Recent Trends and Applications of Molecular Modeling in GPCR–Ligand Recognition and Structure-Based Drug Design

Abstract
:1. Introduction
2. Using Molecular Modeling in the Investigation of Mechanisms Underlying GPCR-Ligand Recognition
2.1. Decoding the Mechanism of Ligand-GPCR Binding Using MD Simulations
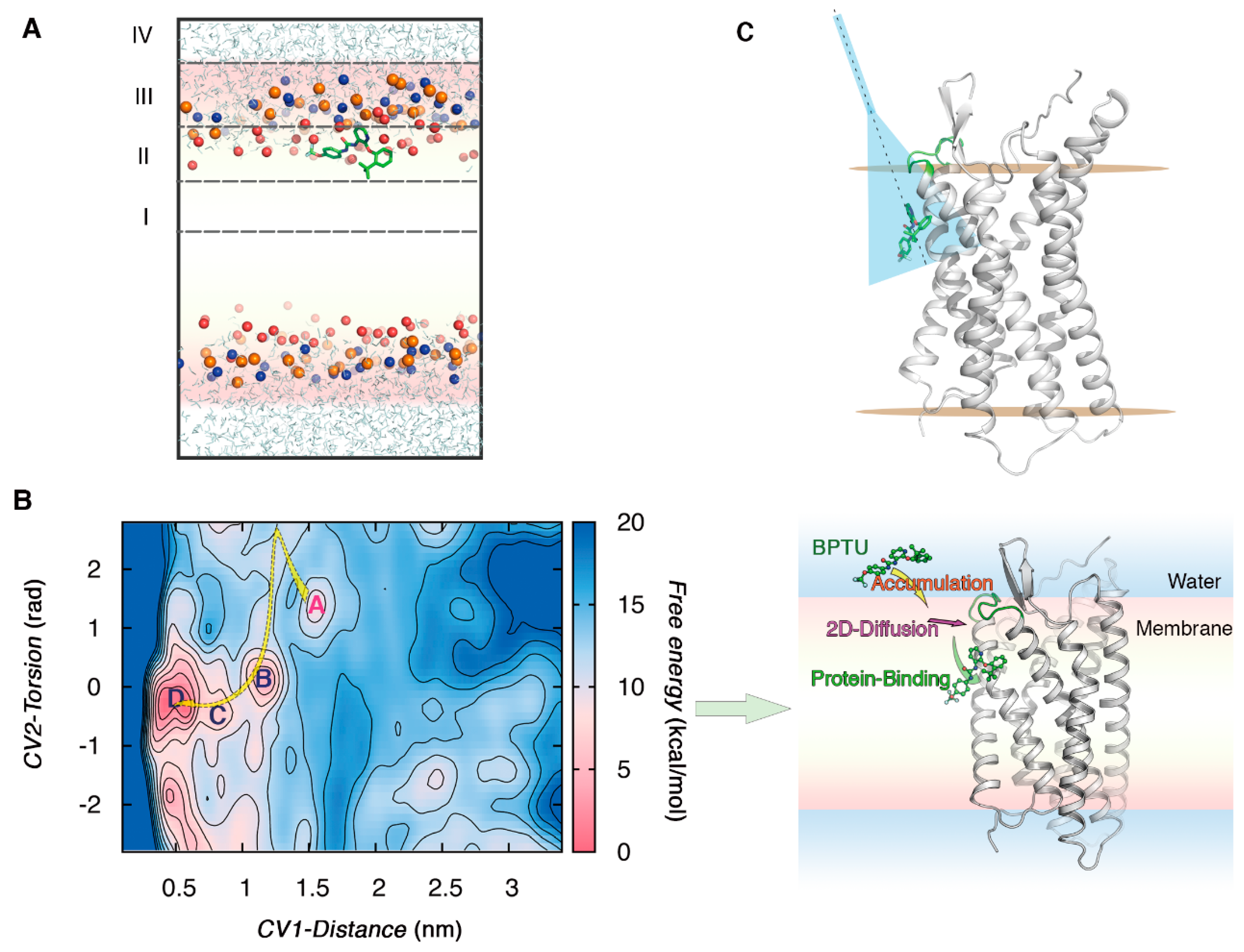
2.2. The Role of the Membrane in Ligand-GPCR Recognition
2.3. Case Study 1: The Mechanism Underlying an Antagonist Binding to the Extra-Helical Site of P2Y1R
3. Applications of Molecular Modeling in Structure-Based Drug Design of GPCRs
3.1. The Most Widely Used Method: Molecular Docking
3.2. The Most Promising Methods: Free-Energy-Calculation Methods
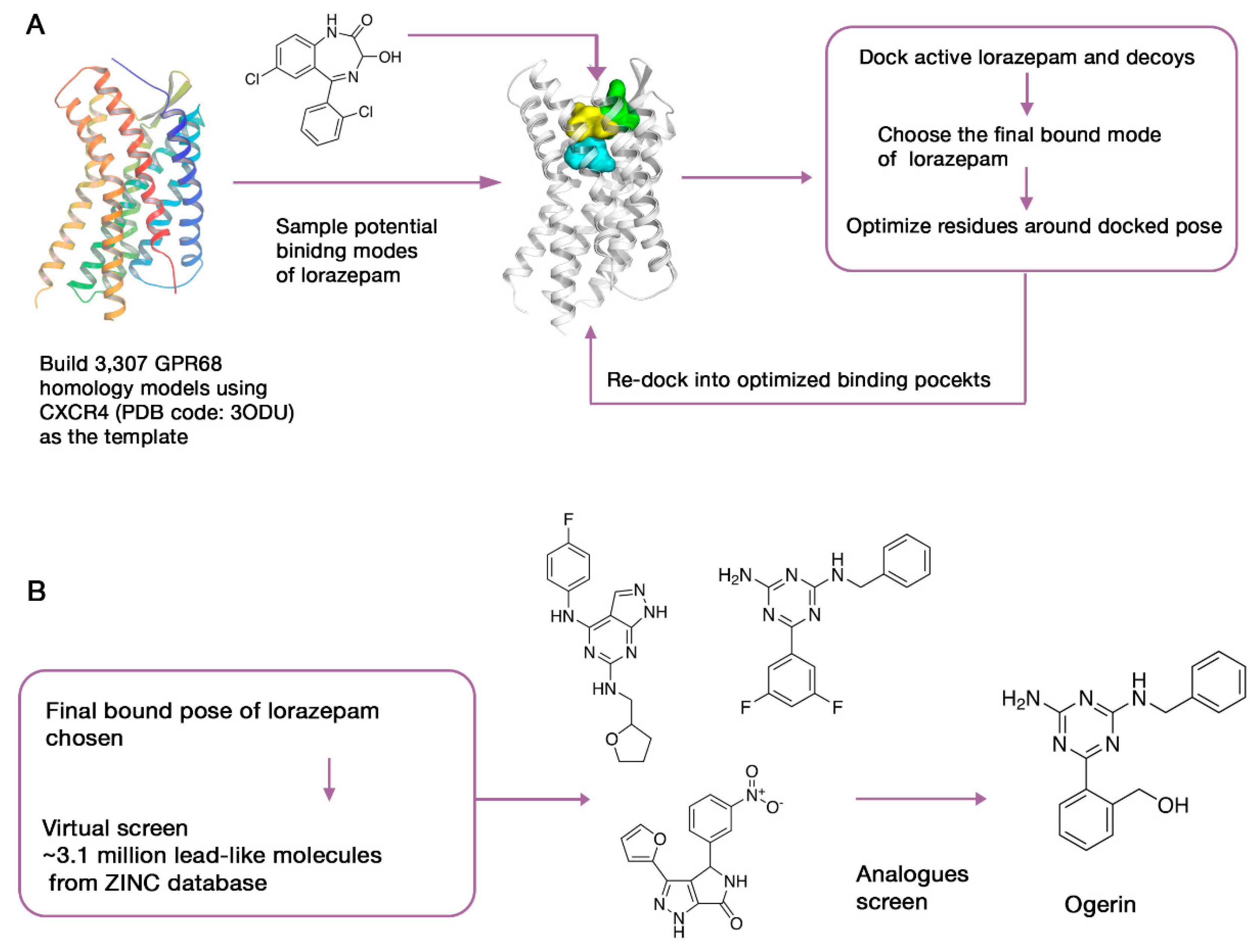
3.3. Case Study 2: Computer-Aided SBDD as a Useful Tool for Probing the Pharmacological Functions of Dark GPCRs
4. Summary and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-Function of the G Protein–Coupled Receptor Superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geppetti, P.; Veldhuis, N.A.; Lieu, T.; Bunnett, N.W. G Protein-Coupled Receptors: Dynamic Machines for Signaling Pain and Itch. Neuron 2015, 88, 635–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.J. Molecular modelling in structural biology. Micron 2002, 33, 365–384. [Google Scholar] [CrossRef]
- Schlick, T.; Collepardo-Guevara, R.; Halvorsen, L.A.; Jung, S.; Xiao, X. Biomolecular modeling and simulation: A field coming of age. Q. Rev. Biophys. 2011, 44, 191–228. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Zhou, H.-X. Calculation of Protein-Ligand Binding Affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.J.; Hein, P.; Hoffmann, C.; Nikolaev, V.O.; Vilardaga, J.-P.; Bünemann, M. Kinetics of G-protein-coupled receptor signals in intact cells. Br. J. Pharmacol. 2009, 153, S125–S132. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Chao, J.C.; Eastwood, M.P.; Gagliardo, J.; Grossman, J.P.; Ho, C.R.; Lerardi, D.J.; Kolossváry, I.; Klepeis, J.L.; Layman, T.; et al. Anton, a special-purpose machine for molecular dynamics simulation. Commun. ACM 2008, 51, 91–97. [Google Scholar] [CrossRef]
- Friedrichs, M.S.; Eastman, P.; Vaidyanathan, V.; Houston, M.; Legrand, S.; Beberg, A.L.; Ensign, D.L.; Bruns, C.M.; Pande, V.S. Accelerating molecular dynamic simulation on graphics processing units. J. Comput. Chem. 2009, 30, 864–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Green, H.F.; Valant, C.; Borhani, D.W.; Valcourt, J.R.; Pan, A.C.; Arlow, D.H.; Canals, M.; Lane, J.R.; Rahmani, R.; et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 2013, 503, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, N.; Ibrahim, P.; Saladino, G.; Gervasio, F.L.; Clark, T. An Efficient Metadynamics-Based Protocol to Model the Binding Affinity and the Transition State Ensemble of G-Protein-Coupled Receptor Ligands. J. Chem. Inf. Model. 