
Strophanthidin Induces Apoptosis of Human Lung Adenocarcinoma Cells by Promoting TRAIL-DR5 Signaling
 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
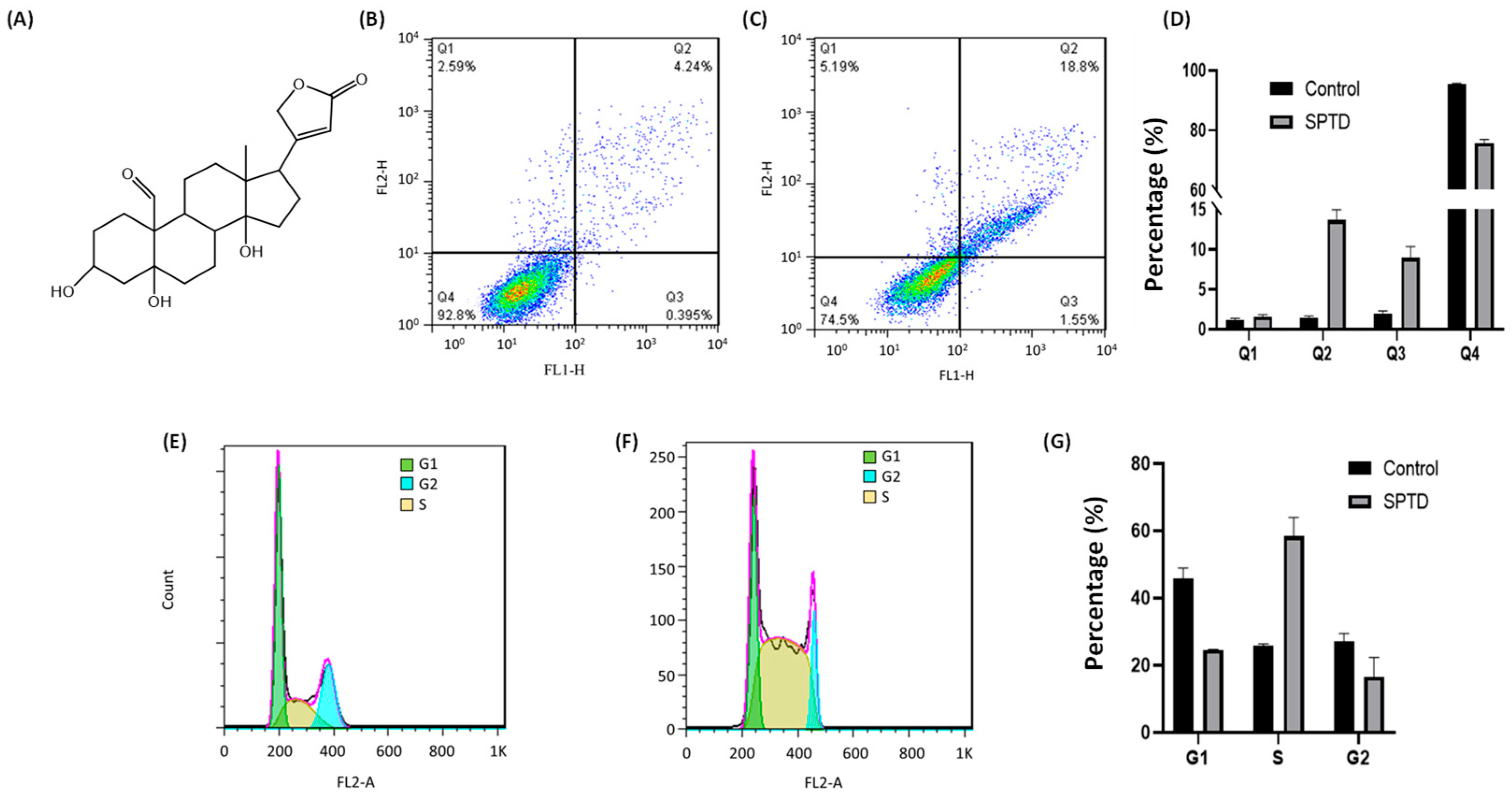
2.1. Cytotoxicity of Strophanthidin
2.2. Cellular Phenotype Characterization of A549 Cells Treated with SPTD
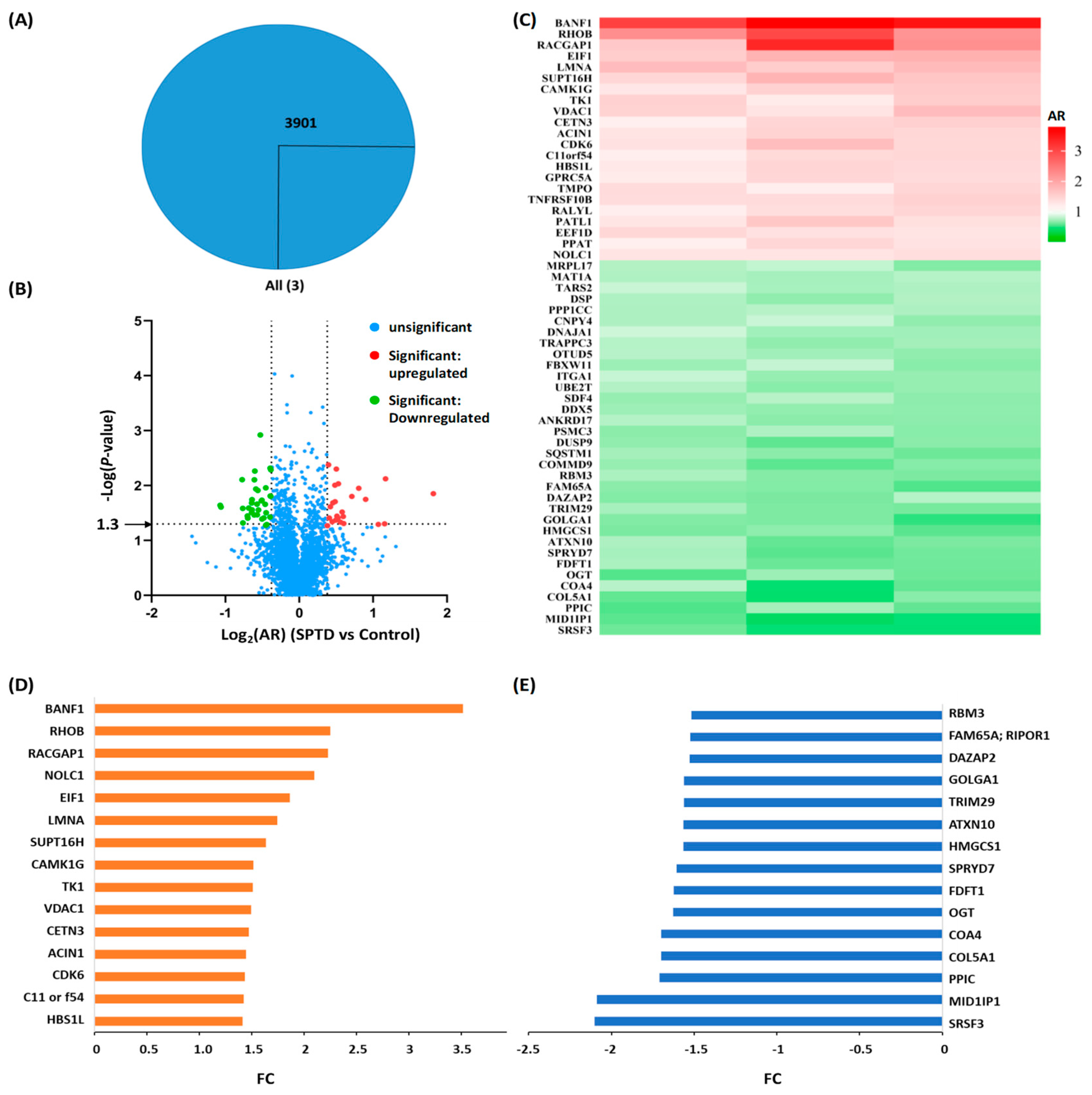
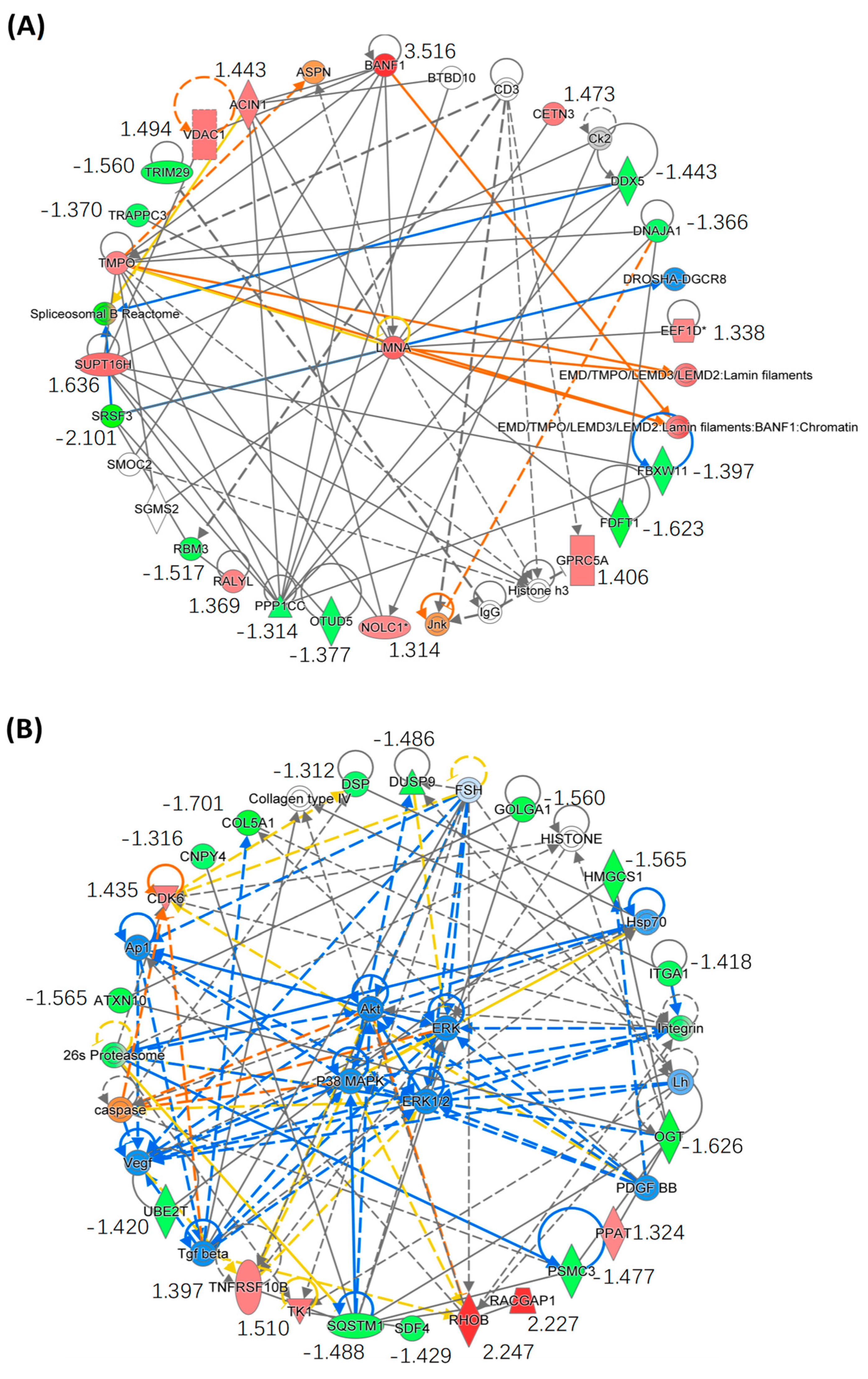
2.3. Quantitative Proteomics Analysis
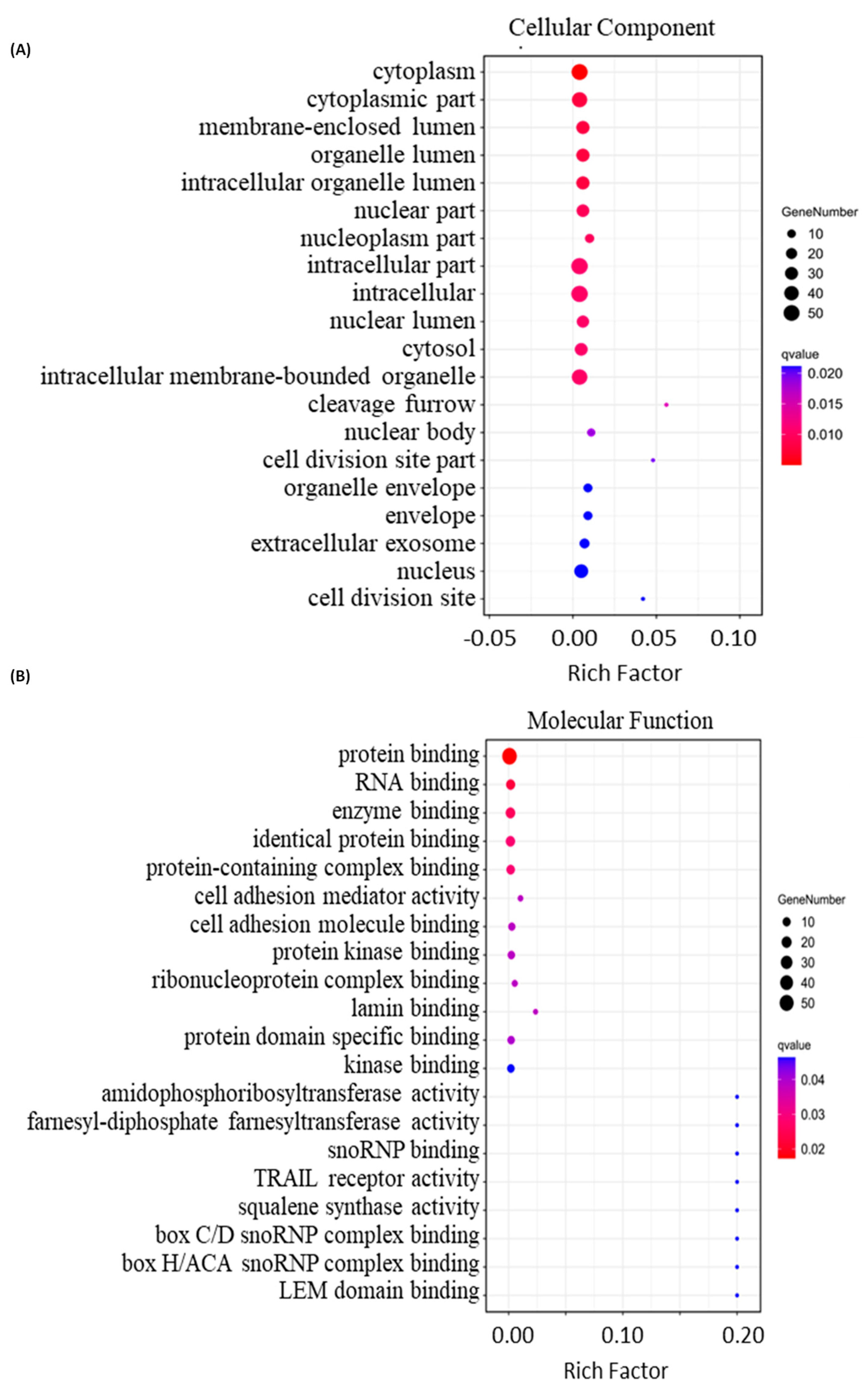
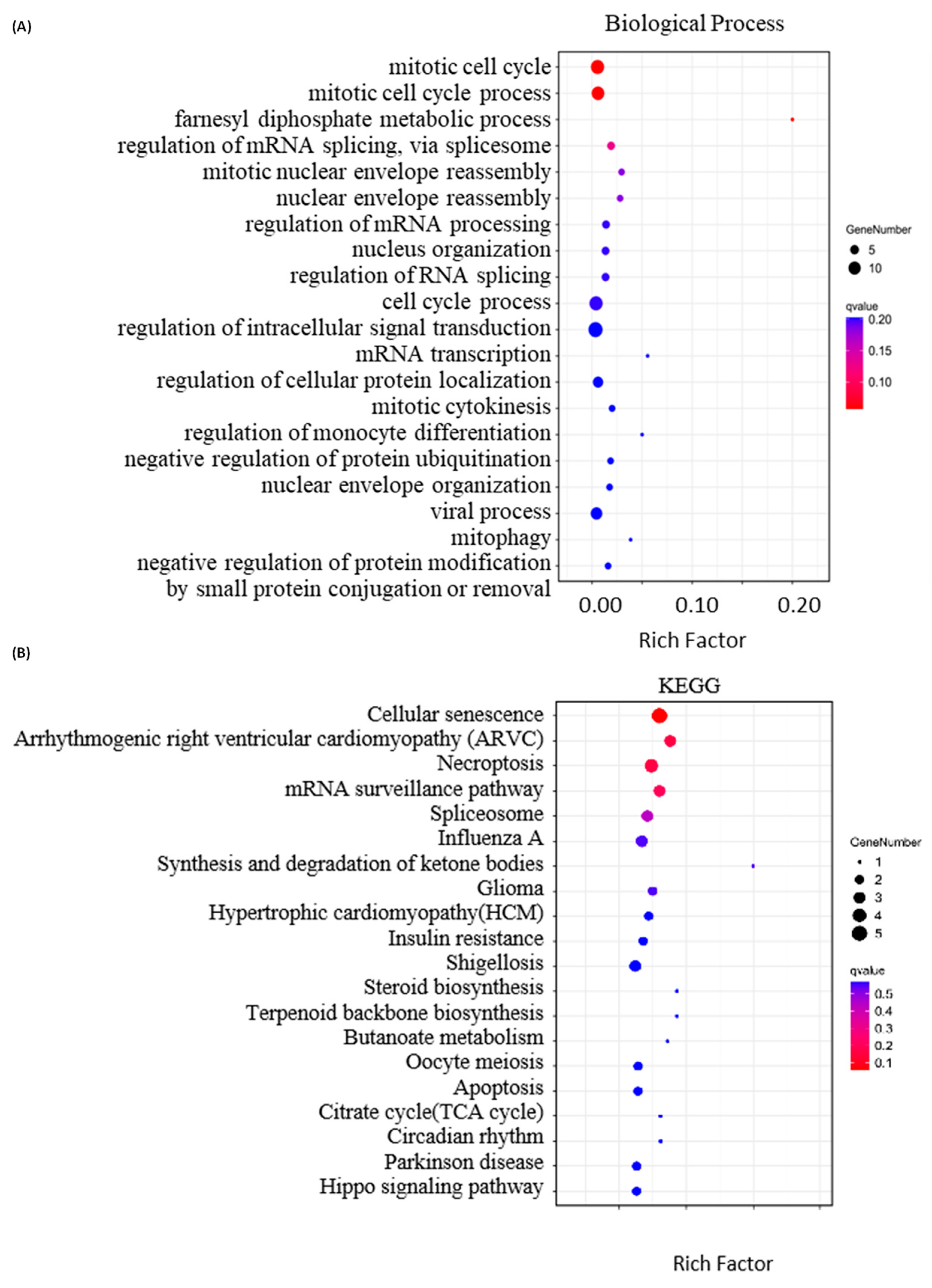
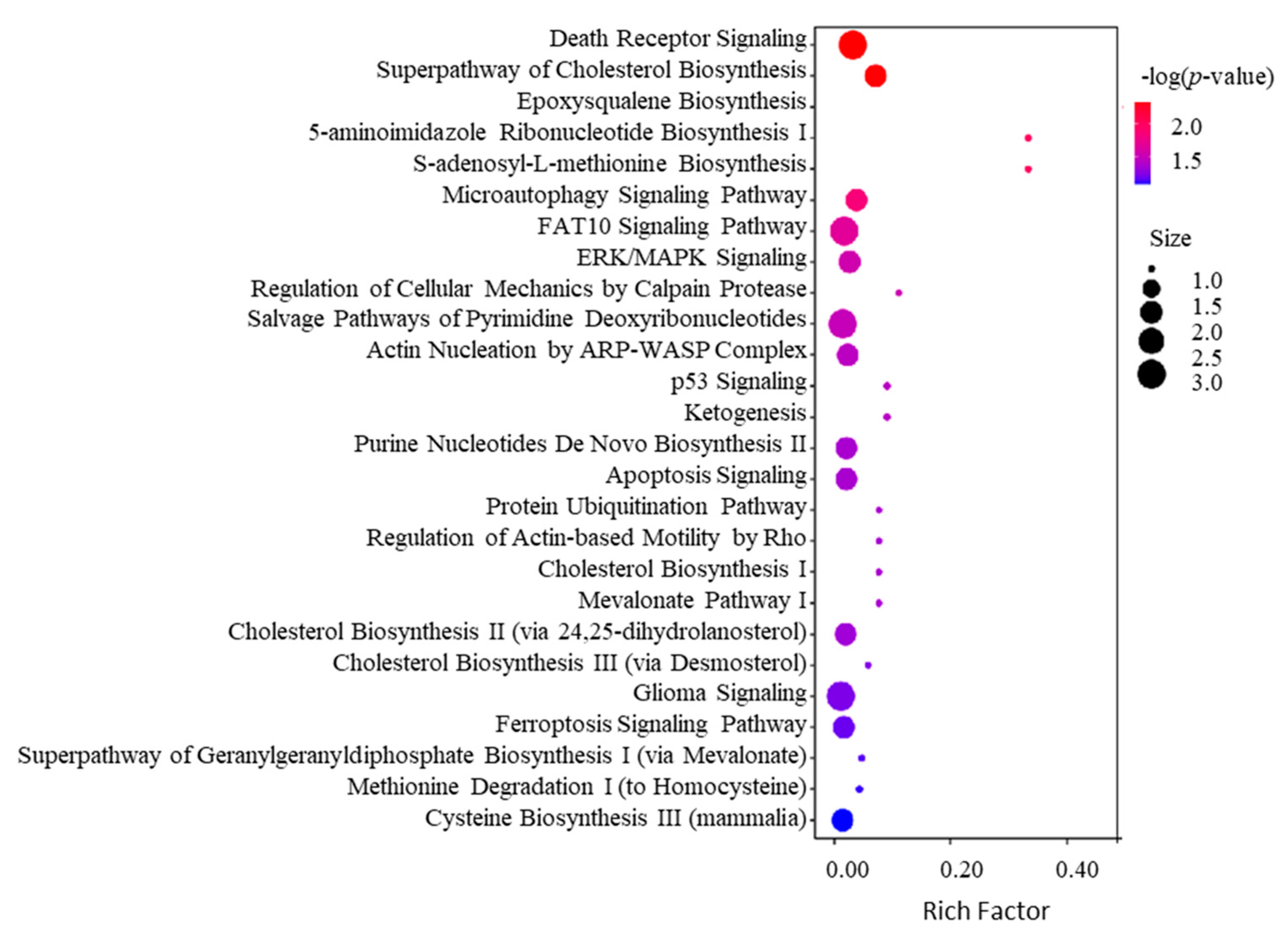
2.4. Bioinformatics Analysis
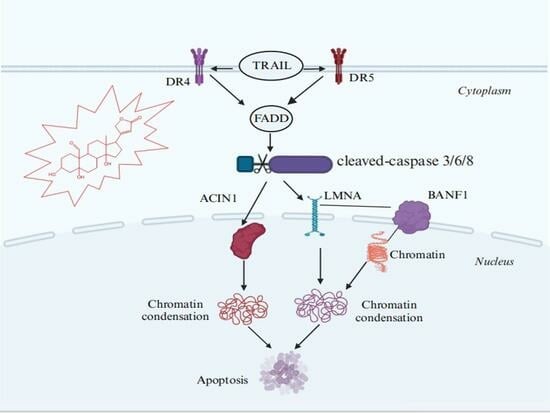
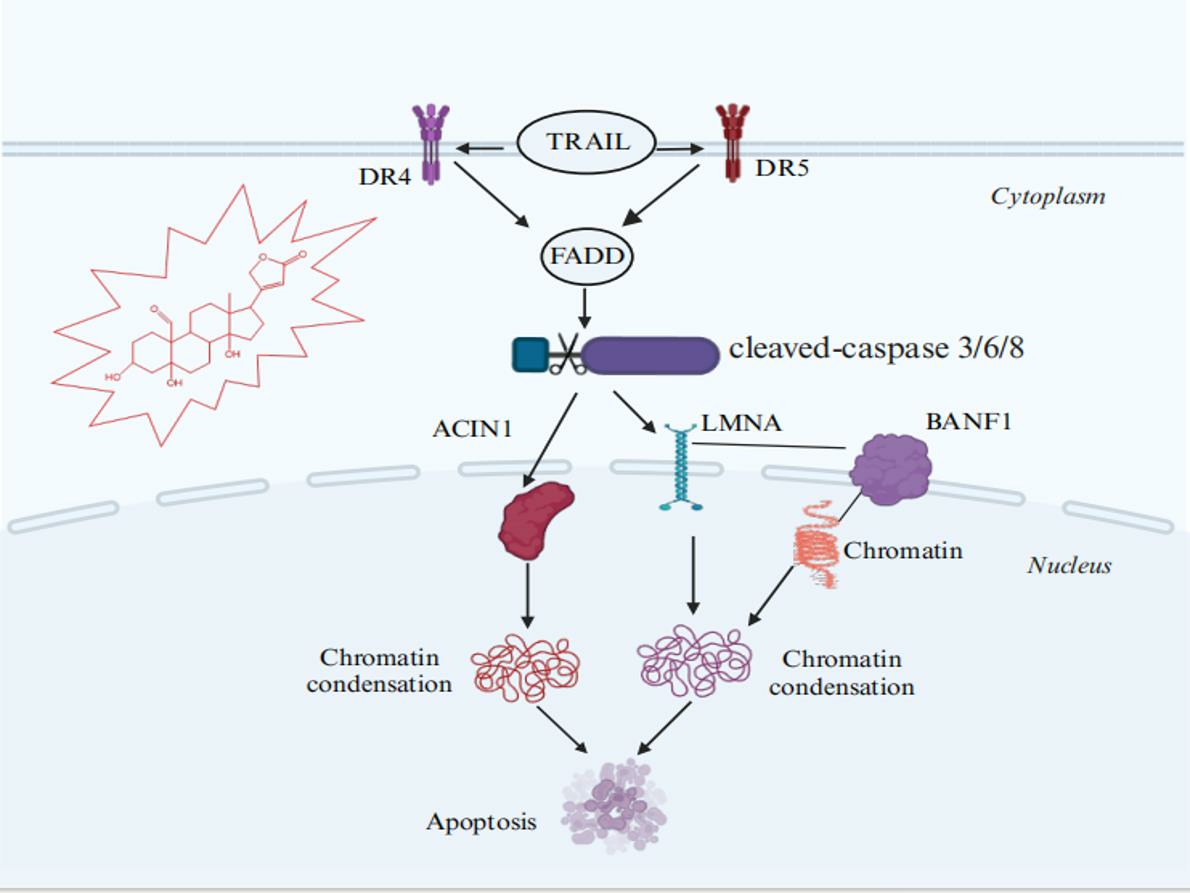
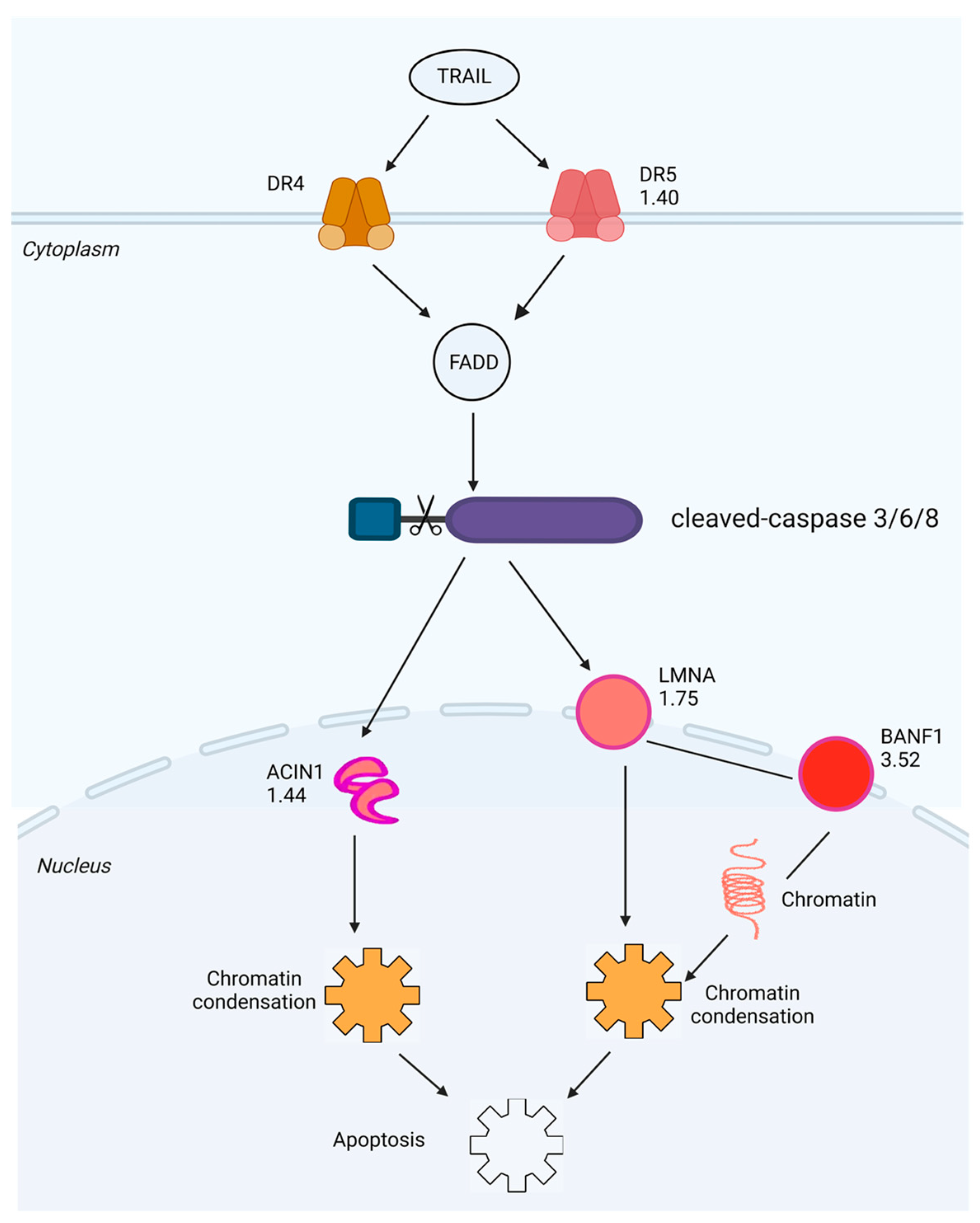
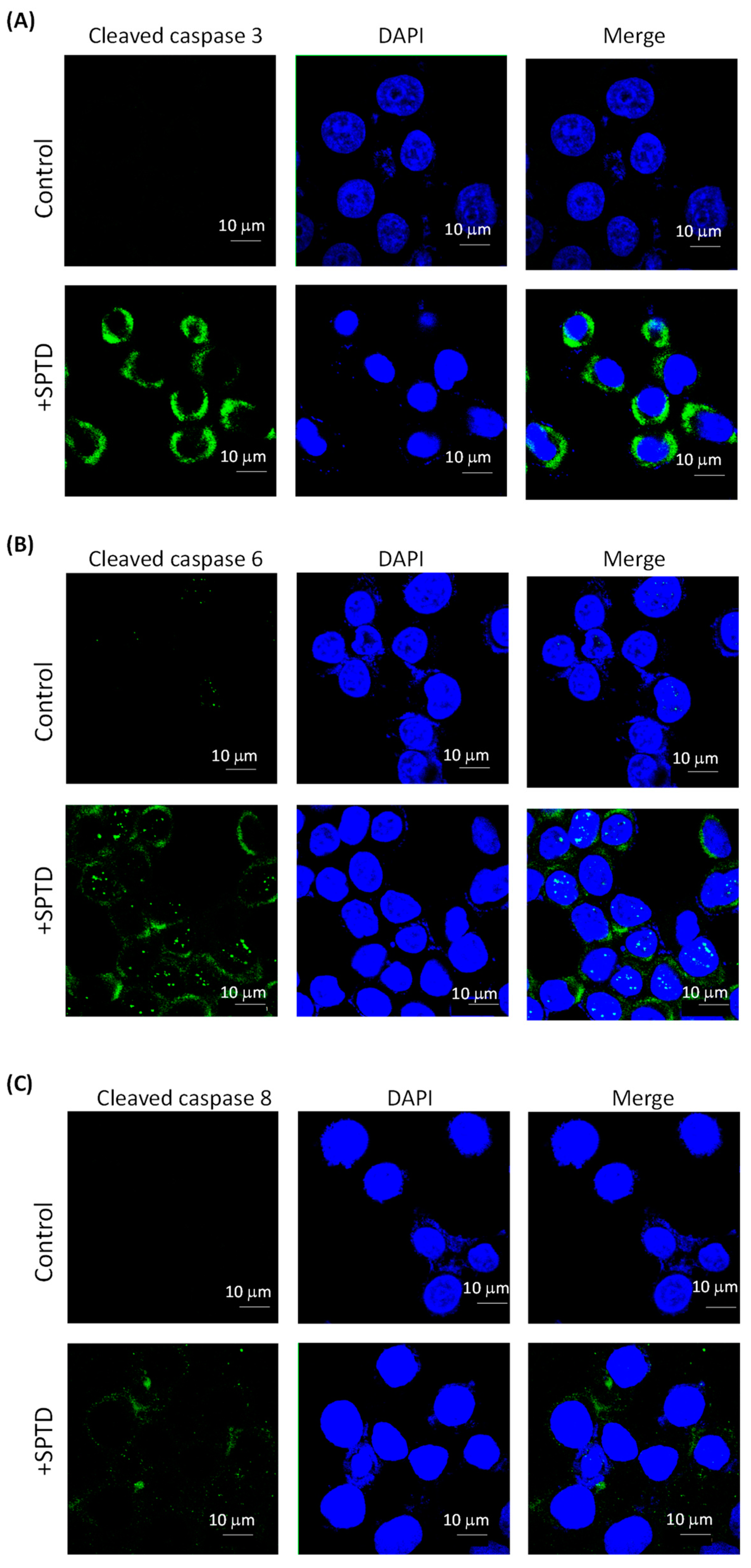
2.5. Investigation of SPTD-Induced Apoptosis via Death Receptor Signaling by Fluorescence Imaging
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Apoptosis Assay
4.5. Cell Cycle Assay
4.6. Protein Extraction
4.7. Protein Digestion
4.8. TMT Labeling
4.9. NanoLC-MS/MS Analysis
4.10. Protein Database Search
4.11. Bioinformatics Analysis
4.12. Immunofluorescence
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef]
- Thomford, N.; Senthebane, D.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef]
- Zhang, J.; Hansen, L.G.; Gudich, O.; Viehrig, K.; Lassen, L.M.M.; Schrübbers, L.; Adhikari, K.B.; Rubaszka, P.; Carrasquer-Alvarez, E.; Chen, L.; et al. A microbial supply chain for production of the anti-cancer drug vinblastine. Nature 2022, 609, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Zhang, Z.; Liao, F.; Jiang, T.; Tu, Y. The birth of artemisinin. Pharmacol. Ther. 2020, 216, 107658. [Google Scholar] [CrossRef]
- Kelly, R.A. Cardiac glycosides and congestive heart failure. Am. J. Cardiol. 1990, 65, 10E–16E; discussion 22E–23E. [Google Scholar] [CrossRef] [PubMed]
- Hancox, J.C.; Levi, A.J. Actions of the digitalis analogue strophanthidin on action potentials and L-type calcium current in single cells isolated from the rabbit atrioventricular node. Br. J. Pharmacol. 1996, 118, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- von Lewinski, D.; Bisping, E.; Elgner, A.; Kockskämper, J.; Pieske, B. Mechanistic insight into the functional and toxic effects of Strophanthidin in the failing human myocardium. Eur. J. Heart Fail. 2007, 9, 1086–1094. [Google Scholar] [CrossRef]
