Iron Overload and Chelation Therapy in Non-Transfusion Dependent Thalassemia

 and
and
Abstract
:1. Introduction
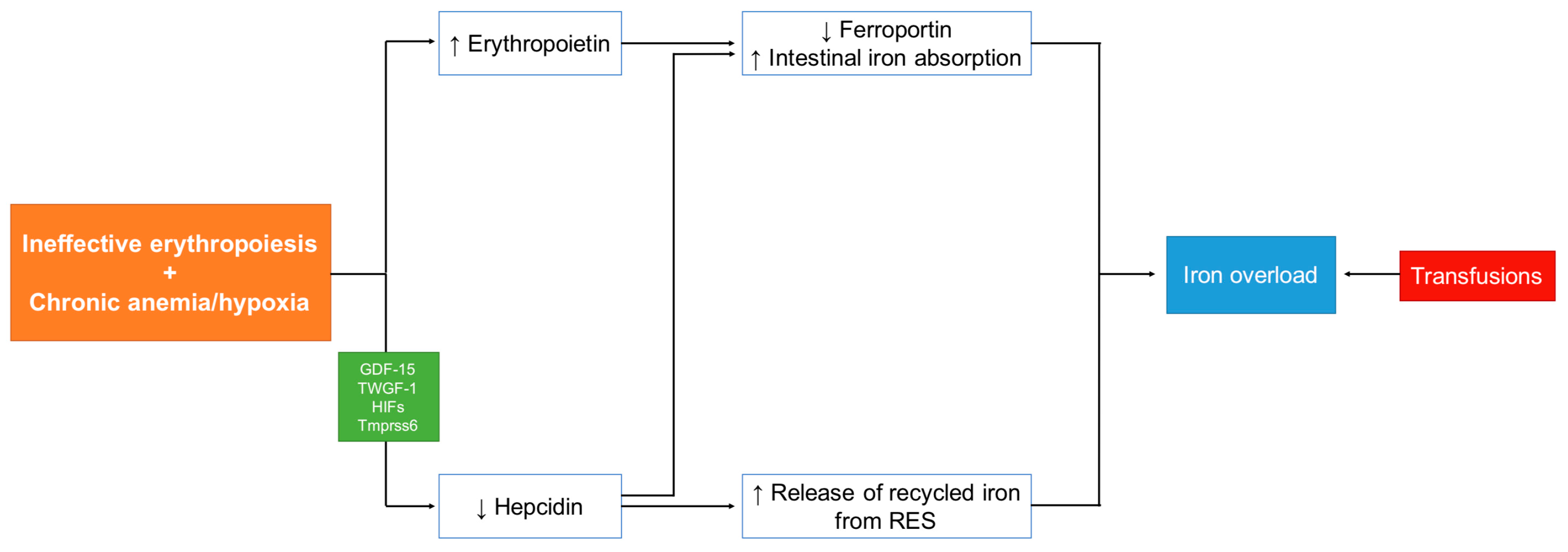
2. Mechanism of Iron Overload (IOL) in Non-Transfusion-Dependent Thalassemia (NTDT)
3. Diagnosis and Quantification of IOL
4. Morbidities Secondary to IOL
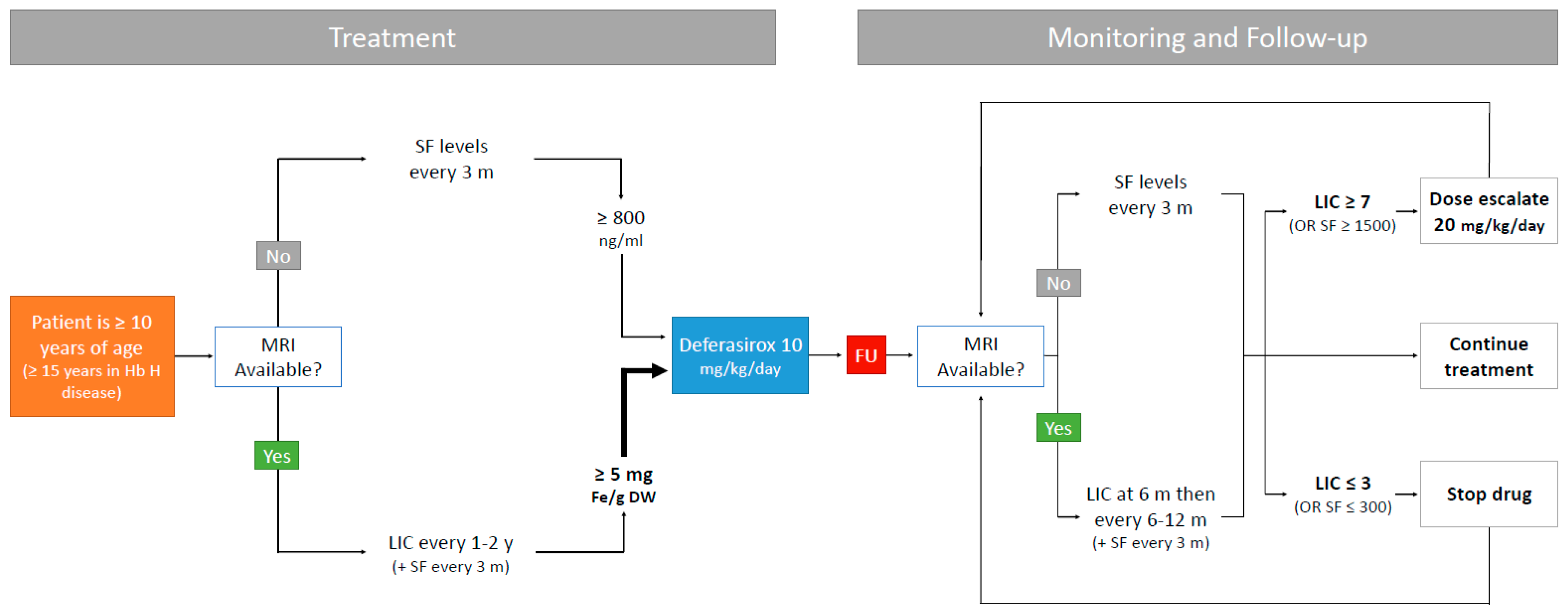
5. Iron Chelation Therapy
6. Recent Advances in ICT
7. Novel Therapies
7.1. Minihepcidins
7.2. TMPRSS6
8. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| IOL | Iron overload |
| NTDT | Non-transfusion dependent thalassemia |
| ICT | Iron chelation therapy |
| TDT | Transfusion-dependent thalassemia |
| β-TI | β-thalassemia intermedia |
| GDF-15 | Growth differentiation factor-15 |
| TGF-β | Transforming growth factor-β |
| SF | Serum ferritin |
| LIC | Liver iron concentration |
| dw | dry weight |
| DFO | Deferoxamine |
| DFP | Deferiprone |
| DFX | Deferasirox |
| DT | Dispersible tablet |
| FCT | Film-coated tablet |
| EMA | European Medicines Agency |
| TIF | Thalassemia International Federation |
| GI | Gastrointestinal |
| PRO | Patient-reported outcomes |
| TMPRSS6 | Transmembrane protein serine 6 |
References
- Taher, A.; Vichinsky, E.; Musallam, K.; Cappellini, M.-D.; Viprakasit, V. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT); Thalassaemia International Federation: Nicosia, Cyprus, 2013. [Google Scholar]
- Musallam, K.M.; Rivella, S.; Vichinsky, E.; Rachmilewitz, E.A. Non-transfusion-dependent thalassemias. Haematologica 2013, 98, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; El-Beshlawy, A.; Karimi, M.; Daar, S.; Belhoul, K.; Saned, M.S.; Graziadei, G.; Cappellini, M.D. Age-related complications in treatment-naive patients with thalassaemia intermedia. Br. J. Haematol. 2010, 150, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Cappellini, M.D.; Daar, S.; Karimi, M.; El-Beshlawy, A.; Graziadei, G.; Magestro, M.; Wulff, J.; Pietri, G.; Taher, A.T. Serum ferritin level and morbidity risk in transfusion-independent patients with β-thalassemia intermedia: The ORIENT study. Haematologica 2014, 99, e218–e221. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Goldrich, M.L.; Haines, D.A.; Azimi, M.; Singer, S.T.; Vichinsky, E.P. Heterogeneity of hemoglobin H disease in childhood. N. Engl. J. Med. 2011, 364, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Pootrakul, P.; Kitcharoen, K.; Yansukon, P.; Wasi, P.; Fucharoen, S.; Charoenlarp, P.; Brittenham, G.; Pippard, M.J.; Finch, C.A. The effect of erythroid hyperplasia on iron balance. Blood 1988, 71, 1124–1129. [Google Scholar] [PubMed]
- Tanno, T.; Miller, J.L. Iron Loading and Overloading due to Ineffective Erythropoiesis. Adv. Hematol. 2010, 2010, 358283. [Google Scholar] [CrossRef] [PubMed]
