
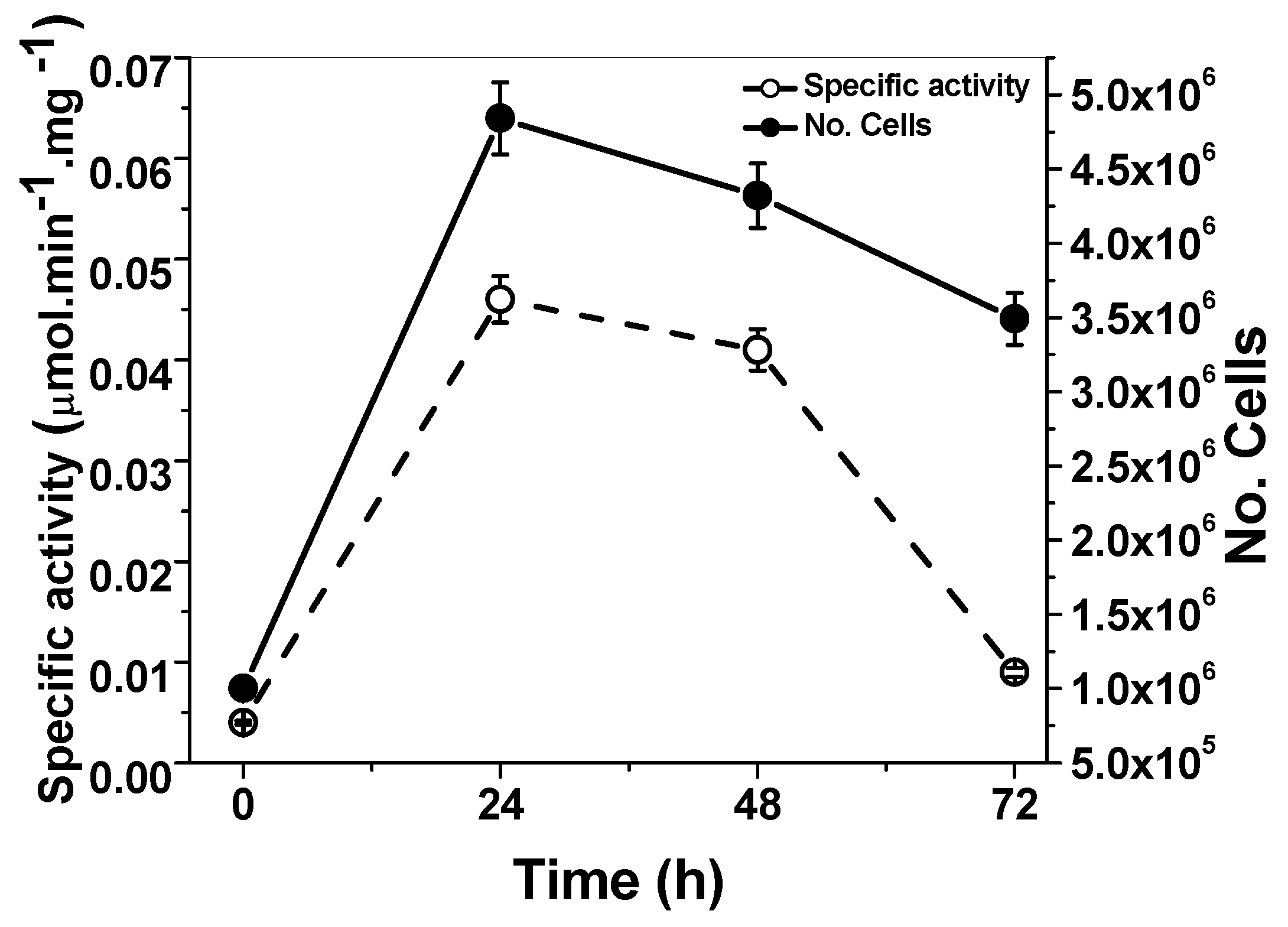
2.1. Quantification of the G6PD Activity from G. lamblia Trophozoites
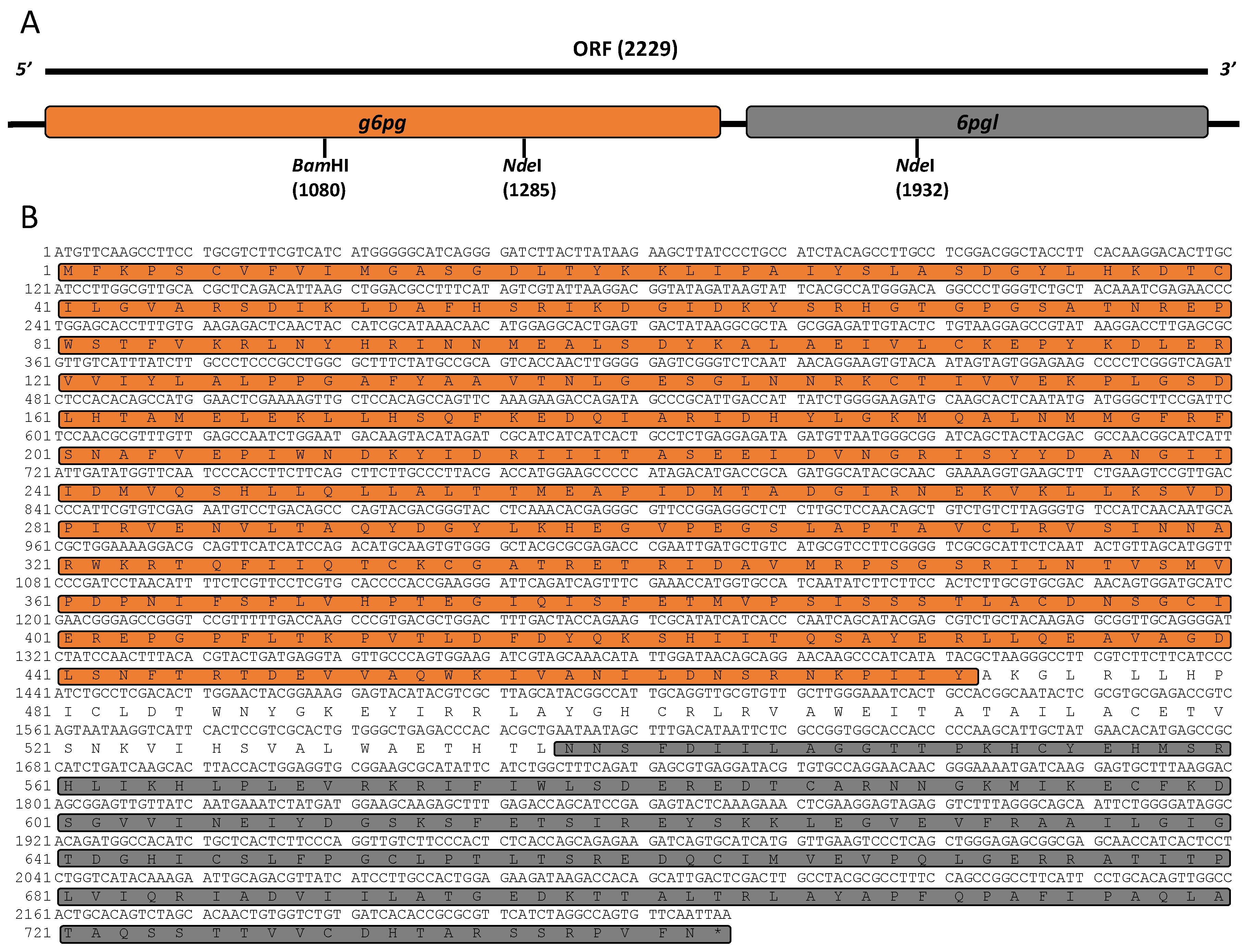
2.2. Isolation, Characterization, and Cloning of g6pd::6pgl cDNA
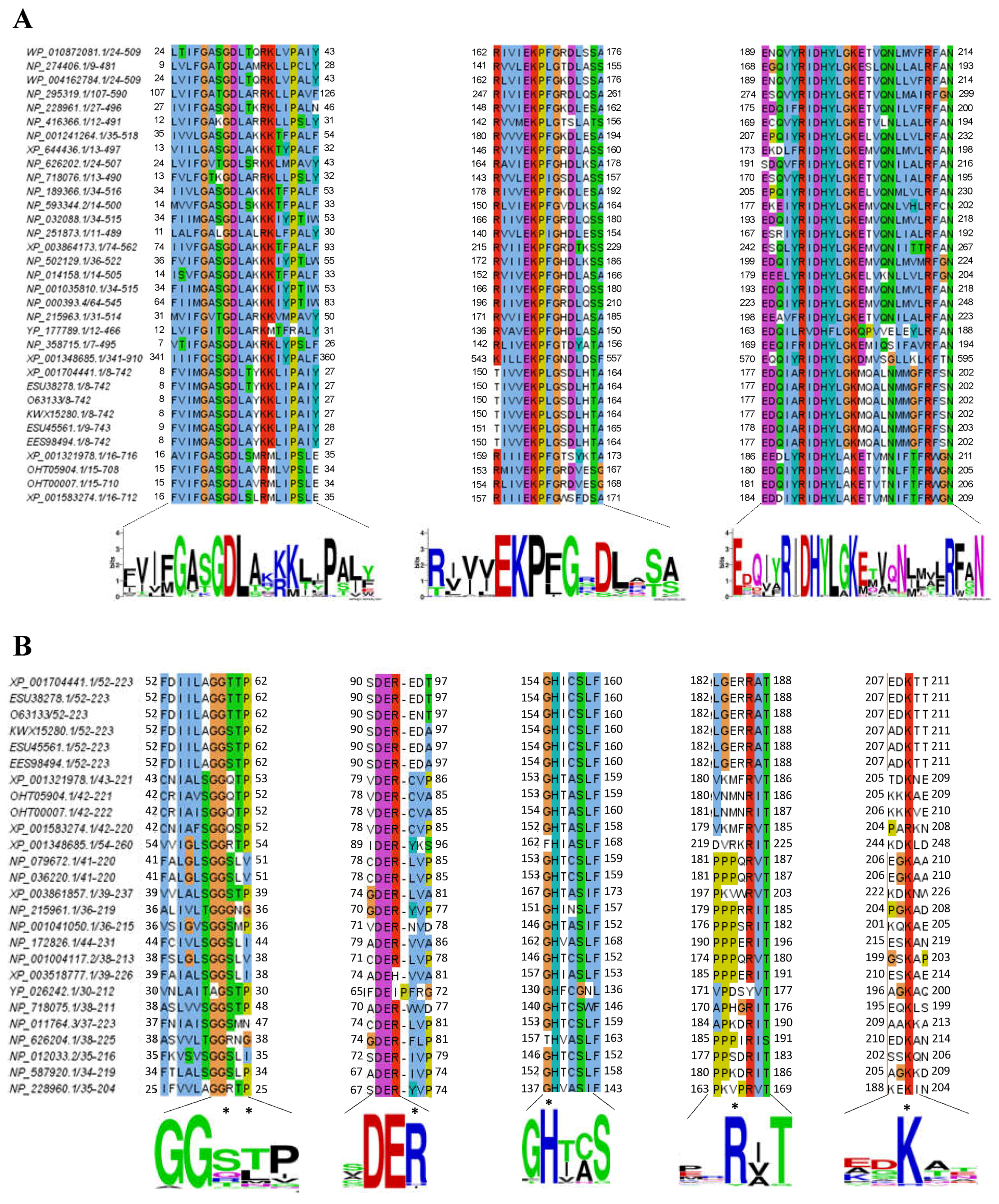
2.3. Alignment of the G6PD::6PGL Protein from G. lamblia
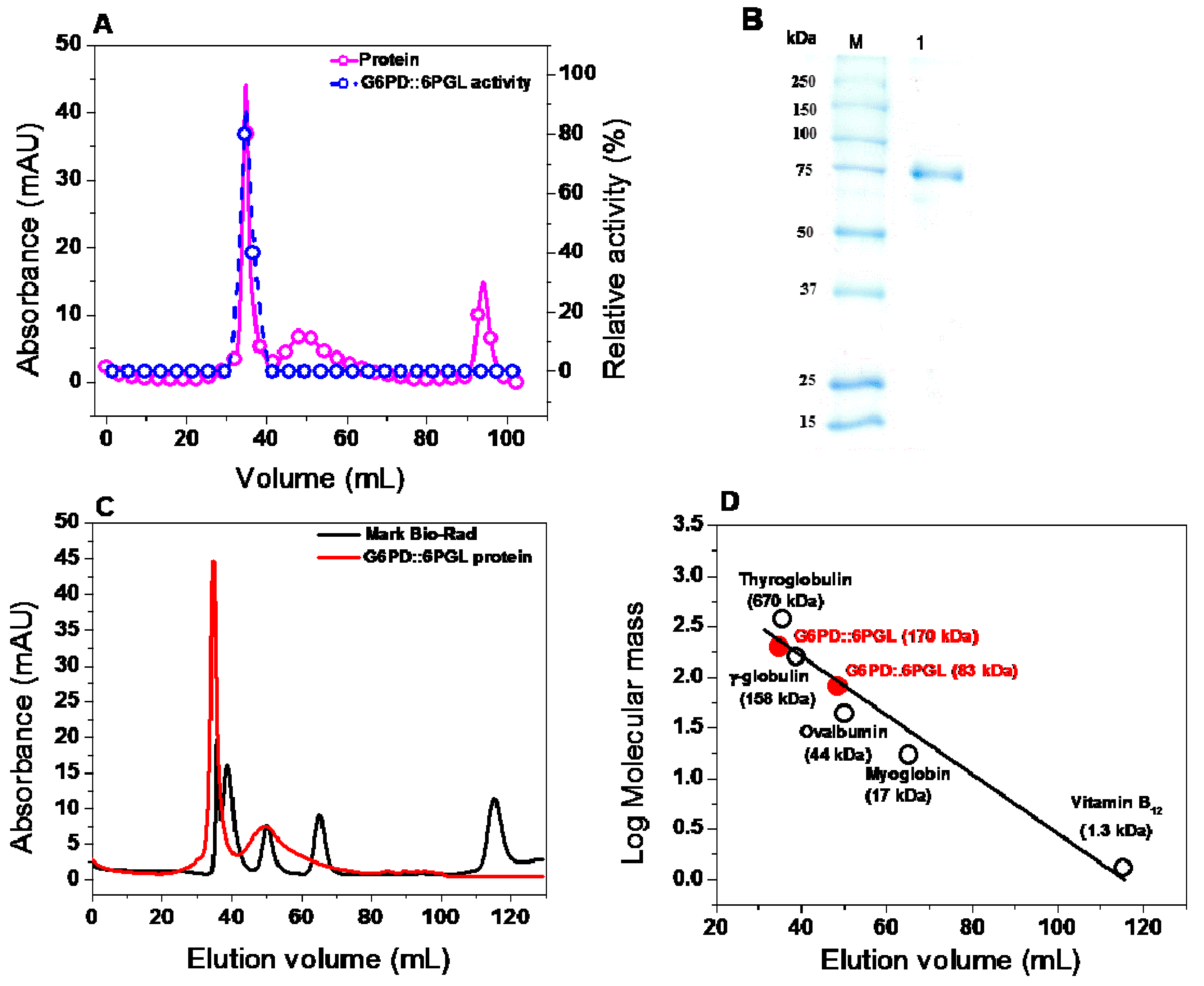
2.4. Expression and Purification of the Recombinant G6PD::6PGL Protein
2.5. Characterization of Functional Properties of Purified G6PD::6PGL Protein
2.5.1. Oligomeric Status of the Recombinant Protein
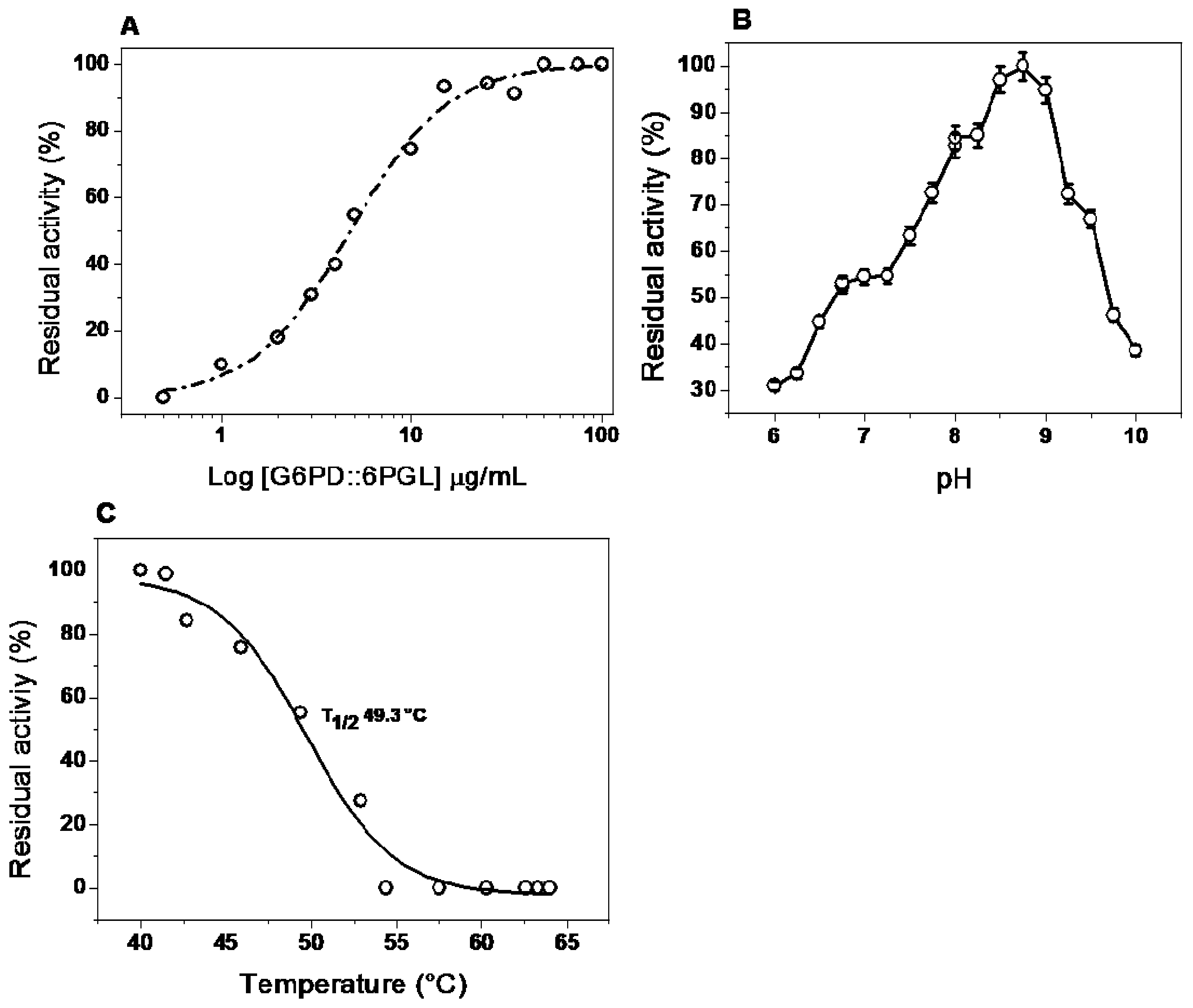
2.5.2. Effect of Dilution and pH on Activity
2.5.3. Effect of Temperature on Activity and Stability
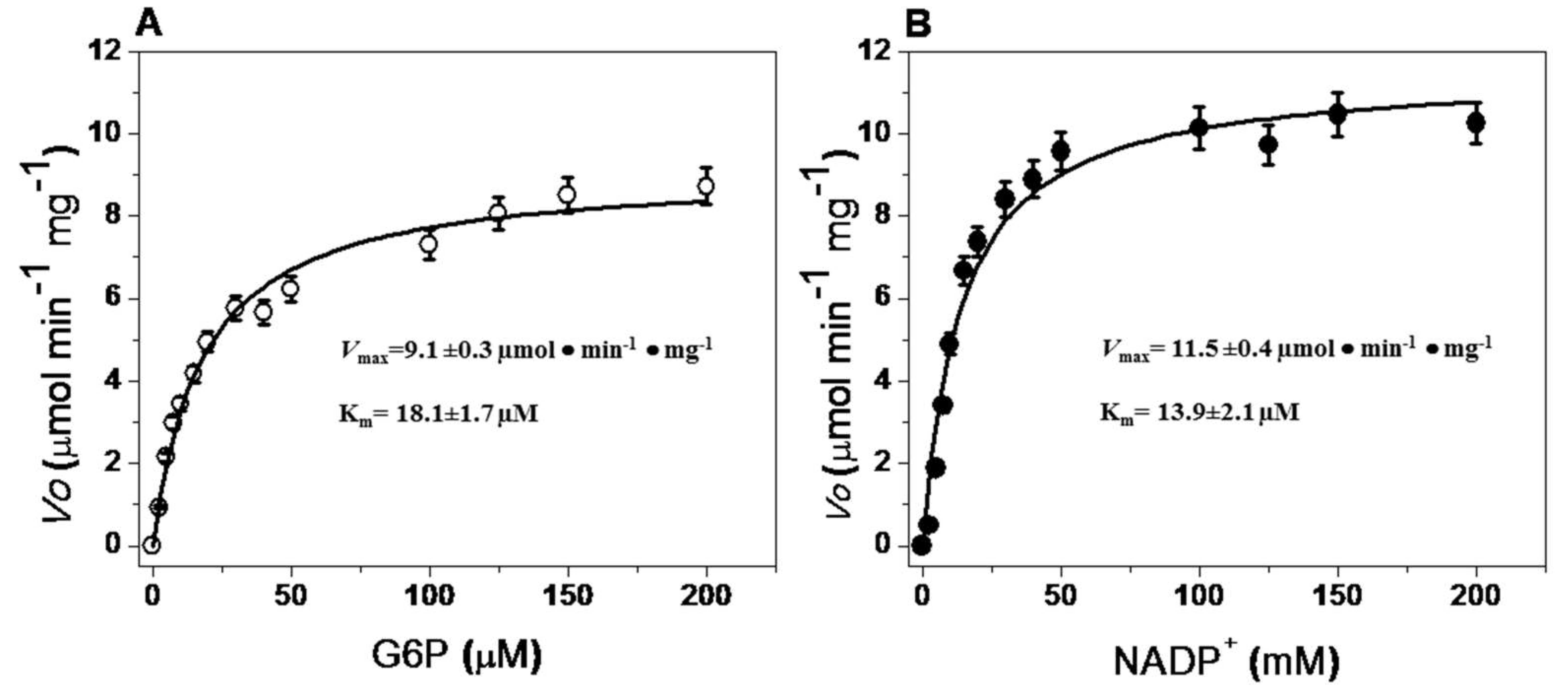
2.5.4. Steady-State Kinetic Parameters
2.6. Evaluation of G6PD::6PGL Protein Stability
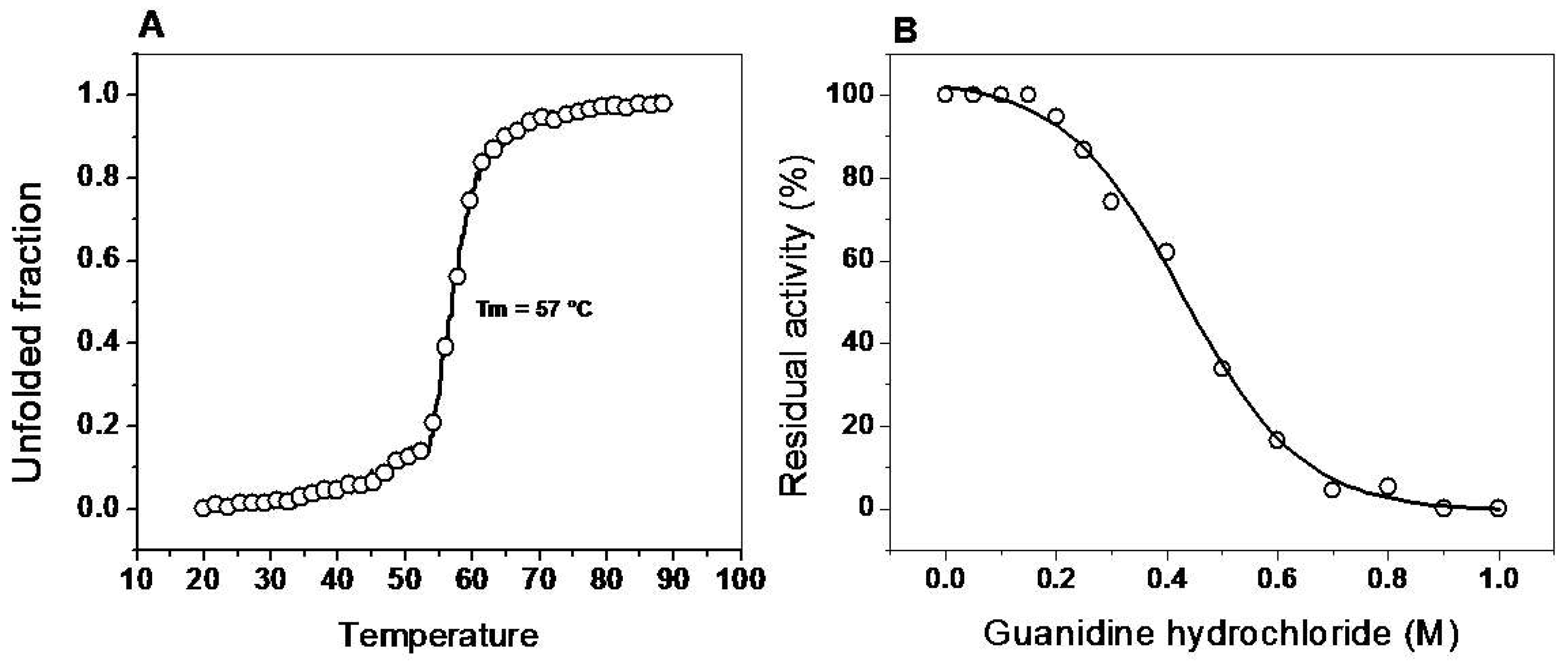
2.6.1. Thermal Stability of the G6PD::6PGL Enzyme
2.6.2. Assay Stability in the Presence of Guanidine Hydrochloride (Gdn-HCl)
2.7. Spectroscopic Characterization of G6PD::6PGL Protein
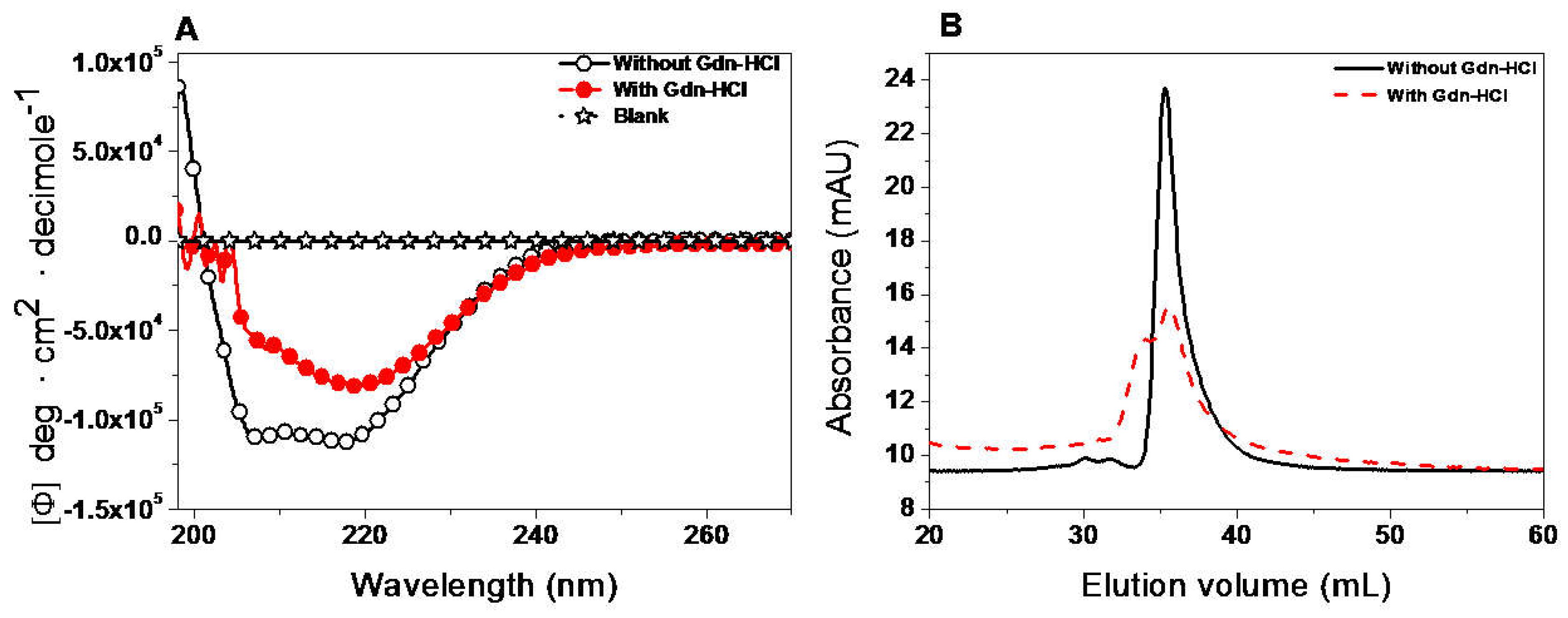
2.7.1. Structural Analysis by Circular Dichroism (CD) and Gel Filtration Column (GFC)
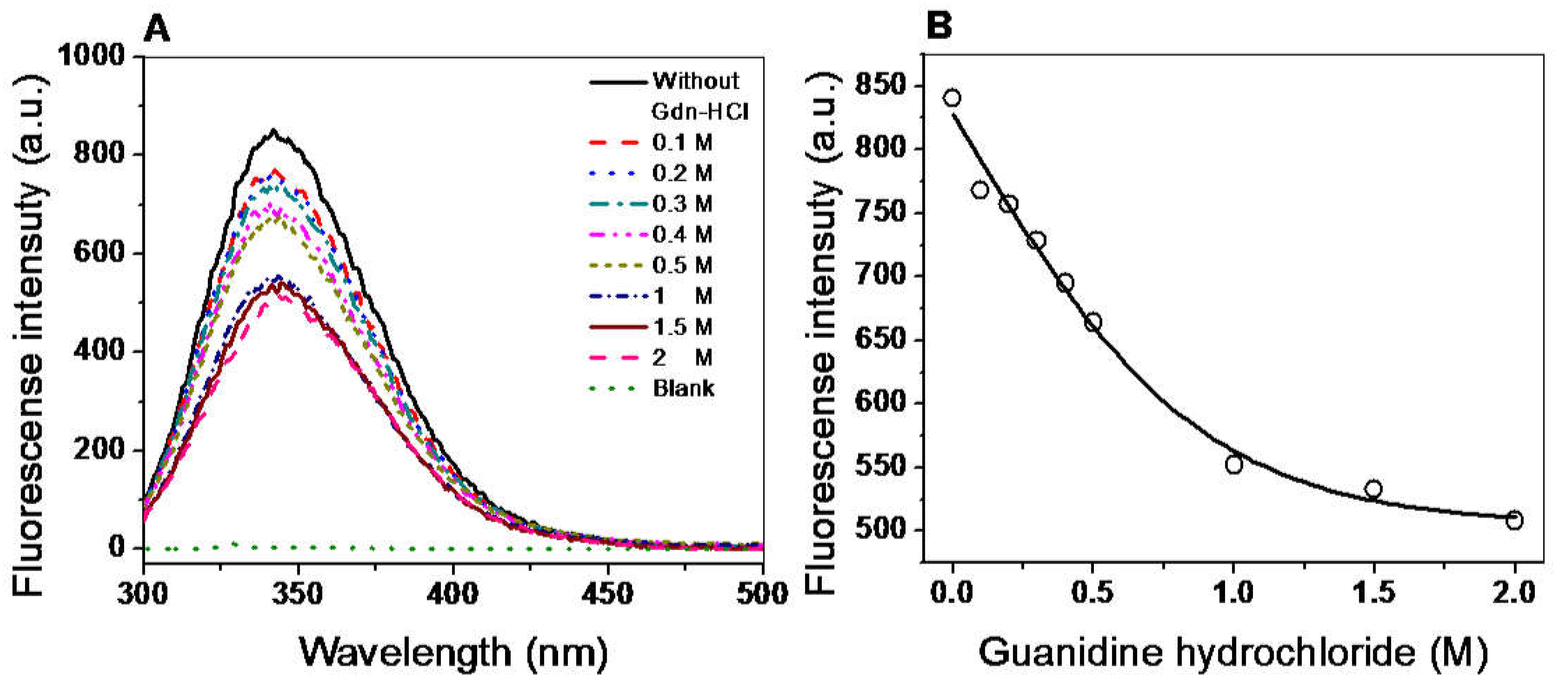
2.7.2. Structural Analysis by Intrinsic Fluorescence
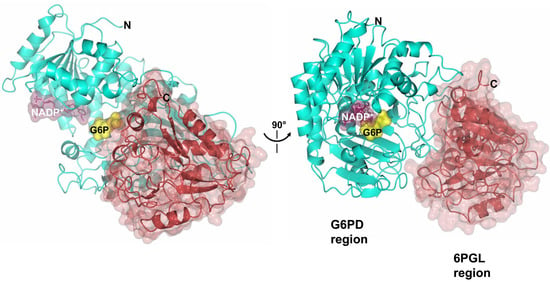
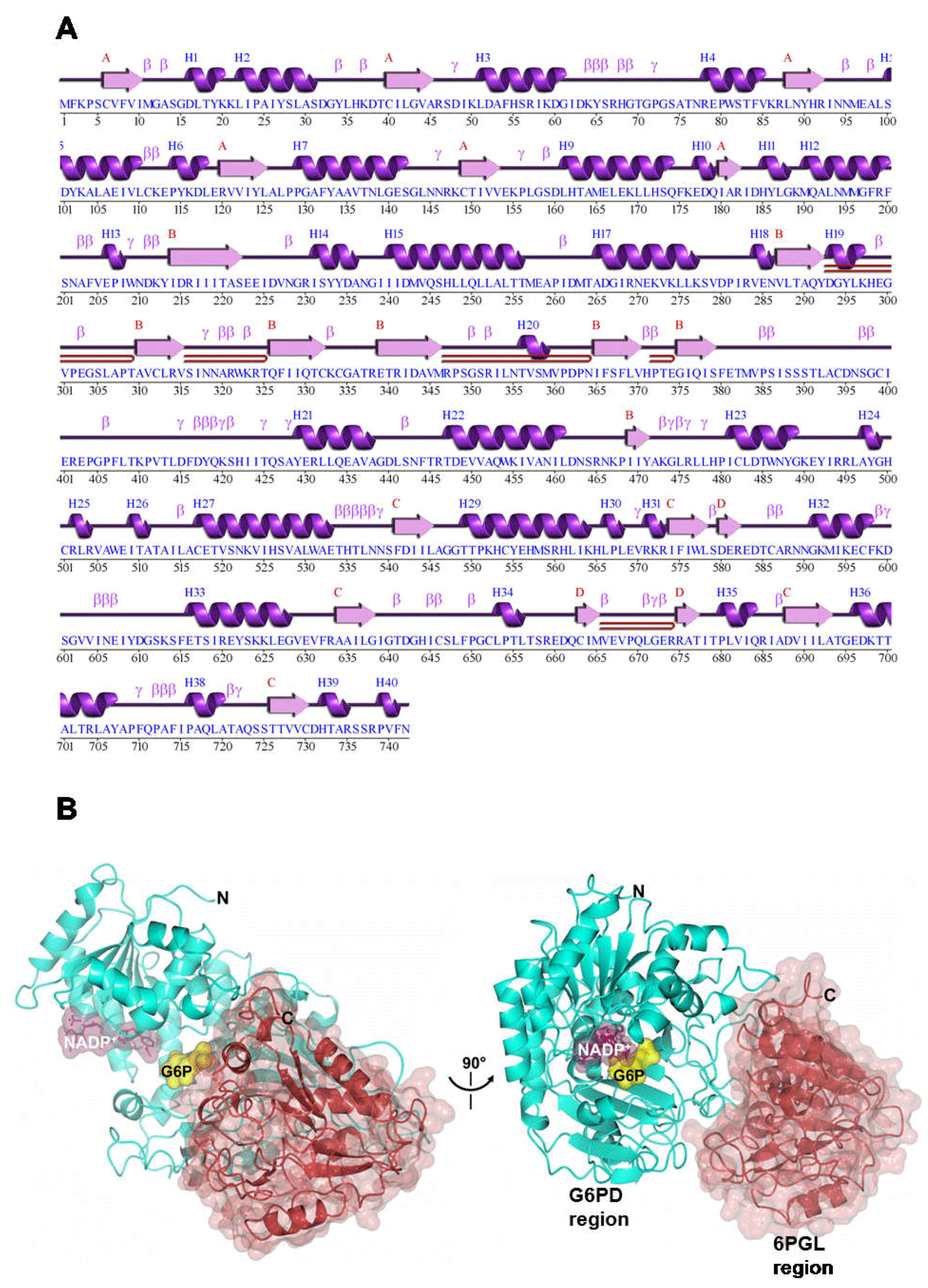
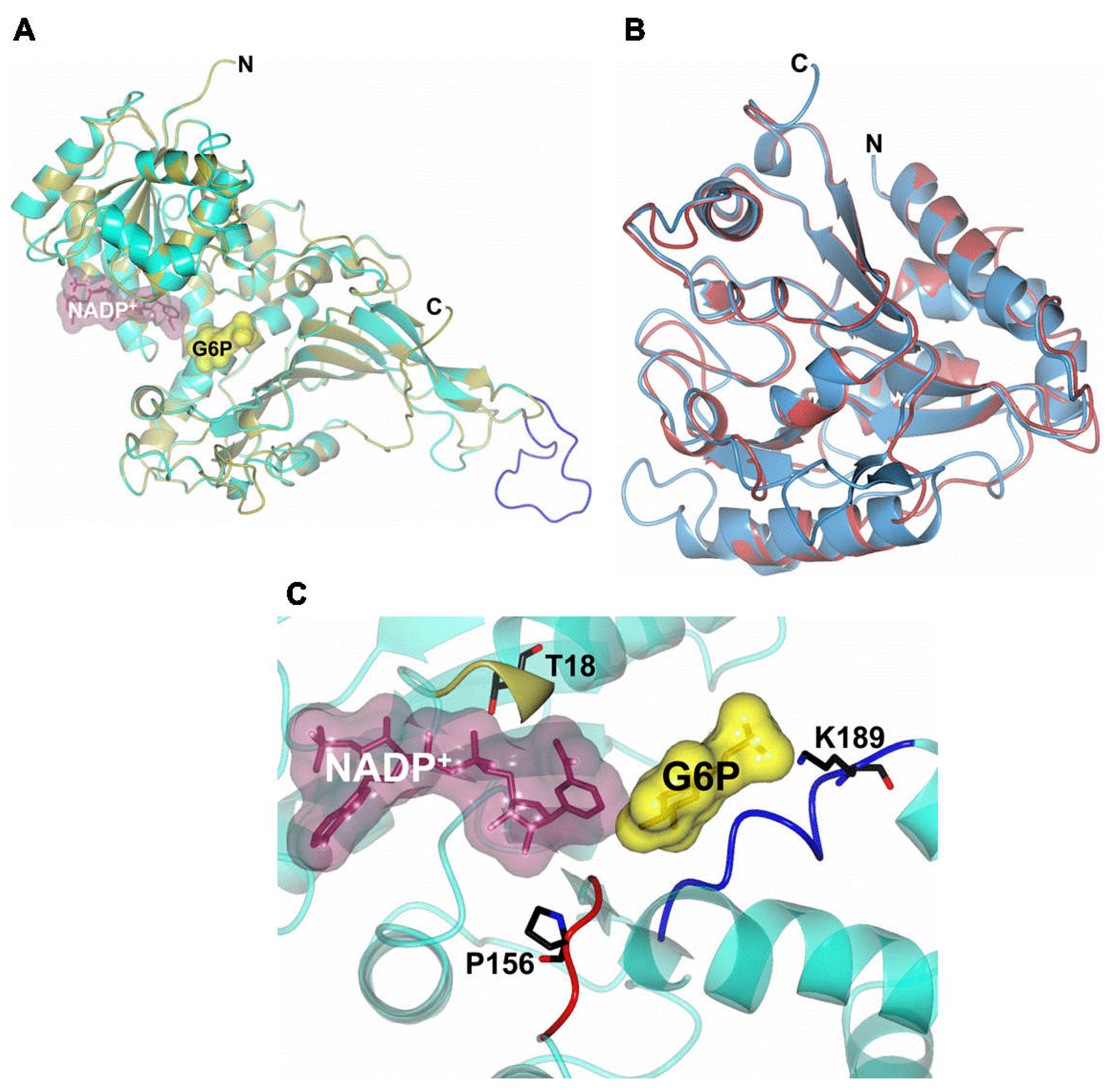
2.8. Homology Modeling of G6PD::6PGL
3. Materials and Methods
3.1. Strain and Experimental Conditions
3.2. Isolation, Characterization, and Cloning of g6pd::6pgl cDNA
3.2.1. RNA Extraction and Synthesis of First Strand cDNA