2017, 57, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Spitaleri, A.; Saladino, G.; Gervasio, F.L. Investigating Drug–Target Association and Dissociation Mechanisms Using Metadynamics-Based Algorithms. Acc. Chem. Res. 2015, 48, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Provasi, D.; Bortolato, A.; Filizola, M. Exploring Molecular Mechanisms of Ligand Recognition by Opioid Receptors with Metadynamics. Biochemistry 2009, 48, 10020–10029. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Yeatman, H.R.; Provasi, D.; Alt, A.; Christopoulos, A.; Canals, M.; Filizola, M. Proposed Mode of Binding and Action of Positive Allosteric Modulators at Opioid Receptors. ACS Chem. Biol. 2016, 11, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Raniolo, S.; Limongelli, V.; Xu, Y. The Molecular Mechanism Underlying Ligand Binding to the Membrane-Embedded Site of a G-Protein-Coupled Receptor. J. Chem. Theory Comput. 2018, 14, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Kappel, K.; Miao, Y.; McCammon, J.A. Accelerated molecular dynamics simulations of ligand binding to a muscarinic G-protein-coupled receptor. Q. Rev. Biophys. 2015, 48, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, D.P.; Grossfield, A.; Lynch, D.L.; Feller, S.; Romo, T.D.; Gawrisch, K.; Pitman, M.C.; Reggio, P.H. A Lipid Pathway for Ligand Binding Is Necessary for a Cannabinoid G Protein-coupled Receptor. J. Biol. Chem. 2010, 285, 17954–17964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pande, V.S.; Beauchamp, K.; Bowman, G.R. Everything you wanted to know about Markov State Models but were afraid to ask. Methods 2010, 52, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buch, I.; Harvey, M.J.; Giorgino, T.; Anderson, D.P.; De Fabritiis, G. High-Throughput All-Atom Molecular Dynamics Simulations Using Distributed Computing. J. Chem. Inf. Model. 2010, 50, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Stanley, N.; Pardo, L.; Fabritiis, G.D. The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci. Rep. 2016, 6, 22639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vauquelin, G. On the ‘micro’-pharmacodynamic and pharmacokinetic mechanisms that contribute to long-lasting drug action. Expert Opin. Drug Discov. 2015, 10, 1085–1098. [Google Scholar] [CrossRef] [PubMed]
- Vauquelin, G. Cell membranes… and how long drugs may exert beneficial pharmacological activity in vivo. Br. J. Clin. Pharmacol. 2016, 82, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Hornak, V.; Pearlstein, R.A.; Duca, J.S. Structure–Kinetic Relationships of Passive Membrane Permeation from Multiscale Modeling. J. Am. Chem. Soc. 2017, 139, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Hornak, V.; Velez-Vega, C.; McKay, D.J.J.; Reilly, J.; Sandham, D.A.; Shaw, D.; Fairhurst, R.A.; Charlton, S.J.; Sykes, D.A.; et al. Uncoupling the Structure–Activity Relationships of β2 Adrenergic Receptor Ligands from Membrane Binding. J. Med. Chem. 2016, 59, 5780–5789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, Z.