- Pan, L.; Zhang, Y.; Zhao, W.; Zhou, X.; Wang, C.; Deng, F. The cardiac glycoside oleandrin induces apoptosis in human colon cancer cells via the mitochondrial pathway. Cancer Chemother. Pharmacol. 2017, 80, 91–100. [Google Scholar] [CrossRef]
- Reddy, D.; Ghosh, P.; Kumavath, R. Strophanthidin Attenuates MAPK, PI3K/AKT/mTOR, and Wnt/β-Catenin Signaling Pathways in Human Cancers. Front. Oncol. 2020, 9, 1469. [Google Scholar] [CrossRef] [PubMed]
- Rix, U.; Superti-Furga, G. Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 2009, 5, 616–624. [Google Scholar] [CrossRef]
- Barglow, K.T.; Cravatt, B.F. Activity-based protein profiling for the functional annotation of enzymes. Nat. Methods 2007, 4, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Meissner, F.; Geddes-McAlister, J.; Mann, M.; Bantscheff, M. The emerging role of mass spectrometry-based proteomics in drug discovery. Nat. Rev. Drug Discov. 2022, 21, 637–654. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Ma, N.; Tian, J.; Shao, Y.; Zhu, B.; Wong, Y.K.; Liang, Z.; Zou, C.; Wang, J. Target identification of natural medicine with chemical proteomics approach: Probe synthesis, target fishing and protein identification. Signal Transduct. Target. Ther. 2020, 5, 72. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, C.-J.; Chia, W.N.; Loh, C.C.Y.; Li, Z.; Lee, Y.M.; He, Y.; Yuan, L.-X.; Lim, T.K.; Liu, M.; et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat. Commun. 2015, 6, 10111. [Google Scholar] [CrossRef]
- Ismail, H.M.; Barton, V.; Phanchana, M.; Charoensutthivarakul, S.; Wong, M.H.L.; Hemingway, J.; Biagini, G.A.; O’Neill, P.M.; Ward, S.A. Artemisinin activity-based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proc. Natl. Acad. Sci. USA 2016, 113, 2080–2085. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.Z.; Liang, Z.Q.; Qi, L.Y.; Tang, C.; Liu, X.K.; Tang, J.L.; Zhao, Y.; Zhang, Y.Y.; Fang, T.T.; Luo, Q.; et al. Baicalin Targets HSP70/90 to Regulate PKR/PI3K/AKT/eNOS Signaling Pathways. Molecules 2022, 27, 1432. [Google Scholar] [CrossRef]