3.2.2. Primers and Amplification of the g6pd::6pgl gene by PCR
3.2.3. Site-Directed Mutagenesis and Cloning of the g6pd::6pgl Gene
3.3. Alignment of the G6PD::6PGL Protein from G. lamblia
3.4. Expression and Purification of Recombinant G6PD::6PGL Protein
3.5. Characterization of Functional Properties of Purified G6PD::6PGL Protein
3.5.1. Oligomeric Status of the Recombinant Protein
3.5.2. Effect of Dilution and pH on Activity
3.5.3. Effect of Temperature on Activity and Stability
3.5.4. Enzymatic Activity Assay
3.6. Evaluation of G6PD::6PGL Protein Stability
3.6.1. Thermal Stability of Recombinant Protein
3.6.2. Stability of Protein in the Presence of Guanidine Hydrochloride (Gdn-HCl)
3.7. Spectroscopic Characterization of G6PD::6PGL Protein
3.7.1. Structural Analysis by CD and GFC
3.7.2. Structural Analysis by Intrinsic Fluorescence
3.8. Homology Modeling and Comparison of G6PD::6PGL
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plutzer, J.; Ongerth, J.; Karanis, P. Giardia taxonomy, phylogeny and epidemiology: Facts and open questions. Int. J. Hyg. Environ. Health 2010, 213, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Morrison, H.G.; McArthur, A.G.; Gillin, F.D.; Aley, S.B.; Adam, R.D.; Olsen, G.J.; Best, A.A.; Cande, W.Z.; Chen, F.; Cipriano, M.J.; et al. Genomic minimalism in the early diverging intestinal parasite Giardia lamblia. Science 2007, 317, 1921–1926. [Google Scholar] [CrossRef] [PubMed]