-G.; Zhang, K.; Kiselev, E.; Crane, S.; Wang, J.; Paoletta, S.; Yi, C.; Ma, L.; Zhang, W.; et al. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature 2015, 520, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, V.; Bonomi, M.; Parrinello, M. Funnel metadynamics as accurate binding free-energy method. Proc. Natl. Acad. Sci. USA 2013, 110, 6358–6363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charifson, P.S. Practical Application of Computer-Aided Drug Design, 1st ed.; Marcel Dekker, Inc.: New York, NY, USA, 1997; ISBN 978-0-8247-9885-7. [Google Scholar]
- Greer, J.; Erickson, J.W.; Baldwin, J.J.; Varney, M.D. Application of the Three-Dimensional Structures of Protein Target Molecules in Structure-Based Drug Design. J. Med. Chem. 1994, 37, 1035–1054. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L. The Many Roles of Computation in Drug Discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Itzstein, M.; Wu, W.-Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Phan, T.V.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Hazuda, D.J.; Anthony, N.J.; Gomez, R.P.; Jolly, S.M.; Wai, J.S.; Zhuang, L.; Fisher, T.E.; Embrey, M.; Guare, J.P.; Egbertson, M.S.; et al. A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integrase. Proc. Natl. Acad. Sci. USA 2004, 101, 11233–11238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schames, J.R.; Henchman, R.H.; Siegel, J.S.; Sotriffer, C.A.; Ni, H.; McCammon, J.A. Discovery of a Novel Binding Trench in HIV Integrase. J. Med. Chem. 2004, 47, 1879–1881. [Google Scholar] [CrossRef] [PubMed]
- Liverton, N.J.; Holloway, M.K.; McCauley, J.A.; Rudd, M.T.; Butcher, J.W.; Carroll, S.S.; DiMuzio, J.; Fandozzi, C.; Gilbert, K.F.; Mao, S.-S.; et al. Molecular Modeling Based Approach to Potent P2−P4 Macrocyclic Inhibitors of Hepatitis C NS3/4A Protease. J. Am. Chem. Soc. 2008, 130, 4607–4609. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, S.R.; Stanton, M.G.; Gregro, A.R.; Steinbeiser, M.A.; Shaffer, J.R.; Nantermet, P.G.; Barrow, J.C.; Rittle, K.E.; Collusi, D.; Espeseth, A.S.; et al. Discovery and SAR of isonicotinamide BACE-1 inhibitors that bind β-secretase in a N-terminal 10s-loop down conformation. Bioorgan. Med. Chem. Lett. 2007, 17, 1788–1792. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Åqvist, J.; Medina, C.; Samuelsson, J.-E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. Des. Sel. 1994, 7, 385–391. [Google Scholar] [CrossRef]
- Cheatham, T.E., III; Srinivasan, J.; Case, D.A.; Kollman, P.A. Molecular Dynamics and Continuum Solvent Studies of the Stability of PolyG-PolyC and PolyA-PolyT DNA Duplexes in Solution. J. Biomol. Struct. Dyn. 1998, 16, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate−DNA Helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Vorobjev, Y.N.; Hermans, J. ES/IS: Estimation of conformational free energy by combining dynamics simulations with explicit solvent with an implicit solvent continuum model. Biophys. Chem. 1999, 78, 195–205. [Google Scholar] [CrossRef]