- Hou, Y.Z.; Gan, T.L.; Fang, T.T.; Zhao, Y.; Luo, Q.; Liu, X.K.; Qi, L.Y.; Zhang, Y.Y.; Jia, F.F.; Han, J.J.; et al. G-quadruplex inducer/stabilizer pyridostatin targets SUB1 to promote cytotoxicity of a transplatinum complex. Nucleic. Acids Res. 2022, 50, 3070–3082. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, H.; Dou, Y.; Luo, Q.; Gu, L.; Liu, X.; Zhou, Q.; Han, J.; Wang, F. A Photoactivated Ru (II) Polypyridine Complex Induced Oncotic Necrosis of A549 Cells by Activating Oxidative Phosphorylation and Inhibiting DNA Synthesis as Revealed by Quantitative Proteomics. Int. J. Mol. Sci. 2023, 24, 7756. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, Q.; Zheng, W.; Wang, Z.; Lin, Y.; Zhang, E.; Lu, S.; Xiang, J.; Zhao, Y.; Wang, F. Luminescent cyclometallated platinum(II) complexes: Highly promising EGFR/DNA probes and dual-targeting anticancer agents. Inorg. Chem. Front. 2018, 5, 413–424. [Google Scholar] [CrossRef]
- Samwer, M.; Schneider, M.W.G.; Hoefler, R.; Schmalhorst, P.S.; Jude, J.G.; Zuber, J.; Gerlich, D.W. DNA Cross-Bridging Shapes a Single Nucleus from a Set of Mitotic Chromosomes. Cell 2017, 170, 956–972.e923. [Google Scholar] [CrossRef] [PubMed]
- Bolderson, E.; Burgess, J.T.; Li, J.; Gandhi, N.S.; Boucher, D.; Croft, L.V.; Beard, S.; Plowman, J.J.; Suraweera, A.; Adams, M.N.; et al. Barrier-to-autointegration factor 1 (Banf1) regulates poly [ADP-ribose] polymerase 1 (PARP1) activity following oxidative DNA damage. Nat. Commun. 2019, 10, 5501. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Luo, G.-Z.; Zhang, Z.; Wang, X.; Zhou, T.; Cui, Y.; Sha, J.; Huang, X.; Guerrero, L.; Xie, P.; et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. eLife 2017, 6, e31311. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.-S.; Hao, Y.-J.; Sun, B.-F.; Sun, H.-Y.; Li, A.; Ping, X.-L.; Lai, W.-Y.; et al. Nuclear m 6 A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wan, R.; Zou, Z.; Lao, L.; Shao, G.; Zheng, Y.; Tang, L.; Yuan, Y.; Ge, Y.; He, C.; et al. O-GlcNAcylation determines the translational regulation and phase separation of YTHDF proteins. Nat. Cell Biol. 2023, 25, 1676–1690. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.; Kumavath, R.; Tan, T.Z.; Ampasala, D.R.; Kumar, A.P. Peruvoside targets apoptosis and autophagy through MAPK Wnt/β-catenin and PI3K/AKT/mTOR signaling pathways in human cancers. Life Sci. 2020, 241, 117147. [Google Scholar] [CrossRef] [PubMed]
- Hymowitz, S.G.; Christinger, H.W.; Fuh, G.; Ultsch, M.; O’Connell, M.; Kelley, R.F.; Ashkenazi, A.; De Vos, A.M. Triggering Cell Death. Mol. Cell 1999, 4, 563–571. [Google Scholar] [CrossRef]