- Einarsson, E.; Ma’ayeh, S.; Svärd, S.G. An up-date on Giardia and giardiasis. Curr. Opin. Microbiol. 2016, 34, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Rosado-García, F.M.; Guerrero-Flórez, M.; Karanis, G.; Hinojosa, M.D.C.; Karanis, P. Water-borne protozoa parasites: The Latin American perspective. Int. J. Hyg. Environ. Health 2017, 220, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Monis, P.T.; Caccio, S.M.; Thompson, R.A. Variation in Giardia: Towards a taxonomic revision of the genus. Trends Parasitol. 2009, 25, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xiao, L. Zoonotic potential and molecular epidemiology of Giardia species and giardiasis. Clin. Microbiol. Rev. 2011, 24, 110–140. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, L.A.; Platts-Mills, J.A. Giardia: A pathogen or commensal for children in high-prevalence settings? Curr. Opin. Infect. Dis. 2016, 29, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Waldram, A.; Vivancos, R.; Hartley, C.; Lamden, K. Prevalence of Giardia infection in households of Giardia cases and risk factors for househod transmission. BMC Infect. Dis. 2017, 11, 486. [Google Scholar] [CrossRef]
- Jedelský, P.L.; Doležal, P.; Rada, P.; Pyrih, J.; Smíd, O.; Hrdý, I.; Sedinová, M.; Marcinčiková, M.; Voleman, L.; Perry, A.J.; et al. The minimal proteome in the reduced mitochondrion of the parasitic protist Giardia Intestinalis. PLoS ONE 2011, 6, e17285. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Lam, V.M.S.; Engel, P.C. Functional properties of two mutants of human glucose 6-phosphate dehydrogenase, R393G and R393H, corresponding to the clinical variants G6PD Wisconsin and Nashville. Biochim. Biophys. Acta 2006, 1762, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Fiorelli, G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008, 371, 64–74. [Google Scholar] [CrossRef]