- Tembre, B.L.; Mc Cammon, J.A. Ligand-receptor interactions. Comput. Chem. 1984, 8, 281–283. [Google Scholar] [CrossRef]
- Boresch, S.; Tettinger, F.; Leitgeb, M.; Karplus, M. Absolute Binding Free Energies: A Quantitative Approach for Their Calculation. J. Phys. Chem. B 2003, 107, 9535–9551. [Google Scholar] [CrossRef]
- Hermans, J.; Wang, L. Inclusion of Loss of Translational and Rotational Freedom in Theoretical Estimates of Free Energies of Binding. Application to a Complex of Benzene and Mutant T4 Lysozyme. J. Am. Chem. Soc. 1997, 119, 2707–2714. [Google Scholar] [CrossRef]
- Venhorst, J.; ter Laak, A.M.; Commandeur, J.N.M.; Funae, Y.; Hiroi, T.; Vermeulen, N.P.E. Homology Modeling of Rat and Human Cytochrome P450 2D (CYP2D) Isoforms and Computational Rationalization of Experimental Ligand-Binding Specificities. J. Med. Chem. 2003, 46, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Varady, J.; Wu, X.; Fang, X.; Min, J.; Hu, Z.; Levant, B.; Wang, S. Molecular Modeling of the Three-Dimensional Structure of Dopamine 3 (D3) Subtype Receptor: Discovery of Novel and Potent D3 Ligands through a Hybrid Pharmacophore- and Structure-Based Database Searching Approach. J. Med. Chem. 2003, 46, 4377–4392. [Google Scholar] [CrossRef] [PubMed]
- Evers, A.; Klebe, G. Successful Virtual Screening for a Submicromolar Antagonist of the Neurokinin-1 Receptor Based on a Ligand-Supported Homology Model. J. Med. Chem. 2004, 47, 5381–5392. [Google Scholar] [CrossRef] [PubMed]
- Becker, O.M.; Marantz, Y.; Shacham, S.; Inbal, B.; Heifetz, A.; Kalid, O.; Bar-Haim, S.; Warshaviak, D.; Fichman, M.; Noiman, S. G protein-coupled receptors: In silico drug discovery in 3D. Proc. Natl. Acad. Sci. USA 2004, 101, 11304–11309. [Google Scholar] [CrossRef] [PubMed]
- Evers, A.; Klabunde, T. Structure-based Drug Discovery Using GPCR Homology Modeling: Successful Virtual Screening for Antagonists of the Alpha1A Adrenergic Receptor. J. Med. Chem. 2005, 48, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, E.; Springael, J.-Y.; Parmentier, M.; Hachet-Haas, M.; Galzi, J.-L.; Rognan, D. Identification of Nonpeptide CCR5 Receptor Agonists by Structure-based Virtual Screening. J. Med. Chem. 2007, 50, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Orry, A.J.W.; Murgolo, N.J.; Czarniecki, M.F.; Kocsi, S.A.; Hawes, B.E.; O’Neill, K.A.; Hine, H.; Burton, M.S.; Voigt, J.H.; et al. Discovery of Novel Chemotypes to a G-Protein-Coupled Receptor through Ligand-Steered Homology Modeling and Structure-Based Virtual Screening. J. Med. Chem. 2008, 51, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikhonova, I.G.; Sum, C.S.; Neumann, S.; Engel, S.; Raaka, B.M.; Costanzi, S.; Gershengorn, M.C. Discovery of Novel Agonists and Antagonists of the Free Fatty Acid Receptor 1 (FFAR1) Using Virtual Screening. J. Med. Chem. 2008, 51, 625–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, S.; Skoumbourdis, A.P.; Childress, J.; Neumann, S.; Deschamps, J.R.; Thomas, C.J.; Colson, A.