- Pigeon, C.; Ilyin, G.; Courselaud, B.; Leroyer, P.; Turlin, B.; Brissot, P.; Loreal, O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 2001, 276, 7811–7819. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Hepcidin and iron regulation, 10 years later. Blood 2011, 117, 4425–4433. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Origa, R.; Galanello, R.; Ganz, T.; Giagu, N.; Maccioni, L.; Faa, G.; Nemeth, E. Liver iron concentrations and urinary hepcidin in β-thalassemia. Haematologica 2007, 92, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Taher, A.T.; Duca, L.; Cesaretti, C.; Halawi, R.; Cappellini, M.D. Levels of growth differentiation factor-15 are high and correlate with clinical severity in transfusion-independent patients with β thalassemia intermedia. Blood Cells Mol. Dis. 2011, 47, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.; Wang, R.H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Porayette, P.; Sripichai, O.; Noh, S.J.; Byrnes, C.; Bhupatiraju, A.; Lee, Y.T.; Goodnough, J.B.; Harandi, O.; Ganz, T.; et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009, 114, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Nai, A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Haematol. 2016, 172, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Puliyel, M.; Sposto, R.; Berdoukas, V.A.; Hofstra, T.C.; Nord, A.; Carson, S.; Wood, J.; Coates, T.D. Ferritin trends do not predict changes in total body iron in patients with transfusional iron overload. Am. J. Hematol. 2014, 89, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.-D.; Cohen, A.; Porter, J.; Taher, A.; Viprakasit, V. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT); TIF Publication: Nicosia, Cyprus, 2014. [Google Scholar]
- Taher, A.; El Rassi, F.; Isma’eel, H.; Koussa, S.; Inati, A.; Cappellini, M.D. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica 2008, 93, 1584–1586. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Cappellini, M.D.; Taher, A.T. Evaluation of the 5 mg/g liver iron concentration threshold and its association with morbidity in patients with β-thalassemia intermedia. Blood Cells Mol. Dis. 2013, 51, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Porter, J.B.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Origa, R.; Karakas, Z.; Habr, D.; et al. Defining serum ferritin thresholds to predict clinically relevant liver iron concentrations for guiding deferasirox therapy when MRI is unavailable in patients with non-transfusion-dependent thalassaemia. Br. J. Haematol. 2015, 168, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Karimi, M.; El-Beshlawy, A.; Belhoul, K.; Daar, S.; Saned, M.S.; El-Chafic, A.H.; Fasulo, M.R.; Cappellini, M.D. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: The OPTIMAL CARE study. Blood 2010, 115, 1886–1892. [Google Scholar] [CrossRef] [PubMed]