- Mongkolsapaya, J.; Grimes, J.M.; Chen, N.; Xu, X.-N.; Stuart, D.I.; Jones, E.Y.; Screaton, G.R. Structure of the TRAIL–DR5 complex reveals mechanisms conferring specificity in apoptotic initiation. Nat. Struct. Biol. 1999, 6, 1048–1053. [Google Scholar]
- Bonne, G.; Barletta, M.R.D.; Varnous, S.; Bécane, H.-M.; Hammouda, E.-H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.-A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Digitalis Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N. Engl. J. Med. 1997, 336, 525–533. [Google Scholar] [CrossRef]
- Prassas, I.; Diamandis, E.P. Novel therapeutic applications of cardiac glycosides. Nat. Rev. Drug Discov. 2008, 7, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, O. Growth inhibitory effect of cardiac glycosides and aglycones on neoplastic cells: In vitro and in vivo studies. Gan 1967, 58, 521–528. [Google Scholar] [PubMed]
- Slingerland, M.; Cerella, C.; Guchelaar, H.J.; Diederich, M.; Gelderblom, H. Cardiac glycosides in cancer therapy: From preclinical investigations towards clinical trials. Investig. New Drugs 2013, 31, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Stenkvist, B. Is digitalis a therapy for breast carcinoma? Oncol. Rep. 1999, 6, 493–496. [Google Scholar] [CrossRef]
- Chao, M.-W.; Chen, T.-H.; Huang, H.-L.; Chang, Y.-W.; Huangfu, W.-C.; Lee, Y.-C.; Teng, C.-M.; Pan, S.-L. Lanatoside C, a cardiac glycoside, acts through protein kinase Cδ to cause apoptosis of human hepatocellular carcinoma cells. Sci. Rep. 2017, 7, 46134. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.; Kumavath, R.; Ghosh, P.; Barh, D. Lanatoside C Induces G2/M Cell Cycle Arrest and Suppresses Cancer Cell Growth by Attenuating MAPK, Wnt, JAK-STAT, and PI3K/AKT/mTOR Signaling Pathways. Biomolecules 2019, 9, 792. [Google Scholar] [CrossRef]
- Feng, Q.; Leong, W.S.; Liu, L.; Chan, W.-I. Peruvoside, a Cardiac Glycoside, Induces Primitive Myeloid Leukemia Cell Death. Molecules 2016, 21, 534. [Google Scholar] [CrossRef]