- Winiarska, K.; Drozak, J.; Wegrzynowicz, M.; Jagielski, A.K.; Bryla, J. Relationship between gluconeogenesis and glutathion redox state in rabbit kidney-cortex tubules. Metabolism 2003, 52, 739–746. [Google Scholar] [CrossRef]
- Stover, N.A.; Dixon, T.A.; Cavalcanti, A.R. Multiple Independent Fusions of Glucose-6-Phosphate Dehydrogenase with Enzymes in the Pentose Phosphate Pathway. PLoS ONE 2011, 6, e22269. [Google Scholar] [CrossRef] [PubMed]
- Jortzik, E.; Mailu, B.M.; Preuss, J.; Fischer, M.; Bode, L.; Rahlfs, S.; Becker, K. Glucose-6-phosphate dehydrogenase–6-phosphogluconolactonase: A unique bifunctional enzyme from Plasmodium falciparum. Biochem. J. 2011, 436, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Kotaka, M.; Gover, S.; Vandeputte-Rutten, L.; Au, S.W.N.; Lam, V.M.S.; Adams, M.J. Structural studies of glucose-6-phosphate and NADP+ binding to human glucose-6-phosphate dehydrogenase. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Rowland, P.; Basak, A.K.; Gover, S.; Levy, H.R.; Adams, M.J. The three-dimensional structure of glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides refined at 2.0 Å resolution. Structure 1994, 2, 1073–1087. [Google Scholar] [CrossRef]
- Bautista, J.M.; Mason, P.J.; Luzzatto, L. Human glucose-6-phosphate dehydrogenase: Lysine 205 is dispensable for substrate binding but essential for catalysis. FEBS Lett. 1995, 366, 61–64. [Google Scholar] [CrossRef]