-O.; Costanzi, S.; Gershengorn, M.C. A Virtual Screen for Diverse Ligands: Discovery of Selective G Protein-Coupled Receptor Antagonists. J. Am. Chem. Soc. 2008, 130, 5115–5123. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Rosenbaum, D.M.; Irwin, J.J.; Fung, J.J.; Kobilka, B.K.; Shoichet, B.K. Structure-based discovery of β2-adrenergic receptor ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 6843–6848. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Jaakola, V.-P.; Lane, J.R.; Lin, J.; IJzerman, A.P.; Yeager, M.; Kufareva, I.; Stevens, R.C.; Abagyan, R. Structure-Based Discovery of Novel Chemotypes for Adenosine A2A Receptor Antagonists. J. Med. Chem. 2010, 53, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, J.; Coleman, R.G.; Setola, V.; Irwin, J.J.; Fan, H.; Schlessinger, A.; Sali, A.; Roth, B.L.; Shoichet, B.K. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat. Chem. Biol. 2011, 7, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, D.; Gao, Z.-G.; Moss, S.M.; Jacobson, K.A.; Carlsson, J. Molecular Docking Screening Using Agonist-Bound GPCR Structures: Probing the A2A Adenosine Receptor. J. Chem. Inf. Model. 2015, 55, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wacker, D.; Levit, A.; Che, T.; Betz, R.M.; McCorvy, J.D.; Venkatakrishnan, A.J.; Huang, X.-P.; Dror, R.O.; Shoichet, B.K.; et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 2017, 358, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Lansu, K.; Karpiak, J.; Liu, J.; Huang, X.-P.; McCorvy, J.D.; Kroeze, W.K.; Che, T.; Nagase, H.; Carroll, F.I.; Jin, J.; et al. In silico design of novel probes for the atypical opioid receptor MRGPRX2. Nat. Chem. Biol. 2017, 13, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korczynska, M.; Clark, M.J.; Valant, C.; Xu, J.; Moo, E.V.; Albold, S.; Weiss, D.R.; Torosyan, H.; Huang, W.; Kruse, A.C.; et al. Structure-based discovery of selective positive allosteric modulators of antagonists for the M2 muscarinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA 2018, 115, E2419–E2428. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem. 2016, 59, 4103–4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-Based Virtual Screening for Drug Discovery: A Problem-Centric Review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Pati, S.P.; Kumar, P.P.; Pradeep, H.N.; Sastry, G.N. Virtual Screening in Drug Discovery—A Computational Perspective. Curr. Protein Pept. Sci. 2007, 8, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhang, J. Small Molecule Allosteric Modulators of G-Protein-Coupled Receptors: Drug–Target Interactions. J. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wu, B. Structural studies of G protein-coupled receptors. IUBMB Life 2016, 68, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Graaf, C.; Song, G.; Cao, C.; Zhao, Q.; Wang, M.-W.; Wu, B.; Stevens, R.C. Extending the Structural View of Class B GPCRs. Trends Biochem. Sci. 2017, 42, 946–960. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Lesk, A.M. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986, 5, 823–826. [Google Scholar] [PubMed]