- Bazarbachi, A.; Moukhadder, H.M.; Bou Fakhredin, R.I.; Roumi, J.E.; Chaya, B.F.; Taher, A.T. How I treat and monitor non-transfusion-dependent thalassaemia. Haematologica 2017, 102 (Suppl. 1), 20–27. [Google Scholar]
- Moukhadder, H.M.; Halawi, R.; Cappellini, M.D.; Taher, A.T. Hepatocellular carcinoma as an emerging morbidity in the thalassemia syndromes: A comprehensive review. Cancer 2017, 123, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Kurtoglu, A.U.; Kurtoglu, E.; Temizkan, A.K. Effect of iron overload on endocrinopathies in patients with β-thalassaemia major and intermedia. Endokrynol. Polska 2012, 63, 260–263. [Google Scholar]
- Inati, A.; Noureldine, M.A.; Mansour, A.; Abbas, H.A. Endocrine and bone complications in β-thalassemia intermedia: Current understanding and treatment. BioMed. Res. Int. 2015, 2015, 813098. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.T.; Johnson, V.L.; Kim, H.Y.; Trachtenberg, F.; Vogiatzi, M.G.; Kwiatkowski, J.L.; Neufeld, E.J.; Fung, E.; Oliveri, N.; Kirby, M.; et al. Renal dysfunction in patients with thalassaemia. Br. J. Haematol. 2011, 153, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Rivella, S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012, 26 (Suppl. 1), S12–S15. [Google Scholar] [CrossRef]
- Haidar, R.; Mhaidli, H.; Musallam, K.M.; Taher, A.T. The spine in β-thalassemia syndromes. Spine 2012, 37, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Beydoun, A.; Hourani, R.; Nasreddine, W.; Raad, R.; Koussa, S.; Taher, A.T. Brain magnetic resonance angiography in splenectomized adults with β-thalassemia intermedia. Eur. J. Haematol. 2011, 87, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Nasreddine, W.; Beydoun, A.; Hourani, R.; Hankir, A.; Koussa, S.; Haidar, M.; Taher, A.T. Brain positron emission tomography in splenectomized adults with β-thalassemia intermedia: Uncovering yet another covert abnormality. Ann. Hematol. 2012, 91, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Pennell, D.J.; Porter, J.B.; Piga, A.; Lai, Y.; El-Beshlawy, A.; Belhoul, K.M.; Elalfy, M.; Yesilipek, A.; Kilinc, Y.; Lawniczek, T.; et al. A 1-year randomized controlled trial of deferasirox vs deferoxamine for myocardial iron removal in β-thalassemia major (CORDELIA). Blood 2014, 123, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Weatherall, D.J. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012, 26 (Suppl. 1), S3–S6. [Google Scholar] [CrossRef]