- Camardella, L.; Caruso, C.; Rutigliano, B.; Romano, M.; Di Prisco, G.; Descalzi-Cancedda, F. Human erythrocyte glucose 6-phosphate dehydrogenase. Identification of a reactive lysyl residue labeled with pyridoxal 5’-phosphate. Eur. J. Biochem. 1988, 171, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, M.S.; Gover, S.; Naylor, C.E.; Vandeputte-Rutten, L.; Adams, M.J.; Levy, H.R. An examination of the role of asp-177 in the His-Asp catalytic dyad of Leuconostoc mesenteroides glucose 6-phosphate dehydrogenase: X-ray structure and pH dependence of kinetic parameters of the D177N mutant enzyme. Biochemistry 2000, 39, 15002–15011. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Terrón-Hernández, J.; De la Mora-De la Mora, I.; González-Valdez, A.; Marcial-Quino, J.; García-Torres, I.; Vanoye-Carlo, A.; López-Velázquez, G.; Hernández-Alcantara, G.; Oria-Hernández, J.; et al. The stability of G6PD is affected by mutations with different clinical phenotypes. Int. J. Mol. Sci. 2014, 15, 21179–21201. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Lam, V.M.; Engel, P.C. Marked decrease in specific activity contributes to disease phenotype in two human glucose-6-phosphate dehydrogenase mutants, G6PDUnion and G6PDAndalus. Hum. Mutat. 2005, 26, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Terrón-Hernández, J.; de la Mora-de la Mora, I.; García-Torres, I.; López-Velázquez, G.; Reyes-Vivas, H.; Oria-Hernández, J. Cloning, expression, purification and characterization of His-tagged human glucose-6-phosphate dehydrogenase: A simplified method for protein yield. Protein J. 2013, 32, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.; Schlichting, B.; Schonheit, P. Glucose-6-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima: Expression of the g6pd gene and characterization of an extremely thermophilic enzyme. FEMS Microbiol. Lett. 