- Nowak, M.; Kołaczkowski, M.; Pawłowski, M.; Bojarski, A.J. Homology Modeling of the Serotonin 5-HT1A Receptor Using Automated Docking of Bioactive Compounds with Defined Geometry. J. Med. Chem. 2006, 49, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Evers, A.; Klebe, G. Ligand-Supported Homology Modeling of G-Protein-Coupled Receptor Sites: Models Sufficient for Successful Virtual Screening. Angew. Chem. Int. Ed. 2003, 43, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Štular, T.; Lešnik, S.; Rožman, K.; Schink, J.; Zdouc, M.; Ghysels, A.; Liu, F.; Aldrich, C.C.; Haupt, V.J.; Salentin, S.; et al. Discovery of Mycobacterium tuberculosis InhA Inhibitors by Binding Sites Comparison and Ligands Prediction. J. Med. Chem. 2016, 59, 11069–11078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, B.L.; Irwin, J.J.; Shoichet, B.K. Discovery of new GPCR ligands to illuminate new biology. Nat. Chem. Biol. 2017, 13, 1143–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertzen, C.G.W.; Spomer, L.; Smits, S.H.J.; Häussinger, D.; Keitel, V.; Gohlke, H. Mutational mapping of the transmembrane binding site of the G-protein coupled receptor TGR5 and binding mode prediction of TGR5 agonists. Eur. J. Med. Chem. 2015, 104, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.Q.; Calhoun, S.; Fan, H.; Kalyanaraman, C.; Branch, M.C.; Mashiyama, S.T.; London, N.; Jacobson, M.P.; Babbitt, P.C.; Shoichet, B.K.; et al. Prediction of Substrates for Glutathione Transferases by Covalent Docking. J. Chem. Inf. Model. 2014, 54, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Shacham, S.; Topf, M.; Avisar, N.; Glaser, F.; Marantz, Y.; Bar-Haim, S.; Noiman, S.; Naor, Z.; Becker, O.M. Modeling the 3D structure of GPCRs from sequence. Med. Res. Rev. 2001, 21, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Shacham, S.; Marantz, Y.; Bar-Haim, S.; Kalid, O.; Warshaviak, D.; Avisar, N.; Inbal, B.; Heifetz, A.; Fichman, M.; Topf, M.; et al. PREDICT modeling and in-silico screening for G-protein coupled receptors. Proteins Struct. Funct. Bioinform. 2004, 57, 51–86. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabuurs, S.B.; Wagener, M.; de Vlieg, J. A Flexible Approach to induced Fit Docking. J. Med. Chem. 2007, 50, 6507–6518. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.; Jaśkowska, J.; Bojarski, A.J.; Duszyńska, B.; Bucki, A.; Kołaczkowski, M. Evaluation of 1-arylpiperazine derivative of hydroxybenzamides as 5-HT1A and 5-HT7 serotonin receptor ligands: An experimental and molecular modeling approach. J. Heterocycl. Chem. 2010, 48, 192–198. [Google Scholar] [CrossRef]