- Hossan, M.S.; Chan, Z.-Y.; Collins, H.M.; Shipton, F.N.; Butler, M.S.; Rahmatullah, M.; Lee, J.B.; Gershkovich, P.; Kagan, L.; Khoo, T.-J.; et al. Cardiac glycoside cerberin exerts anticancer activity through PI3K/AKT/mTOR signal transduction inhibition. Cancer Lett. 2019, 453, 57–73. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Jin, Z.; El-Deiry, W.S. Overview of cell death signaling pathways. Cancer Biol. Ther. 2005, 4, 147–171. [Google Scholar] [CrossRef]
- Wu, G.S. TRAIL as a target in anti-cancer therapy. Cancer Lett. 2009, 285, 1–5. [Google Scholar] [CrossRef]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Lowry, S.F.; Cerami, A. Cachetin/TNF-alpha in septic shock and septic adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1988, 138, 1377–1379. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Lowry, S.F.; Fahey, T.J., 3rd; Albert, J.D.; Fong, Y.; Hesse, D.; Beutler, B.; Manogue, K.R.; Calvano, S.; Wei, H.; et al. Cachectin/tumor necrosis factor induces lethal shock and stress hormone responses in the dog. Surg. Gynecol. Obs. 1987, 164, 415–422. [Google Scholar]
- Lemke, J.; Von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor–related apoptosis–inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Lim, B.; Allen, J.E.; Prabhu, V.V.; Talekar, M.K.; Finnberg, N.K.; El-Deiry, W.S. Targeting TRAIL in the treatment of cancer: New developments. Expert Opin. Ther. Targets 2015, 19, 1171–1185. [Google Scholar] [CrossRef]
- Fox, J.L.; Hughes, M.A.; Meng, X.; Sarnowska, N.A.; Powley, I.R.; Jukes-Jones, R.; Dinsdale, D.; Ragan, T.J.; Fairall, L.; Schwabe, J.W.R.; et al. Cryo-EM structural analysis of FADD:Caspase-8 complexes defines the catalytic dimer architecture for coordinated control of cell fate. Nat. Commun. 2021, 12, 819. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.-T.; Yue, P.; Wang, D.; Tong, J.-S.; Chen, Z.G.; Khuri, F.R.; Sun, S.-Y. Suppression of death receptor 5 enhances cancer cell invasion and metastasis through activation of caspase-8/TRAF2-mediated signaling. Oncotarget 2015, 6, 41324–41338. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Yuan, C.; Wei, F.; Wang, G.; Zhang, J.; Bellail, A.C.; Zhang, Z.; Olson, J.J.; Hao, C. Cisplatin Restores TRAIL Apoptotic Pathway in Glioblastoma-Derived Stem Cells through Up-regulation of DR5 and Down-regulation of c-FLIP. Cancer Investig. 2011, 29, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Altucci, L.; Rossin, A.; Raffelsberger, W.; Reitmair, A.; Chomienne, C.; Gronemeyer, H. Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat. Med. 2001, 7, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2005, 11, 77–84. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, N. On the TRAIL of death. Nat. Rev. Cancer 2005, 5, 89. [Google Scholar] [CrossRef]