2002, 216, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Wennekes, L.M.; Goosen, T.; van den Broek, P.J.; van den Broek, H.W. Purification and characterization of glucose-6-phosphate dehydrogenase from Aspergillus niger and Aspergillus nidulans. J. Gen. Microbiol. 1993, 139, 2793–2800. [Google Scholar] [CrossRef] [PubMed]
- Pickl, A.; Schönheit, P. The oxidative pentose phosphate pathway in the haloarchaeon Haloferax volcanii involves a novel type of glucose-6-phosphate dehydrogenase—The archaeal Zwischenferment. FEBS Lett. 2015, 589, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Mercaldi, G.F. The structure of a Trypanosoma cruzi glucose-6-phosphate dehydrogenase reveals differences from the mammalian enzyme. FEBS Lett. 2016, 590, 2776–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.; Chandra, S.; Suthar, M.K.; Doharey, P.K.; Siddiqi, M.I.; Saxena, J.K. NADP+ binding effects tryptophan accessibility, folding and stability of recombinant B. malayi G6PD. Int. J. Biol. Macromol. 2016, 85, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Ghazy, A.H.; Salem, A.M.; Ghazy, M.A.; Abdel-Monsef, M.M. Biochemical characterization of buffalo liver glucose-6-phosphate dehydrogenase isoforms. Protein J. 2015, 34, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Ghazy, A.M.; Salem, A.M.H.; Ghazy, M.A.; Abdel-Monsef, M.M. Purification and characterization of glucose-6-phosphate dehydrogenase from camel liver. Enzym. Res. 2014, 2014, 714054. [Google Scholar] [CrossRef] [PubMed]
- Özer, N.; Bilgi, C.; Ögüsa, I.H. Dog liver glucose-6-phosphate dehydrogenase: Purification and kinetic properties. Int. J. Biochem. Cell Biol. 2002, 34, 253–262. [Google Scholar] [CrossRef]