- Galanello, R.; Cao, A. Relationship between genotype and phenotype. Thalassemia intermedia. Ann. N. Y. Acad. Sci. 1998, 850, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with β-thalassemia. Blood 2006, 107, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Porter, J.B.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Galanello, R.; Karakas, Z.; Lawniczek, T.; et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann. Hematol. 2013, 92, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Cappellini, M.D.; Aydinok, Y.; Porter, J.B.; Karakas, Z.; Viprakasit, V.; Siritanaratkul, N.; Kattamis, A.; Wang, C.; Zhu, Z.; et al. Optimising iron chelation therapy with deferasirox for non-transfusion-dependent thalassaemia patients: 1-year results from the THETIS study. Blood Cells Mol. Dis. 2016, 57, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Calvaruso, G.; Vitrano, A.; Di Maggio, R.; Lai, E.; Colletta, G.; Quota, A.; Gerardi, C.; Rigoli, L.C.; Sacco, M.; Pitrolo, L.; et al. Deferiprone versus deferoxamine in thalassemia intermedia: Results from a 5-year long-term Italian multicenter randomized clinical trial. Am. J. Hematol. 2015, 90, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Brittenham, G.M.; Griffith, P.M.; Nienhuis, A.W.; McLaren, C.E.; Young, N.S.; Tucker, E.E.; Allen, C.J.; Farrell, D.E.; Harris, J.W. Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. N. Engl. J. Med. 1994, 331, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Gabutti, V.; Piga, A. Results of long-term iron-chelating therapy. Acta Haematol. 1996, 95, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Delea, T.E.; Edelsberg, J.; Sofrygin, O.; Thomas, S.K.; Baladi, J.F.; Phatak, P.D.; Coates, T.D. Consequences and costs of noncompliance with iron chelation therapy in patients with transfusion-dependent thalassemia: A literature review. Transfusion 2007, 47, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Origa, R.; Perrotta, S.; Kourakli, A.; Ruffo, G.B.; Kattamis, A.; Goh, A.S.; Cortoos, A.; Huang, V.; Weill, M.; et al. New film-coated tablet formulation of deferasirox is well tolerated in patients with thalassemia or lower-risk MDS: Results of the randomized, phase II ECLIPSE study. Am. J. Hematol. 2017, 92, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Casu, C.; Oikonomidau, R.; Shah, Y.; Nemeth, E.; Ganz, T.; MacDonald, B.; Rivella, S. Concurrent Treatment with Minhepcidin and Deferiprone Improves Anemia and Enhances Reduction of Spleen Iron in a Mouse Model of Non-Transfusion Dependent Thalassemia. Blood 2014, 124, 748. [Google Scholar]
- Casu, C.; Goldberg, A.; Nemeth, E.; Ganz, T.; Gardenghi, S.; MacDonald, B.; Rivella, S. Treatment with Minihepcidin Peptide Improves Anemia and Iron Overload in a Mouse Model of Thalassemia Intermedia. Blood 2013, 122, 431. [Google Scholar]