- Wyllie, A.H.; Beattie, G.J.; Hargreaves, A.D. Chromatin changes in apoptosis. Histochem. J. 1981, 13, 681–692. [Google Scholar] [CrossRef]
- Sahara, S.; Aoto, M.; Eguchi, Y.; Imamoto, N.; Yoneda, Y.; Tsujimoto, Y. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature 1999, 401, 168–173. [Google Scholar] [CrossRef]
- Rappsilber, J.; Ryder, U.; Lamond, A.I.; Mann, M. Large-scale proteomic analysis of the human splicesome. Genome Res. 2002, 12, 1231–1245. [Google Scholar] [CrossRef]
- Zhou, Z.; Licklider, L.J.; Gygi, S.P.; Reed, R. Comprehensive proteomic analysis of the human spliceosome. Nature 2002, 419, 182–185. [Google Scholar] [CrossRef]
- Earnshaw, W.C.; Martins, L.M.; Kaufmann, S.H. Mammalian Caspases: Structure, Activation, Substrates, and Functions During Apoptosis. Annu. Rev. Biochem. 1999, 68, 383–424. [Google Scholar] [CrossRef]
- Kidd, V.J. Proteolytic Activities that Mediate Apoptosis. Annu. Rev. Physiol. 1998, 60, 533–573. [Google Scholar] [CrossRef]
- Li, W.; Bai, X.; Li, J.; Zhao, Y.; Liu, J.; Zhao, H.; Liu, L.; Ding, M.; Wang, Q.; Shi, F.-Y.; et al. The nucleoskeleton protein IFFO1 immobilizes broken DNA and suppresses chromosome translocation during tumorigenesis. Nat. Cell Biol. 2019, 21, 1273–1285. [Google Scholar] [CrossRef]
- Zheng, R.; Ghirlando, R.; Lee, M.S.; Mizuuchi, K.; Krause, M.; Craigie, R. Barrier-to-autointegration factor (BAF) bridges DNA in a discrete, higher-order nucleoprotein complex. Proc. Natl. Acad. Sci. USA 2000, 97, 8997–9002. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 1619–1633. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Yoshizumi, M.; Kyotani, Y.; Zhao, J.; Nagayama, K.; Ito, S.; Tsuji, Y.; Ozawa, K. The Role of Big Mitogen-Activated Protein Kinase 1 (BMK1)/Extracellular Signal-Regulated Kinase 5 (ERK5) in the Pathogenesis and Progression of Atherosclerosis. J. Pharmacol. Sci. 2012, 120, 259–263. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Hendrikse, C.S.E.; Theelen, P.M.M.; van der Ploeg, P.; Westgeest, H.M.; Boere, I.A.; Thijs, A.M.J.; Ottevanger, P.B.; van de Stolpe, A.; Lambrechts, S.; Bekkers, R.L.M.; et al. The potential of RAS/RAF/MEK/ERK (MAPK) signaling pathway inhibitors in ovarian cancer: A systematic review and meta-analysis. Gynecol. Oncol. 2023, 171, 83–94. [Google Scholar] [CrossRef]