- Ninfali, P.; Malatesta, M.; Biagiotti, E.; Aluigi, G.; Gazzanelli, G. Glucose-6-phosphate dehydrogenase in small intestine of rabit: Biochemical properties and subcellular localization. Acta Histochem. 2001, 103, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Boonyuen, U.; Chamchoy, K.; Swangsri, T.; Saralamba, T.; Day, N.P.J.; Imwong, M. Detailed functional analysis of two clinical glucose-6-phosphate dehydrogenase (G6PD) variants, G6PDViangchan and G6PDViangchan + Mahidol: Decreased stability and catalytic efficiency contribute to the clinical phenotype. Mol. Genet. Metab. 2016, 2, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Boonyuen, U.; Chamchoy, K.; Swangsri, T.; Junkree, T.; Day, M.; White, N.; Imwong, M. A trade-off between catalytic activity and protein stability determines the clinical manifestations of glucose-6-phosphate dehydrogenase (G6PD) deficiency. Int. J. Biol. Macromol. 2017, 104, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Serrano-Posada, H.; Ortega-Cuellar, D.; González-Valdez, A.; Castillo-Rodríguez, R.A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Rodríguez-Bustamante, E.; et al. Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World. Int. J. Mol. Sci. 2016, 17, 2069. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Painter, J.; Merrit, E.A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, T.L.; Merritt, E.A. 6-Phosphogluconolactonase from Leishmania braziliensis. Structural Genomics of Pathogenic Protozoa Consortium (SGPP). 2008. Available online: https://www.rcsb.org/structure/3ch7 (accessed on 10 April 2008).