- Preza, G.C.; Ruchala, P.; Pinon, R.; Ramos, E.; Qiao, B.; Peralta, M.A.; Sharma, S.; Waring, A.; Ganz, T.; Nemeth, E. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J. Clin. Investig. 2011, 121, 4880–4888. [Google Scholar] [CrossRef] [PubMed]
- Gardenghi, S.; Ramos, P.; Marongiu, M.F.; Melchiori, L.; Breda, L.; Guy, E.; Muirhead, K.; Rao, N.; Roy, C.N.; Andrews, N.C.; et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in β-thalassemic mice. J. Clin. Investig. 2010, 120, 4466–4477. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.; Ruchala, P.; Goodnough, J.B.; Kautz, L.; Preza, G.C.; Nemeth, E.; Ganz, T. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood 2012, 120, 3829–3836. [Google Scholar] [CrossRef] [PubMed]
- Nai, A.; Pagani, A.; Mandelli, G.; Lidonnici, M.R.; Silvestri, L.; Ferrari, G.; Camaschella, C. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of β-thalassemia. Blood 2012, 119, 5021–5029. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Casu, C.; Gardenghi, S.; Booten, S.; Aghajan, M.; Peralta, R.; Watt, A.; Freier, S.; Monia, B.P.; Rivella, S. Reducing TMPRSS6 ameliorates hemochromatosis and β-thalassemia in mice. J. Clin. Investig. 2013, 123, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Toudjarska, I.; Sendamarai, A.K.; Racie, T.; Milstein, S.; Bettencourt, B.R.; Hettinger, J.; Bumcrot, D.; Fleming, M.D. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine β-thalassemia intermedia. Blood 2013, 121, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Aghajan, M.; Casu, C.; Io Presti, V.; Booten, S.; Monia, B.P.; Rivella, S.; Guo, S. Developing a Galnac-Conjugated TMPRSS6 Antisense Therapy for the Treatment of β-Thalassemia. Blood 2016, 128, 1013. [Google Scholar]


{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bou-Fakhredin, R.; Bazarbachi, A.-H.; Chaya, B.; Sleiman, J.; Cappellini, M.D.; Taher, A.T. Iron Overload and Chelation Therapy in Non-Transfusion Dependent Thalassemia. Int. J. Mol. Sci. 2017, 18, 2778. https://doi.org/10.3390/ijms18122778
Bou-Fakhredin R, Bazarbachi A-H, Chaya B, Sleiman J, Cappellini MD, Taher AT. Iron Overload and Chelation Therapy in Non-Transfusion Dependent Thalassemia. International Journal of Molecular Sciences. 2017; 18(12):2778. https://doi.org/10.3390/ijms18122778
Chicago/Turabian StyleBou-Fakhredin, Rayan, Abdul-Hamid Bazarbachi, Bachar Chaya, Joseph Sleiman, Maria Domenica Cappellini, and Ali T. Taher. 2017. "Iron Overload and Chelation Therapy in Non-Transfusion Dependent Thalassemia" International Journal of Molecular Sciences 18, no. 12: 2778. https://doi.org/10.3390/ijms18122778