- Kao, T.J.; Wu, C.C.; Phan, N.N.; Liu, Y.H.; Ta, H.D.K.; Anuraga, G.; Wu, Y.F.; Lee, K.H.; Chuang, J.Y.; Wang, C.Y. Prognoses and genomic analyses of proteasome 26S subunit, ATPase (PSMC) family genes in clinical breast cancer. Aging 2021, 13, 18946–18977. [Google Scholar] [CrossRef]
- Perissi, V.; Aggarwal, A.; Glass, C.K.; Rose, D.W.; Rosenfeld, M.G. A Corepressor/Coactivator Exchange Complex Required for Transcriptional Activation by Nuclear Receptors and Other Regulated Transcription Factors. Cell 2004, 116, 511–526. [Google Scholar] [CrossRef]
- Wang, R.; Wei, B.; Wei, J.; Li, Z.; Tian, Y.; Du, C. Caspase-related apoptosis genes in gliomas by RNA-seq and bioinformatics analysis. J. Clin. Neurosci. 2016, 33, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Jongsma, M.L.M.; Berlin, I.; Wijdeven, R.H.M.; Janssen, L.; Janssen, G.M.C.; Garstka, M.A.; Janssen, H.; Mensink, M.; van Veelen, P.A.; Spaapen, R.M.; et al. An ER-Associated Pathway Defines Endosomal Architecture for Controlled Cargo Transport. Cell 2016, 166, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef]
- Sánchez-Martín, P.; Saito, T.; Komatsu, M. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-Based Polyubiquitination and p62-Dependent Aggregation of Caspase-8 Mediate Extrinsic Apoptosis Signaling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef]
- Emanuele, S.; Lauricella, M.; D’Anneo, A.; Carlisi, D.; De Blasio, A.; Di Liberto, D.; Giuliano, M. p62: Friend or Foe? Evidences for OncoJanus and NeuroJanus Roles. Int. J. Mol. Sci. 2020, 21, 5029. [Google Scholar] [CrossRef]
- Heino, J. The collagen family members as cell adhesion proteins. BioEssays 2007, 29, 1001–1010. [Google Scholar] [CrossRef]
- Martins Cavaco, A.C.; Dâmaso, S.; Casimiro, S.; Costa, L. Collagen biology making inroads into prognosis and treatment of cancer progression and metastasis. Cancer Metastasis Rev. 2020, 39, 603–623. [Google Scholar] [CrossRef]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 309. [Google Scholar] [CrossRef]