- Duclert-Savatier, N.; Poggi, L.; Miclet, E.; Lopes, P.; Ouazzani, J.; Chevalier, N.; Nilges, M.; Delarue, M.; Stoven, V. Insights into the enzymatic mechanism of 6-phosphogluconolactonase from Trypanosoma brucei using structural data and molecular dynamics simulation. J. Mol. Biol. 2009, 388, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Baugh, L.; Phan, I.; Begley, D.W.; Clifton, M.C.; Armour, B.; Dranow, D.M.; Taylor, B.M.; Muruthi, M.M.; Abendroth, J.; Fairman, J.W.; et al. Increasing the structural coverage of tuberculosis drug targets. Tuberculosis 2015, 95, 142–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collard, F.; Collet, J.F.; Gerin, I.; Veiga-da-Cunha, M.; Van Schaftingen, E. Identification of the cDNA encoding human 6-phosphogluconolactonase, the enzyme catalyzing the second step of the pentose phosphate pathway. FEBS Lett. 1999, 459, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Alencar, N.; Sola, I.; Linares, M.; Juarez-Jiménez, J.; Pont, C.; Viayna, A.; Vílchez, D.; Sampedro, D.; Abad, P.; Perez-Benavente, S.; et al. First homology model of Plasmodium falciparum glucose-6-phosphate dehydrogenase: Discovery of selective substrate analog-based inhibitors as novel antimalarial agents a European J. Med. Chem. 2018, 146, 108–122. [Google Scholar] [CrossRef] [PubMed]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Carlton, J.M.; Dommer, J.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; et al. GiardiaDB and TrichDB: Integrated genomic resources for the eukaryotic protist pathogens Giardia lamblia and Trichomonas vaginalis. Nucleic Acids Res. 2009, 37, D526–D530. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Enríquez-Flores, S.; De la Mora-De la Mora, I.; González-Valdez, A.; García-Torres, I.; Martínez-Rosas, V.; Sierra-Palacios, E.; Lazcano-Pérez, F.; et al. Mutations of glucose-6-phosphate dehydrogenase Durham, Santa-Maria and A+ variants are associated with loss functional and structural stability of the protein. Int. J. Mol. Sci. 2015, 16, 28657–28668. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Manzo, S.; Marcial-Quino, J.; Ortega-Cuellar, D.; Serrano-Posada, H.; González-Valdez, H.; Vanoye-Carlo, A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Castillo-Villanueva, A.; Reyes-Vivas, H. Functional and Biochemical Analysis of Glucose-6-Phosphate Dehydrogenase (G6PD) Variants: Elucidating the Molecular Basis of G6PD Deficiency. Catalysts 2017, 7, 135. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modelling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- De Beer, T.A.; Berka, K.; Thornton, J.M.; Laskowski, R.A. PDBsum additions. Nucleic Acids Res. 2014, 42, D292–D296. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Total Protein (mg) | Specific Activity (IU·mg−1) | Total Activity (IU) | Yield (%) |
|---|---|---|---|---|
| Crude extract | 432.6 | 0.68 | 297.38 | 100 |
| 2′,5′-ADP Sepharose 4B | 214.4 | 0.73 | 157.28 | 52 |
| Sephacryl 100 | 3.25 | 11.51 | 37.4 | 13 |
| G6PD from Organism | Km G6P (mM) | Km NADP+ (mM) | Vmax (µmol·mg−1·mg −1) | kcat (s−1) | Reference |
|---|---|---|---|---|---|
| G. lamblia | 0.0181±0.0017 | 0.0139 ± 0.0021 | 11.51 ± 0.45 | 31.84 ± 1.5 | This study |
| P. falciparum | 0.019 ± 0.003 | 0.006 ± 0.002 | 5.2 ± 1.6 | 8.6 ± 1.5 | [14] |
| Thermotoga maritima | 0.2 | 0.04 | 20 | 3.5 × 104 | [23] |
| Aspergillus niger | 0.153 ± 0.0010 | 0.026 ± 0.008 | 790 | NR | [24] |
| Haloferax volcanii | 3.7 | 5.2 | 11 | NR | [25] |
| Aspergillus nidulans | 0.092 ± 0.0010 | 0.03 ± 0.008 | 745 | NR | [24] |
| Trypanosoma cruzi | 0.306 ± 0.02 | 0.080 ± 0.05 | NR | 53.6 ± 1 | [26] |
| Brugia malagy | 0.245 ± 0.008 | 0.014 ± 0.0003 | 0.535 | 40 | [27] |
| Liver of buffalo 1 | NR | 0.059 | 6.91 | NR | [28] |
| Liver of buffalo 2 | NR | 0.006 | 8.90 | NR | [28] |
| Camel liver | 0.081 | 0.081 | 1.875 | NR | [29] |
| Dog liver | 0.122 ± 0.18 | 0.010 ± 0.001 | 130 | NR | [30] |
| Rabbit intestine | 0.030 | 0.036 ± 0.008 | NR | NR | [31] |
| Primer | Sequence |
|---|---|
| G6PD Forward | 5′-GCATCATATGTTCAAGCCTTCCTGC-3′ |
| G6PD Reverse | 5′-CTGGGGATCCTTAATTGAACACTGG-3′ |
| BamHI (1083) Forward | 5′-GTTAGCATGGTTCCCGATCCTAACATTT-3′ |
| BamHI (1083) Forward | 5′-AAATGTTAGGATCGGGAACCATGCTAAC-3′ |
| NdeI (1284) Forward | 5′-ACCCAATCAGCATACGAGCGTCTGCTAC-3′ |
| NdeI (1284) Reverse | 5′-GTAGCAGACGCTCGTATGCTGATTGGGT-3′ |
| NdeI (1933) Forward | 5′-ACAGATGGCCACATCTGCTCACTCTTCC-3′ |
| NdeI (1933) Reverse | 5′-GGAAGAGTGAGCAGATGTGGCCATCTGT-3′ |
| pJET Forward | 5′-CGACTCACTATAGGGAGAGCGGC-3′ |
| pJET Reverse | 5′-AAGAACATCGATTTTCCATGGCAG-3′ |
| T7 promoter Forward | 5′-TAATACGACTCACTATAGGG-3′ |
| T7 terminator Reverse | 5′-GCTAGTTATTGCTCAGCGG-3′ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Luna, L.; Serrano-Posada, H.; González-Valdez, A.; Ortega-Cuellar, D.; Vanoye-Carlo, A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Rufino-González, Y.; Castillo-Rodríguez, R.A.; Pérez de la Cruz, V.; et al. Biochemical Characterization and Structural Modeling of Fused Glucose-6-Phosphate Dehydrogenase-Phosphogluconolactonase from Giardia lamblia. Int. J. Mol. Sci. 2018, 19, 2518. https://doi.org/10.3390/ijms19092518
Morales-Luna L, Serrano-Posada H, González-Valdez A, Ortega-Cuellar D, Vanoye-Carlo A, Hernández-Ochoa B, Sierra-Palacios E, Rufino-González Y, Castillo-Rodríguez RA, Pérez de la Cruz V, et al. Biochemical Characterization and Structural Modeling of Fused Glucose-6-Phosphate Dehydrogenase-Phosphogluconolactonase from Giardia lamblia. International Journal of Molecular Sciences. 2018; 19(9):2518. https://doi.org/10.3390/ijms19092518
Chicago/Turabian StyleMorales-Luna, Laura, Hugo Serrano-Posada, Abigail González-Valdez, Daniel Ortega-Cuellar, America Vanoye-Carlo, Beatriz Hernández-Ochoa, Edgar Sierra-Palacios, Yadira Rufino-González, Rosa Angélica Castillo-Rodríguez, Verónica Pérez de la Cruz, and et al. 2018. "Biochemical Characterization and Structural Modeling of Fused Glucose-6-Phosphate Dehydrogenase-Phosphogluconolactonase from Giardia lamblia" International Journal of Molecular Sciences 19, no. 9: 2518. https://doi.org/10.3390/ijms19092518