- Miao, Y.; Goldfeld, D.A.; Moo, E.V.; Sexton, P.M.; Christopoulos, A.; McCammon, J.A.; Valant, C. Accelerated structure-based design of chemically diverse allosteric modulators of a muscarinic G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2016, 113, E5675–E5684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlhoff, K.J.; Shukla, D.; Lawrenz, M.; Bowman, G.R.; Konerding, D.E.; Belov, D.; Altman, R.B.; Pande, V.S. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem. 2014, 6, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.P.; Shivakumar, D.M.; Boyce, S.E.; Jacobson, M.P.; Case, D.A.; Shoichet, B.K. Rescoring Docking Hit Lists for Model Cavity Sites: Predictions and Experimental Testing. J. Mol. Biol. 2008, 377, 914–934. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Andrews, C.W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Zwanzig, R.W. High-Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. J. Chem. Phys. 1954, 22, 1420–1426. [Google Scholar] [CrossRef]
- Hénin, J.; Maigret, B.; Tarek, M.; Escrieut, C.; Fourmy, D.; Chipot, C. Probing a Model of a GPCR/Ligand Complex in an Explicit Membrane Environment: The Human Cholecystokinin-1 Receptor. Biophys. J. 2006, 90, 1232–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukharta, L.; Gutiérrez-de-Terán, H.; Åqvist, J. Computational Prediction of Alanine Scanning and Ligand Binding Energetics in G-Protein Coupled Receptors. PLoS Comput. Biol. 2014, 10, e1003585. [Google Scholar] [CrossRef] [PubMed]
- Rivail, L.; Chipot, C.; Maigret, B.; Bestel, I.; Sicsic, S.; Tarek, M. Large-scale molecular dynamics of a G protein-coupled receptor, the human 5-HT4 serotonin receptor, in a lipid bilayer. J. Mol. Struct. THEOCHEM 2007, 817, 19–26. [Google Scholar] [CrossRef]
- Keränen, H.; Gutiérrez-de-Terán, H.; Åqvist, J. Structural and Energetic Effects of A2A Adenosine Receptor Mutations on Agonist and Antagonist Binding. PLoS ONE 2014, 9, e108492. [Google Scholar] [CrossRef] [PubMed]
- Keränen, H.; Åqvist, J.; Gutiérrez-de-Terán, H. Free energy calculations of A2A adenosine receptor mutation effects on agonist binding. Chem. Commun. 2015, 51, 3522–3525. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ranganathan, A.; IJzerman, A.P.; Siegal, G.; Carlsson, J. Complementarity between in Silico and Biophysical Screening Approaches in Fragment-Based Lead Discovery against the A2A Adenosine Receptor. J. Chem. Inf. Model. 2013, 53, 2701–2714. [Google Scholar] [CrossRef] [PubMed]
- Goldfeld, D.A.; Murphy, R.; Kim, B.; Wang, L.; Beuming, T.; Abel, R.; Friesner, R.A. Docking and Free Energy Perturbation Studies of Ligand Binding in the Kappa Opioid Receptor. J. Phys. Chem. B 2015, 119, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenselink, E.B.; Louvel, J.; Forti, A.F.; van Veldhoven, J.P.D.; de Vries, H.; Mulder-Krieger, T.; McRobb, F.M.; Negri, A.; Goose, J.; Abel, R.; et al. Predicting Binding Affinities for GPCR Ligands Using Free-Energy Perturbation. ACS Omega 2016, 1, 293–304. [Google Scholar] [CrossRef]