- Ma, H.-P.; Chang, H.-L.; Bamodu, O.A.; Yadav, V.K.; Huang, T.-Y.; Wu, A.T.H.; Yeh, C.-T.; Tsai, S.-H.; Lee, W.-H. Collagen 1A1 (COL1A1) Is a Reliable Biomarker and Putative Therapeutic Target for Hepatocellular Carcinogenesis and Metastasis. Cancers 2019, 11, 786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Yano, H.; Sasaki, T.; Matsuo, N.; Yoshioka, H. The pro-α1(V) collagen gene (Col5a1) is coordinately regulated by miR-29b with core promoter in cultured cells. Connect. Tissue Res. 2018, 59, 263–273. [Google Scholar] [CrossRef]
- Liu, W.; Wei, H.; Gao, Z.; Chen, G.; Liu, Y.; Gao, X.; Bai, G.; He, S.; Liu, T.; Xu, W.; et al. COL5A1 may contribute the metastasis of lung adenocarcinoma. Gene 2018, 665, 57–66. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, J.; Wang, F.; Xu, X.; Li, X.; Guan, W.; Men, T.; Xu, G. Overexpressed COL5A1 is correlated with tumor progression, paclitaxel resistance, and tumor-infiltrating immune cells in ovarian cancer. J. Cell. Physiol. 2021, 236, 6907–6919. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-F.; Chang, Y.-C.; Li, C.-H.; Chan, M.-H.; Chen, C.-L.; Tsai, W.-C.; Hsiao, M. Type V collagen alpha 1 chain promotes the malignancy of glioblastoma through PPRC1-ESM1 axis activation and extracellular matrix remodeling. Cell Death Discov. 2021, 7, 313. [Google Scholar] [CrossRef]
- Zheng, W.; Yang, P.; Sun, C.; Zhang, Y. Comprehensive comparison of sample preparation workflows for proteomics. Mol. Omics 2022, 18, 555–567. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, X.; Gu, L.; Zeng, F.; Liu, X.; Zhou, Y.; Dou, Y.; Han, J.; Zhao, Y.; Zhang, Y.; Luo, Q.; et al. Strophanthidin Induces Apoptosis of Human Lung Adenocarcinoma Cells by Promoting TRAIL-DR5 Signaling. Molecules 2024, 29, 877. https://doi.org/10.3390/molecules29040877
Tian X, Gu L, Zeng F, Liu X, Zhou Y, Dou Y, Han J, Zhao Y, Zhang Y, Luo Q, et al. Strophanthidin Induces Apoptosis of Human Lung Adenocarcinoma Cells by Promoting TRAIL-DR5 Signaling. Molecules. 2024; 29(4):877. https://doi.org/10.3390/molecules29040877
Chicago/Turabian StyleTian, Xiao, Liangzhen Gu, Fangang Zeng, Xingkai Liu, Yang Zhou, Yang Dou, Juanjuan Han, Yao Zhao, Yanyan Zhang, Qun Luo, and et al. 2024. "Strophanthidin Induces Apoptosis of Human Lung Adenocarcinoma Cells by Promoting TRAIL-DR5 Signaling" Molecules 29, no. 4: 877. https://doi.org/10.3390/molecules29040877