- Huang, X.-P.; Karpiak, J.; Kroeze, W.K.; Zhu, H.; Chen, X.; Moy, S.S.; Saddoris, K.A.; Nikolova, V.D.; Farrell, M.S.; Wang, S.; et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 2015, 527, 477–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Year/Reference | Target | Method 1 | PDB Code | Known Active Ligand (s) | Receptor Models (Initial/Final) | Screened Compounds | Hit Rate (Hit/Tested) |
|---|---|---|---|---|---|---|---|
| 2003/[52] | D3 dopamine receptor | HM | 1f88 | -- | 4 | 6727 | 0.55 (11/20) |
| 2004/[53] | Neurokinin-1 (NK1) receptor | HM | 1hzx | 1 | 100/1 | 827,000 | 0.14 (1/7) |
| 2004/[54] | 5-HT1A serotonin receptor | PREDICT | -- | 1 | 1 | 40,000 | 0.21 (16/78) |
| NK1 receptor | 1 | 1 | 150,000 | 0.15(5/53) | |||
| 5-HT4 serotonin receptor | 1 | 1 | 150,000 | 0.21 (19/93) | |||
| 2005/[55] | Alpha1A adrenergic receptor | HM | 1f88 | 1 | 100/1 | 22,950 | 0.46 (37/80) |
| 2007/[56] | CCR5 chemokine receptor | HM | 1f88 | 5 | --/1 | 1,620,316 | 0.17 (10/59) |
| 2008/[57] | MCH-R1 | HM | 1l9h | 4 | 20/1 | 187,084 | 0.05 (6/129) |
| 2008/[58] | FFAR1 | HM | 1gzm | 1 | 100/ | 2,600,000 | 0.29 (15/52) |
| 2008/[59] | TRH-R1 | HM | 1f88 | -- | 1 | 1,000,000 | 0.05 (5/100) |
| 2009/[60] | β2 adrenergic receptor | X-ray | 2rh1 | -- | -- | 1,000,000 | 0.24 (6/25) |
| 2010/[61] | Adenosine A2A receptor | X-ray | 3eml | -- | -- | 4,000,000 | 0.41 (23/56) |
| 2011/[62] | D3 dopamine receptor | HM | 2vt4+2rh1 | 1300 | 20,000/1 | 3,300,000 | 0.23 (6/25) |
| X-ray | 3pbl | -- | -- | 3,300,000 | 0.2 (5/25) | ||
| 2015/[63] | Adenosine A2A receptor | X-ray | 3qak/2ydo/2ydv | -- | -- | 6,700,000 | 0.45 (9/20) |
| 2016/[64] | μ-opioid receptor | X-ray | 4dkl/5cm1 | -- | -- | >3 million | 0.30 (7/23) |
| 2017/[65] | D4 dopamine receptor | X-ray | 3pbl | -- | -- | >600,000 | 0.2 (2/10) |
| 2017/[66] | MRGPRX2 opioid receptor | HM | 4djh | 1 | 1080 | ~3.7 million | 0.05 (1/20) |
| 2018/[67] | M2 mAChR | X-ray | 3uon | -- | -- | 4.6 million | 0.23(3/13) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.; Xu, Y. Recent Trends and Applications of Molecular Modeling in GPCR–Ligand Recognition and Structure-Based Drug Design. Int. J. Mol. Sci. 2018, 19, 2105. https://doi.org/10.3390/ijms19072105
Yuan X, Xu Y. Recent Trends and Applications of Molecular Modeling in GPCR–Ligand Recognition and Structure-Based Drug Design. International Journal of Molecular Sciences. 2018; 19(7):2105. https://doi.org/10.3390/ijms19072105
Chicago/Turabian StyleYuan, Xiaojing, and Yechun Xu. 2018. "Recent Trends and Applications of Molecular Modeling in GPCR–Ligand Recognition and Structure-Based Drug Design" International Journal of Molecular Sciences 19, no. 7: 2105. https://doi.org/10.3390/ijms19072105